Comparing the evolutionary dynamics of predominant SARS-CoV-2 virus lineages co-circulating in Mexico
- Consorcio Mexicano de Vigilancia Genómica (CoViGen-Mex), Mexico
- Programa de Investigadoras e Investigadores por México, Consejo Nacional de Ciencia y Tecnología, Mexico
- Departamento de Ingeniería Genética, CINVESTAV-Unidad Irapuato, Mexico
- Department of Biology, University of Oxford, United Kingdom
- Spatial Epidemiology Lab (SpELL), Université Libre de Bruxelles, Belgium
- Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Belgium
- Centro de Investigación en Enfermedades Infecciosas, Instituto Nacional de Enfermedades Respiratorias, Mexico
- Department of Pathobiology, Royal Veterinary College, United Kingdom
- Instituto Nacional de Medicina Genómica, Mexico
- Astronomer LTD, Mexico
- Escuela Nacional de Estudios Superiores, Universidad Nacional Autónoma de México, Mexico
- Departamento de Ciencias de la Computación, CINVESTAV-IPN, Mexico
- Laboratorio de expresión génica y desarrollo en hongos, CINVESTAV-Unidad Irapuato, Mexico
- Unidad Universitaria de Secuenciación Masiva y Bioinformática, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Mexico
- Coordinación de Calidad de Insumos y Laboratorios Especializados, Instituto Mexicano del Seguro Social, Mexico
- Facultad de Medicina y Centro de Investigación en Ciencias de la Salud y Biomedicina, Universidad Autónoma de San Luis Potosí, Mexico
- Centro de Investigación en Alimentación y Desarrollo-CIAD, Unidad Regional Mazatlán en Acuicultura y Manejo Ambiental, Mexico
- Posgrado en Ciencias Genómicas, Universidad Autónoma de la Ciudad de México, Mexico
- Departamento de Genética del Desarrollo y Fisiología Molecular, Universidad Nacional Autónoma de México, Mexico
- Facultad de Ciencias, Universidad Nacional Autónoma de Méxic, Mexico
Figures
Figure 1 with 2 supplements

Overview of the SARS-CoV-2 epidemic in Mexico.
(a) Time-scaled phylogeny of representative SARS-CoV-2 genomes from Mexico within a global context, highlighting the phylogenetic positioning of B.1.1.222, B.1.1.519, B.1.1.7, P.1, and B.1.617.2 sequences. Lineage B.1.1.222 is shown in light green, B.1.1.519 in yellow, P.1 in red (Gamma), B.1.1.7 (Alpha) in dark green, and B.1.617.2 (Delta) in teal (b) The epidemic curve for COVID-19 in Mexico from January 2020 up to November 2021, showing the average number of daily cases (red line) and associated excess mortality (represented by a punctuated grey curve, denoting weekly average values). The peak of the first (July 2020), the second (January 2021), and the third wave (August 2021) of infection are highlighted in yellow shadowing. The dashed red line corresponds to the start date national vaccination campaign (December 2020), whilst the dashed black line represents the implementation date of a systematic genome sampling and sequencing scheme for the surveillance of SARS-CoV-2 in Mexico (February 2021). The period for the implementation of non-pharmaceutical interventions at national scale is highlighted in grey shadowing. The lower panel represents the genome sampling frequency (defined here as the proportion of viral genomes assigned to a specific lineage, relative to the proportion of viral genomes assigned to any other virus lineage in a given time point) of dominant virus lineages detected in the country during the first year of the epidemic. Lineages displaying a lower sampling frequency are jointly shown in purple. (c) Heatmap displaying the volume of trips into a given state from any other state recorded from January 2020 up to November 2021 derived from anonymized mobile device geolocated and time-stamped data.
Figure 1—figure supplement 1
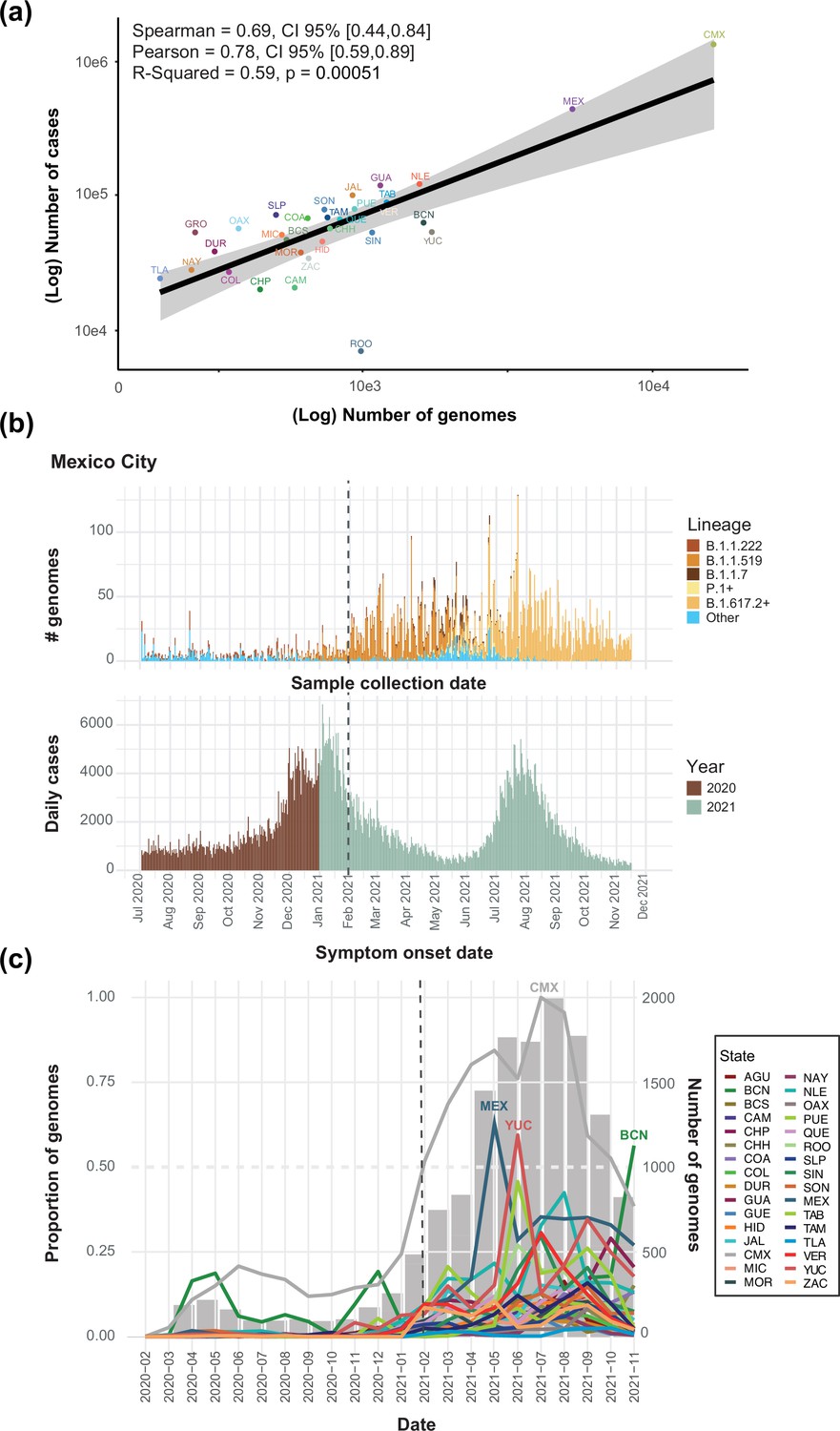
Cumulative number of genome sequences generated per state (data available up to March 2022).
(a) A significant correlation between the cumulative number of cases per state versus the number of viral genome sequences available per state is observed, indicating the estimated Spearman/Pearson coefficients and associated 95% confidence intervals (CI). Mexico City (CMX) displays the highest number of genomes sequenced relative to the reported number of cases. (b) A comparison between the total number of genomes sequenced from Mexico City (CMX) assigned to the lineages of interest plotted against collection date, and the number of daily cases reported for Mexico City (CMX) with symptom onset dates ranging from July 2020 up to November 2021 (colored according to the year of sample collection). The dashed black line represents the implementation date of a broader viral genome sampling and sequencing scheme for the surveillance of SARS-CoV-2 in Mexico (February 2021). (c) The cumulative proportion of genomes sequences generated per state across time (data from February 2020 up to November 2021). The states that generated a proportion of genome sequences above 0.50 (represented by a dashed grey line, relative to other states) are indicated: Mexico City (CMX-grey), State of Mexico (MEX-light blue), Yucatan (YUC-red) and Baja California Norte (BCN-dark green). Once more, the dashed black line represents the implementation date of a broader viral genome sampling and sequencing scheme for the surveillance of SARS-CoV-2 in Mexico (February 2021).
Figure 1—figure supplement 2
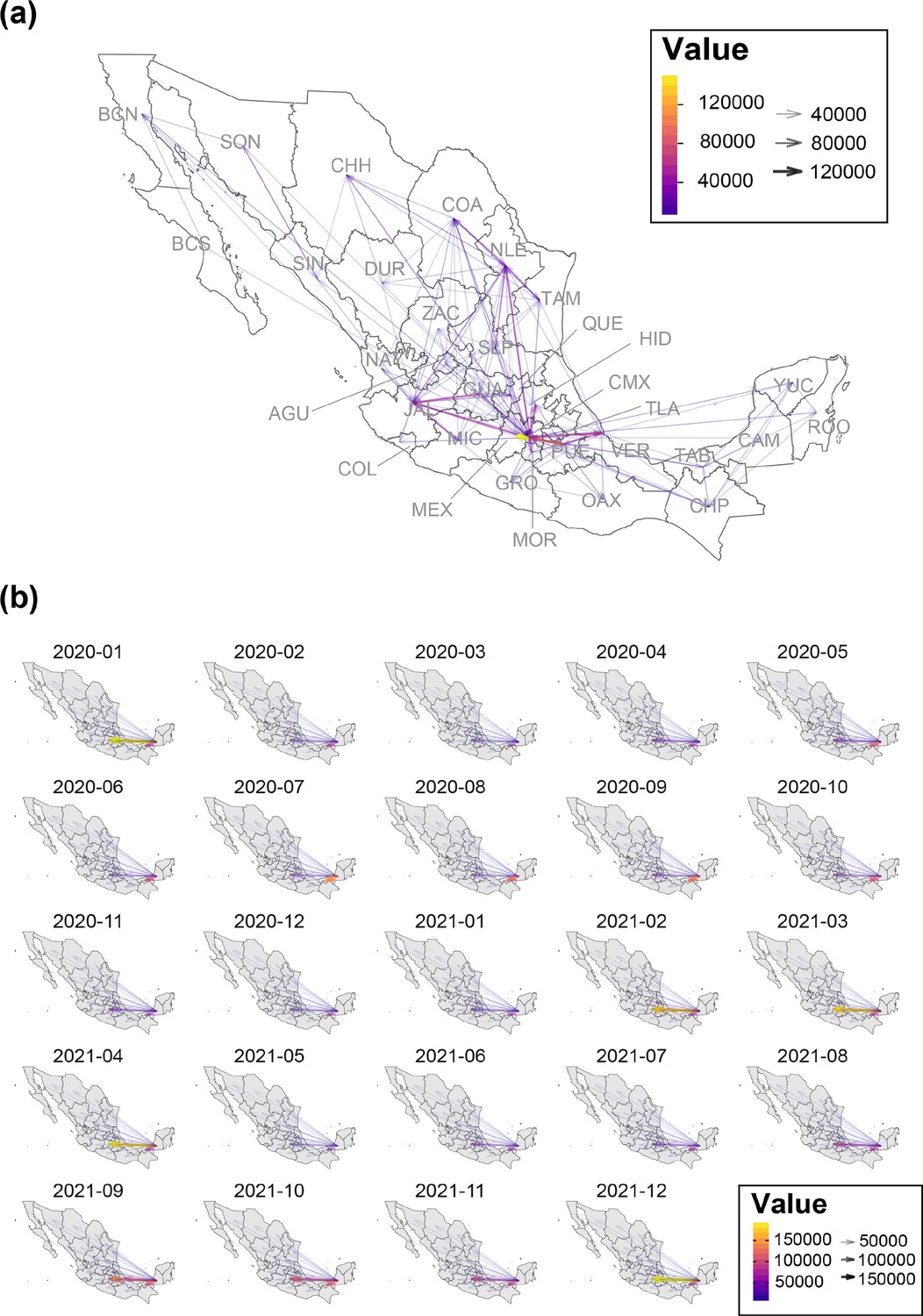
Mean interstate connectivity recorded between 2021 and 2022.
(a) Map graph showing the mean intra-state connectivity recorded within national territory, derived from anonymized mobile device locations collected between 01/01/2020 and 31/12/2021. Values above 4E4 are indicated using a color gradient, whilst arrow thickness within the map represents the total number of bidirectional movements between states. (b) Maps graphs showing the mean inter-state connectivity between the southern region of the country (represented by the states of Yucatán, Quintana Roo, Chiapas and Campeche) and the remaining 28 states (recorded between 01/01/2020 and 31/12/2021). Again, values above 10–4 are indicated using a color gradient, whilst arrow thickness within the map represents the total number of bidirectional movements between states.
Figure 2
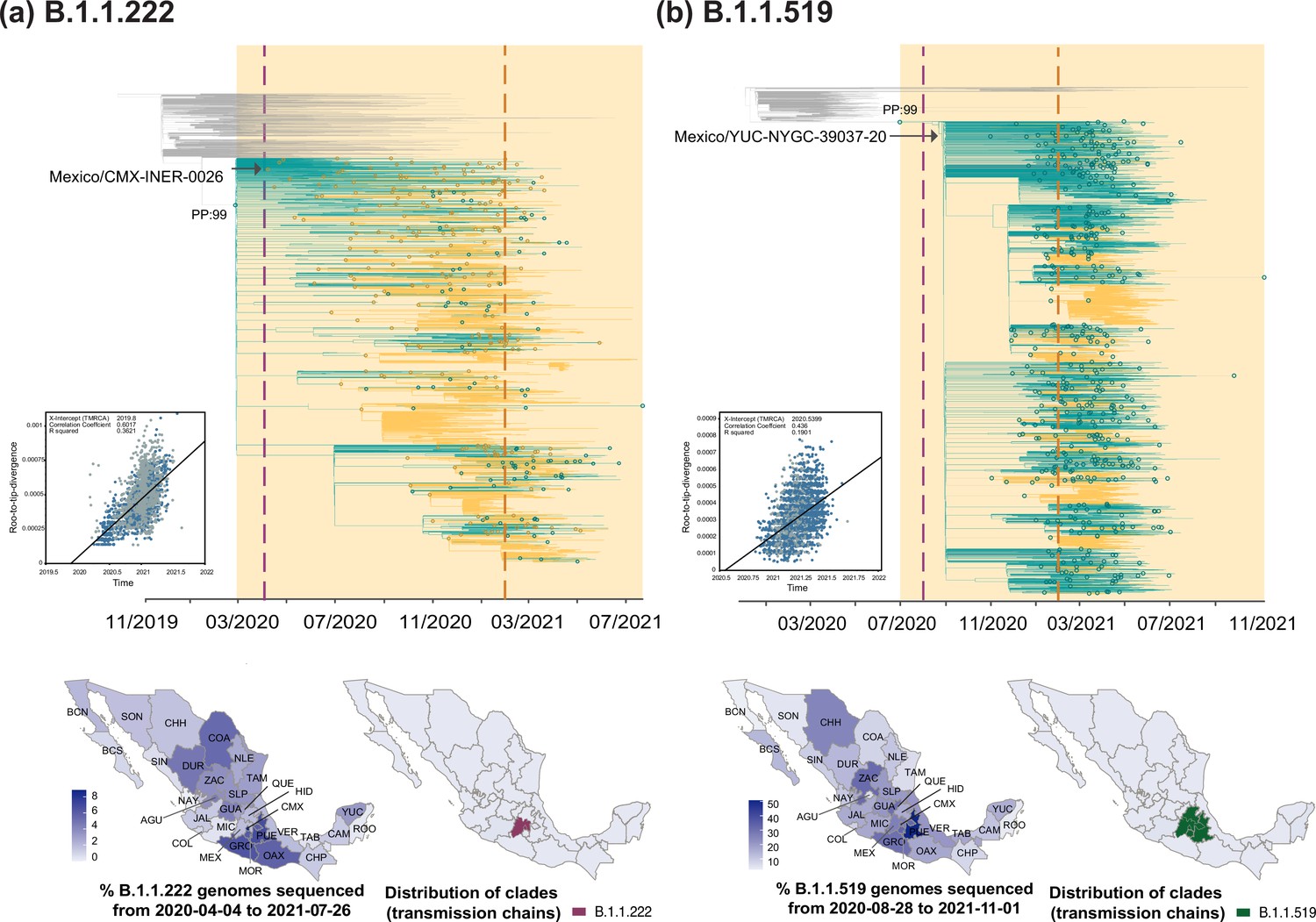
Time-scaled phylogenetic analyses for the B.1.1.222 and B.1.1.519 lineage.
Maximum clade credibility (MCC) trees for the (a) B.1.1.222 and (b) B.1.1.519 lineages, in which clades corresponding to distinct introduction events into Mexico are highlighted. Nodes shown as outline circles correspond to the most recent common ancestor (MRCA) for clades representing independent re-introduction events into Mexico (in teal) or from the USA (in ochre). Based on the earliest and latest MRCAs, the estimated circulation period for each lineage is highlighted in yellow shadowing. The dashed purple line represents the date of the earliest viral genome sampled from Mexico, while its position in the tree indicated. The dashed yellow line represents the implementation date of a systematic virus genome sampling and sequencing scheme for the surveillance of SARS-CoV-2 in Mexico. The corresponding root-to-tip regression plots for each tree are shown, in which genomes sampled from Mexico are shown in blue, whilst genomes sampled elsewhere are shown in grey. Map graphs on the left show the cumulative proportion of genomes sampled across states per lineage of interest, corresponding to the period of circulation of the given lineage (relative to the total number of genomes taken from GISAID, corresponding to raw data before subsampling). Maps on the right represent the geographic distribution of the clades identified.
Figure 3 with 2 supplements
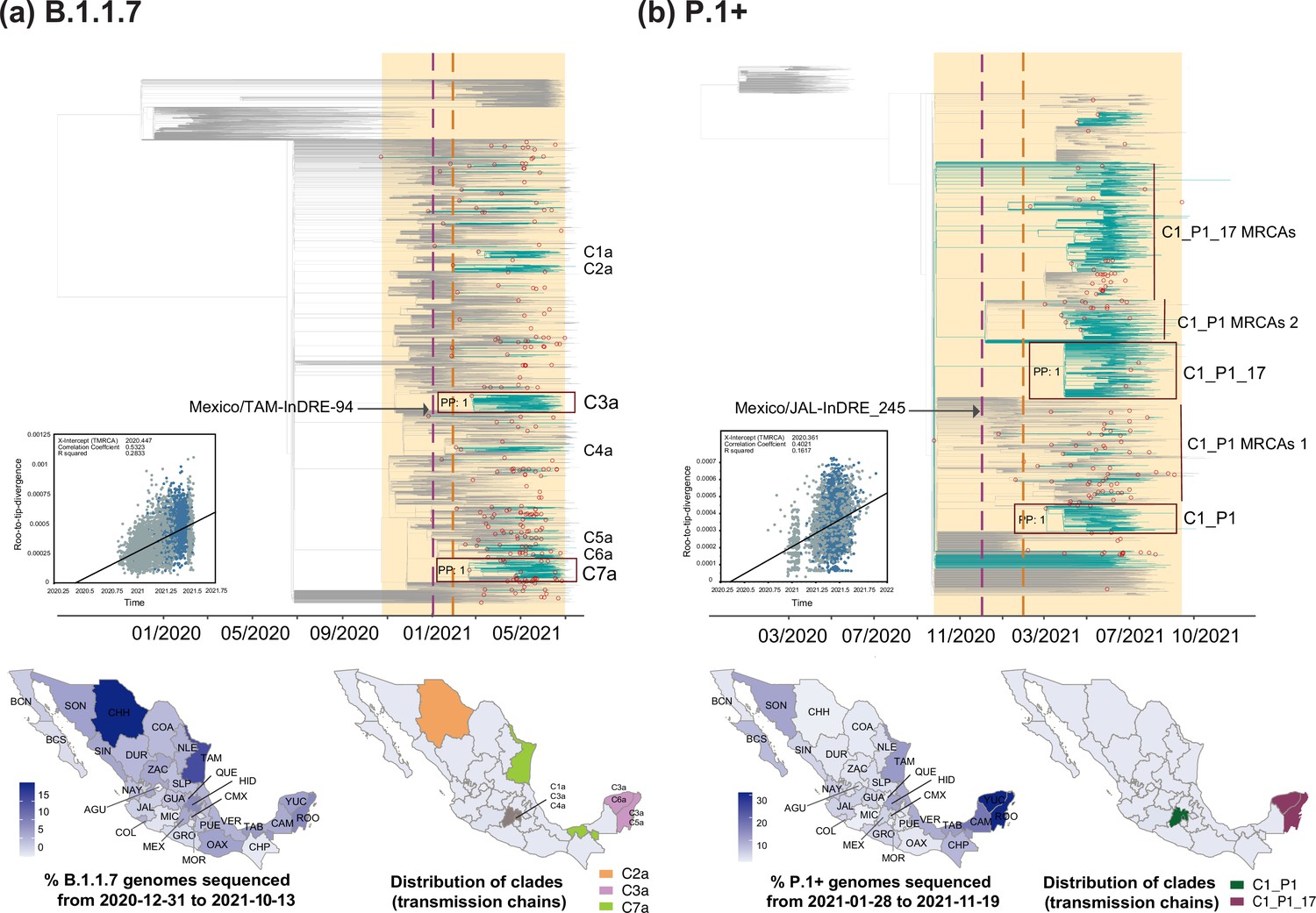
Time-scaled phylogenetic analyses for the B.1.1.7 and P.1 lineages.
Maximum clade credibility (MCC) trees for the (a) B.1.1.7 and the (b) P.1 lineages, in which major clades identified as distinct introduction events into Mexico are highlighted. Nodes shown as red outline circles correspond to the most recent common ancestor (MRCA) for clades representing independent introduction events into Mexico. Based on the earliest and latest MRCAs, the estimated circulation period for each lineage is highlighted in yellow shadowing. The dashed purple line represents the date of the earliest viral genome sampled from Mexico, while its position in the tree indicated. The dashed yellow line represents the implementation date of a systematic virus genome sampling and sequencing scheme for the surveillance of SARS-CoV-2 in Mexico. The corresponding root-to-tip regression plots for each tree are shown, in which genomes sampled from Mexico are shown in blue, whilst genomes sampled elsewhere are shown in grey. Map graphs on the left show the cumulative proportion of genomes sampled across states per lineage of interest, corresponding to the period of circulation of the given lineage (relative to the total number of genomes taken from GISAID, corresponding to raw data before subsampling). Maps on the right represent the geographic distribution of the clades identified.
Figure 3—figure supplement 1

Largest ‘Mexico’ clades within the B.1.1.7 MCC tree.
Figure 3—figure supplement 2
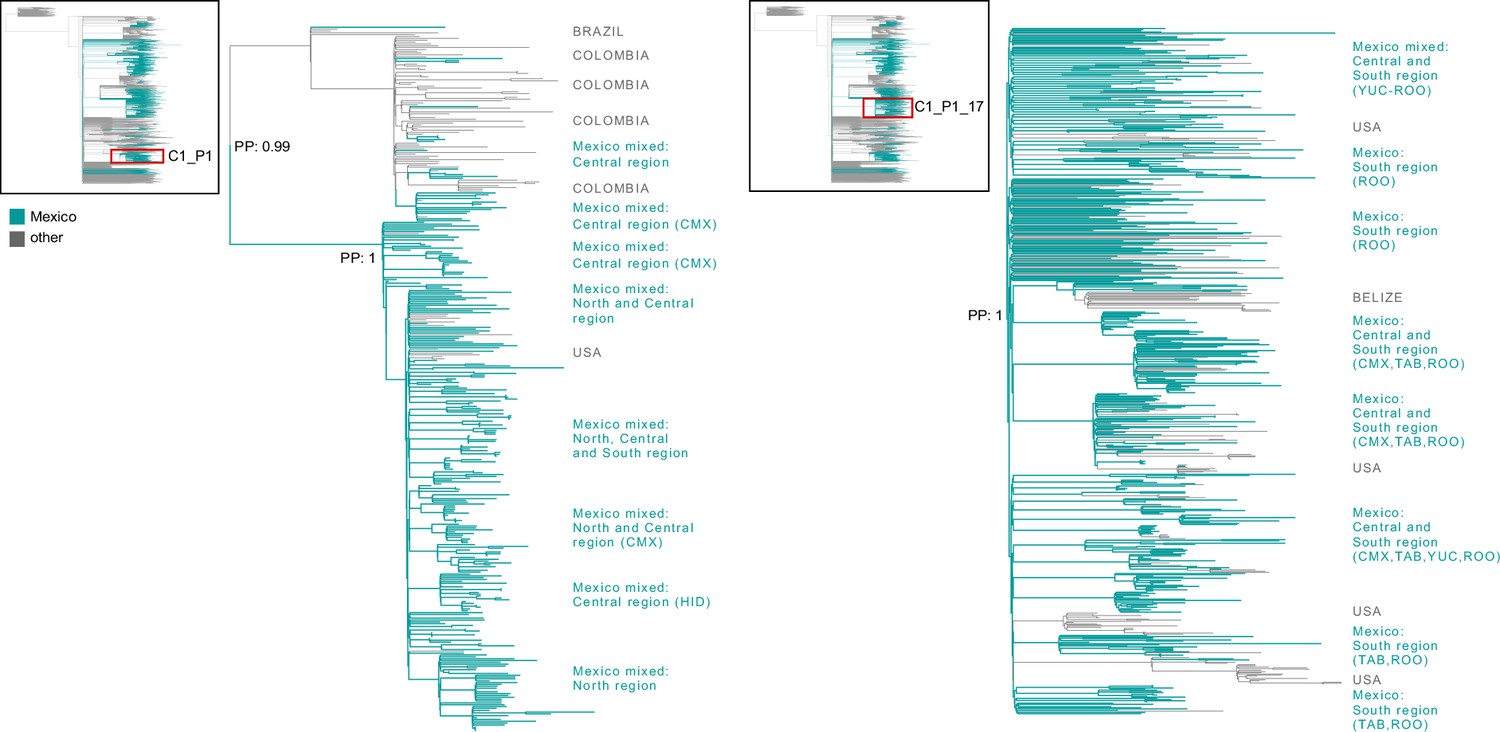
Largest ‘Mexico’ clades within the P.1+MCC tree.
Figure 4 with 2 supplements

Time-scaled and phylogeographic analysis for the B.1.617.2 lineage.
Maximum clade credibility (MCC) tree for the B.1.617.2 lineage, in which major clades identified as distinct introduction events into Mexico are highlighted. Nodes shown as red outline circles correspond to the most recent common ancestor (MRCA) for clades representing independent introduction events into Mexico. Based on the earliest and latest MRCAs, the estimated circulation period for each lineage is highlighted in yellow shadowing. The dashed purple line represents the date of the earliest viral genome sampled from Mexico, while its position in the tree is indicated. The dashed yellow line represents the implementation date of a systematic virus genome sampling and sequencing scheme for the surveillance of SARS-CoV-2 in Mexico. The corresponding root-to-tip regression plot for the tree is shown, in which genomes sampled from Mexico are shown in blue, whilst genomes sampled elsewhere are shown in grey. The map graph on the left show the cumulative proportion of genomes sampled across states per lineage of interest, corresponding to the period of circulation of the given lineage (relative to the total number of genomes taken from GISAID, corresponding to raw data before subsampling). The map on the right represents the geographic distribution of the main clades identified (for further details see Supplementary file 2). On the right, a zoom-in to the C5d and C6d clades showing sub-lineage composition with the most likely location estimated for each node. Geographic spread across Mexico inferred for these clades is further represented on the maps on the right, derived from a discrete phylogeographic analysis (DTA, see Methods section "Time-scaled analysis"). Viral transitions between Mexican states are represented by curved lines colored according to sampling location, showing only well-supported transitions (Bayes Factor >100 and a PP >0.9) (see Table 1).
Figure 4—video 1
Animated visualizations of the spread pattern inferred for the C5d clade across Mexico derived from the DTA phylogeographic analysis.
Figure 4—video 2
Animated visualizations of the spread pattern inferred for the C6d clade across Mexico derived from the DTA phylogeographic analysis.
Appendix 1—figure 1
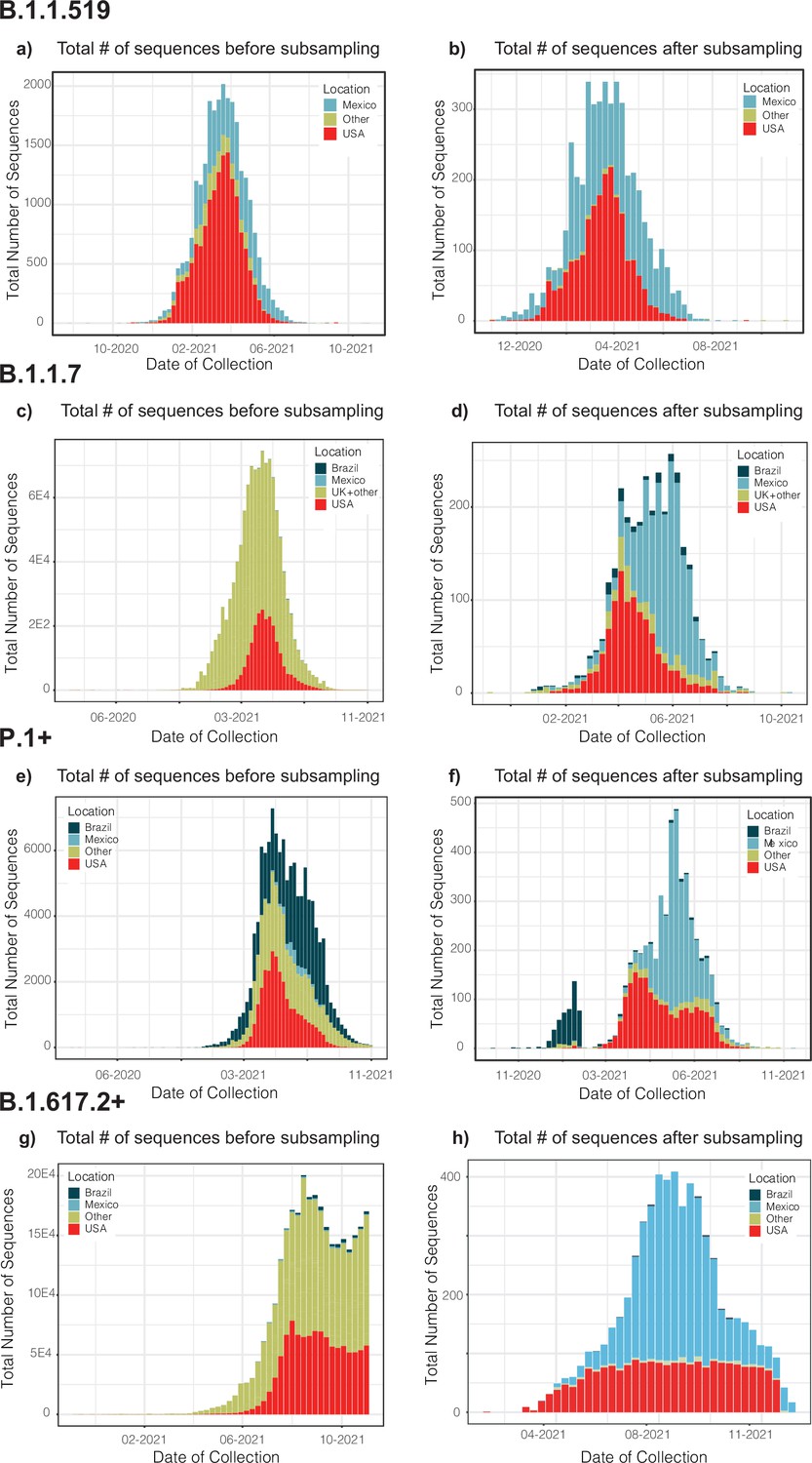
Distribution plots for each genome dataset before and after applying our migration- and phylogenetically-informed subsampling pipeline.
Distribution plots for the number of genomes in the datasets before and after applying our subsampling pipeline. Plots for the B.1.1.519 (a and b), B.1.1.7 (c and d), P.1+ (e and f), and B.1.617.2+ (g and h) show the total number of sampled genomes colored according to location, ranked according to the countries representing the most intense human mobility flow into Mexico derived from anonymized relative human mobility flow into different geographical regions.
Appendix 1—figure 2
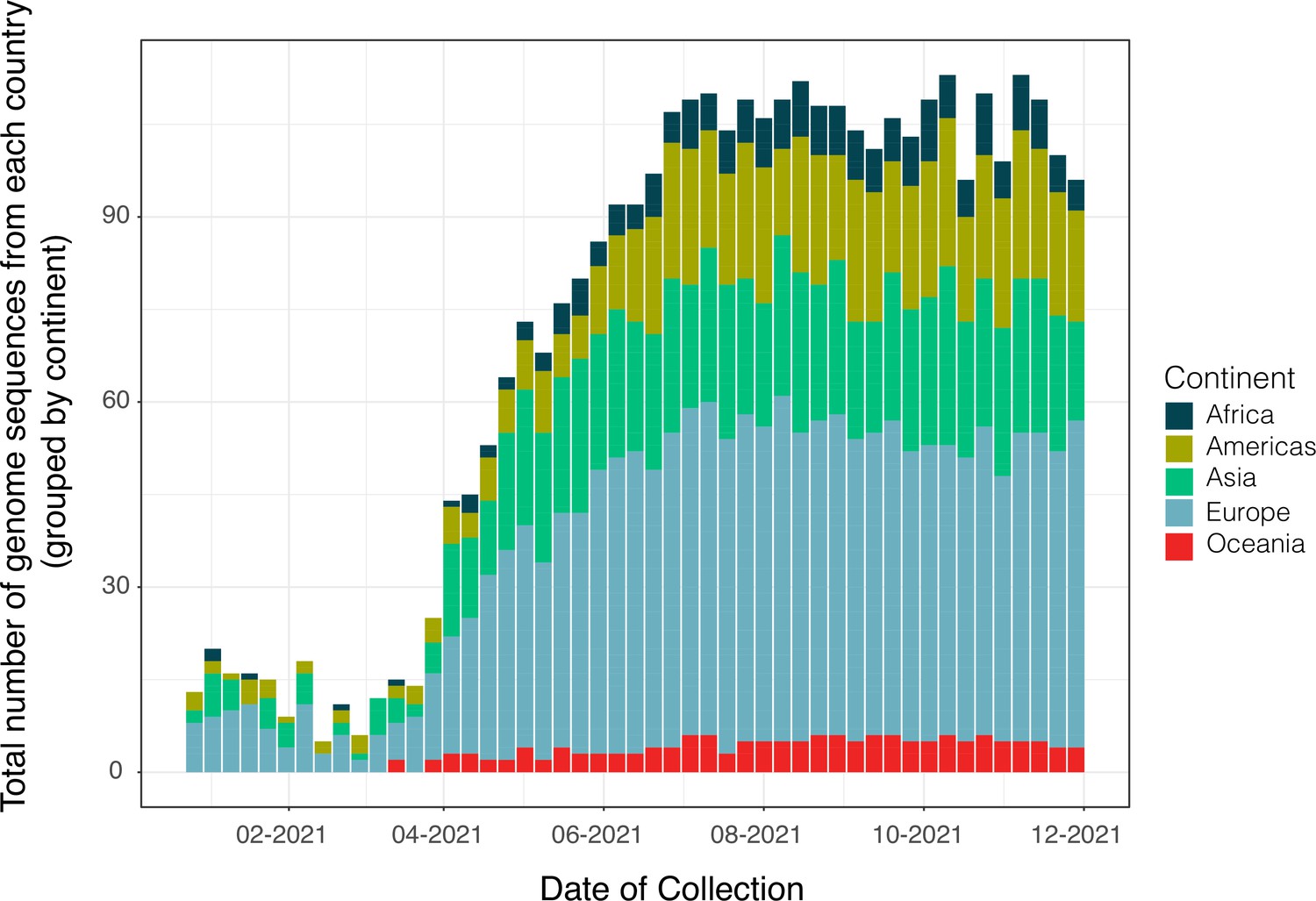
Distribution of genome sequences the new B.1.617.2+dataset after subsampling under a different migration-informed approach (validation).
Distribution of the number of genomes in the dataset corresponding to an alternative sub-sample of B.1.617.2+sequences used for the validation of our migration informed subsampling approach. The dataset was built to obtain a homogeneous and proportional number of genome sequences from all countries sampled in GISAID (relative to their availability in the platform). The total number of genomes sequences sampled per region (represented by countries grouped by continent) are colored according to their continent of origin. To compare to the distribution of genome sequences before subsampling, see Appendix 1—figure 1 above.
Appendix 1—figure 3
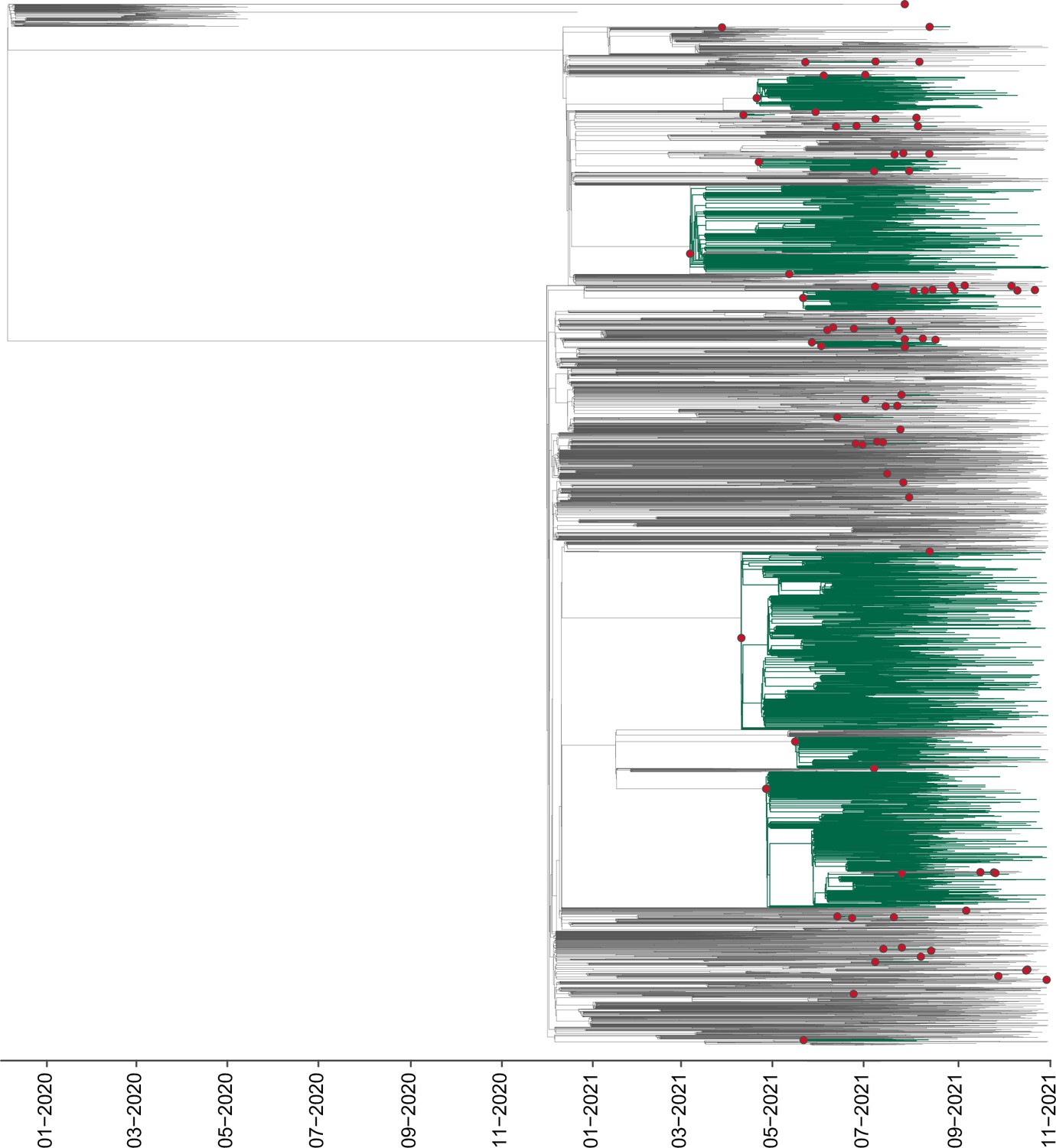
DTA analysis for the new B.1.617.2+dataset (validation).
Maximum clade credibility (MCC) tree for the alternative B.1.617.2+dataset comprising a sub-sampling from all countries, represented by B.1.617.2+sequences deposited in GISAID available up to November 30th 2021, in which major clades identified as distinct introduction events into Mexico are highlighted. Nodes shown as red circles correspond to the inferred most recent common ancestor (MRCA) for clades representing independent introduction events into Mexico.
Tables
Table 1
Bayes Factor (BF) and Posterior Probability (PP) for well-supported transitions observed between locations*.
| C5d | C6d | ||||||
|---|---|---|---|---|---|---|---|
| Location | Location | ||||||
| From | To | BFR | PP | From | To | BFR | PP |
| BCN | CHH | 14535.32494 | 1 | AGU | CHP | 13635.15617 | 1 |
| CAM | CHP | 14535.32494 | 1 | BCN | CHP | 13635.15617 | 1 |
| CAM | CMX | 14535.32494 | 1 | CHP | CMX | 13635.15617 | 1 |
| CAM | MEX | 14535.32494 | 1 | CHP | COA | 13635.15617 | 1 |
| CAM | MIC | 14535.32494 | 1 | CHP | DUR | 13635.15617 | 1 |
| CAM | other | 14535.32494 | 1 | CHP | GRO | 13635.15617 | 1 |
| CAM | QUE | 14535.32494 | 1 | CHP | GUA | 13635.15617 | 1 |
| CAM | ROO | 14535.32494 | 1 | CHP | HID | 13635.15617 | 1 |
| CAM | SLP | 14535.32494 | 1 | CHP | JAL | 13635.15617 | 1 |
| CAM | SON | 14535.32494 | 1 | CHP | MEX | 13635.15617 | 1 |
| CAM | TAB | 14535.32494 | 1 | CHP | MIC | 13635.15617 | 1 |
| CAM | TAM | 14535.32494 | 1 | CHP | NLE | 13635.15617 | 1 |
| CAM | TLA | 14535.32494 | 1 | CHP | OAX | 13635.15617 | 1 |
| CAM | VER | 14535.32494 | 1 | CHP | other | 13635.15617 | 1 |
| CAM | ZAC | 14535.32494 | 1 | CHP | PUE | 13635.15617 | 1 |
| CMX | CHH | 14535.32494 | 1 | CHP | QUE | 13635.15617 | 1 |
| CHH | CHP | 14535.32494 | 1 | CHP | SIN | 13635.15617 | 1 |
| CHH | CMX | 14535.32494 | 1 | CHP | SLP | 13635.15617 | 1 |
| CHH | DUR | 14535.32494 | 1 | CHP | SON | 13635.15617 | 1 |
| CHH | GUA | 14535.32494 | 1 | CHP | TAB | 13635.15617 | 1 |
| CHH | MIC | 14535.32494 | 1 | CHP | TLA | 13635.15617 | 1 |
| CHH | NLE | 14535.32494 | 1 | CHP | VER | 13635.15617 | 1 |
| CHH | QUE | 14535.32494 | 1 | CAM | CHP | 13635.15617 | 0.998890122 |
| CHH | TAB | 14535.32494 | 1 | NLE | TAB | 13635.15617 | 0.998890122 |
| CHH | TAM | 14535.32494 | 1 | CHP | TAM | 6810.002999 | 0.997780244 |
| CHH | VER | 14535.32494 | 1 | CHP | YUC | 2714.911095 | 0.99445061 |
| CHH | ZAC | 14535.32494 | 1 | MEX | PUE | 164.4591205 | 0.915649279 |
| CAM | CMX | 14535.32494 | 0.998890122 | ||||
| CHH | TLA | 14535.32494 | 0.998890122 | ||||
| CAM | SIN | 3621.718465 | 0.995560488 | ||||
| BCS | CHH | 1023.240732 | 0.984461709 | ||||
| MIC | YUC | 468.8988157 | 0.966703663 | ||||
| CHH | other | 399.6060762 | 0.961154273 | ||||
| CAM | COA | 188.7999953 | 0.921198668 | ||||
| MEX | YUC | 126.5111615 | 0.886792453 |
-
*
derived from the phylogeographic analyses for C5d and C6d (B.1.617.2+). Only values of BF >100 and PP >0.9 are shown.
Additional files
-
Supplementary file 1
Virus genome IDs and GISAID accession numbers for the sequences used in each dataset.
- https://cdn.elifesciences.org/articles/82069/elife-82069-supp1-v2.xlsx
-
Supplementary file 2
Full list of names of all genome sequences within each major clade identified for each virus lineage.
- https://cdn.elifesciences.org/articles/82069/elife-82069-supp2-v2.xlsx
-
Supplementary file 3
Mobility matrixes summarizing: 1. Ranking connectivity between the southern region of the country, 2. Pairwise distances between states, 3. Mean intrastate connectivity.
- https://cdn.elifesciences.org/articles/82069/elife-82069-supp3-v2.xls
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/82069/elife-82069-transrepform1-v2.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Comparing the evolutionary dynamics of predominant SARS-CoV-2 virus lineages co-circulating in Mexico
eLife 12:e82069.
https://doi.org/10.7554/eLife.82069




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}