Improved inference of population histories by integrating genomic and epigenomic data
- Professorship for Population Genetics, Department of Life Science Systems, Technical University of Munich, Germany
- Department of Environment and Biodiversity, Paris Lodron University of Salzburg, Austria
- Professorship for Plant Epigenomics, Department of Molecular Life Sciences, Technical University of Munich, Germany
Figures
Figure 1 with 1 supplement
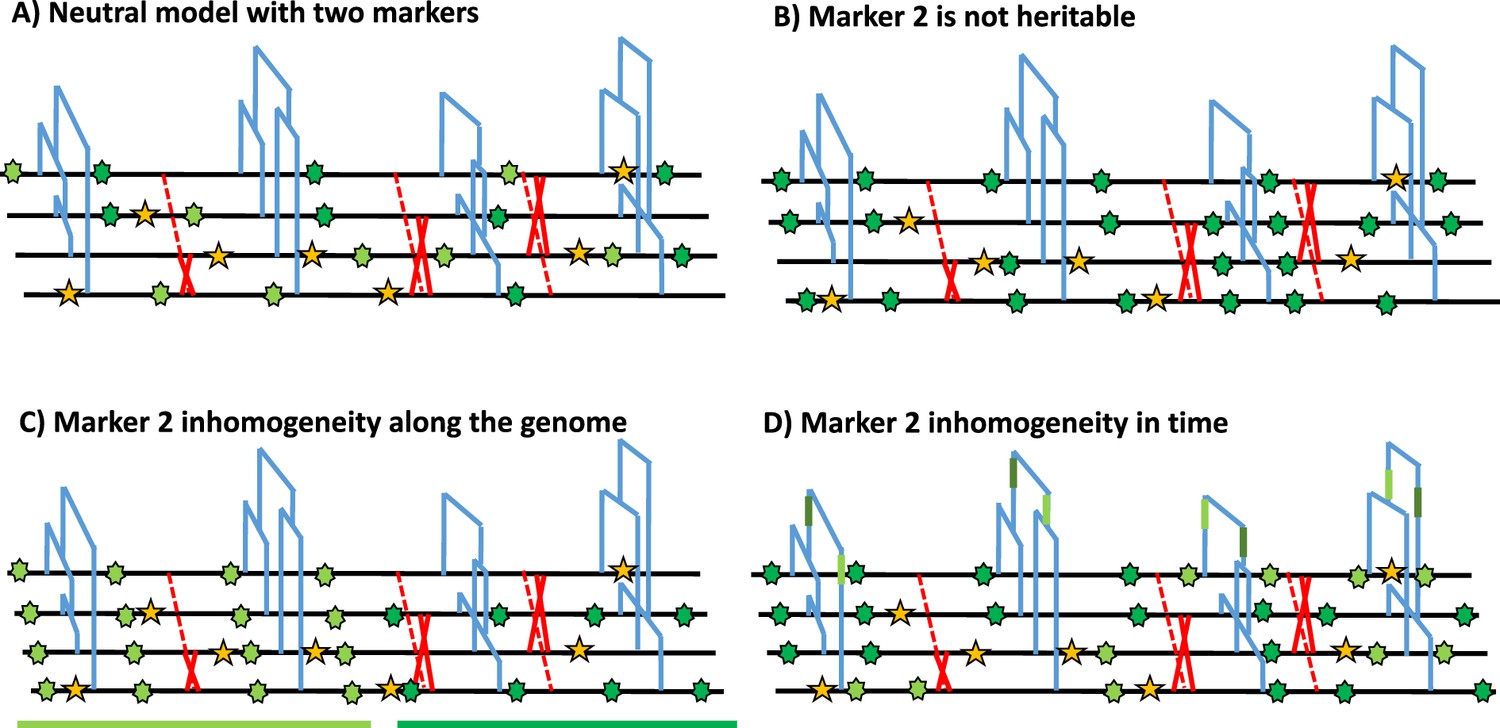
Schematic distribution of two markers along the genealogy and four genomes.
(A) Schematic distribution of marker 1 (yellow star) and marker 2 (green star) along the genealogies in a sample of four genomes, both following a homogeneous Poisson process. (B) The green marker 2 is not heritable so its distribution is independent of the genealogy. (C) The green marker 2 is spatially structured along the genome, violating the distribution of the Poisson process along the genome and conflicting with the genealogy. (D) The green marker 2 does not follow the Poisson process through time, for example burst of mutations at a specific time point represented by given branches of the genealogies in green. The yellow marker 1 has an identical Poisson process along the genome and the genealogy in all four panels, and for readability, marker 2 exhibits light and dark green states.
Figure 1—figure supplement 1

Probability of a site to be segregating in a sample of size two for different mutation rates.
The probability for a site to be segregating in a sample of size two under different mutation rates: 10−2 in red, 10−4 in orange, 10−6 in green, and 10−2 in blue. The marker is assumed here to have possible states.
Figure 2 with 2 supplements
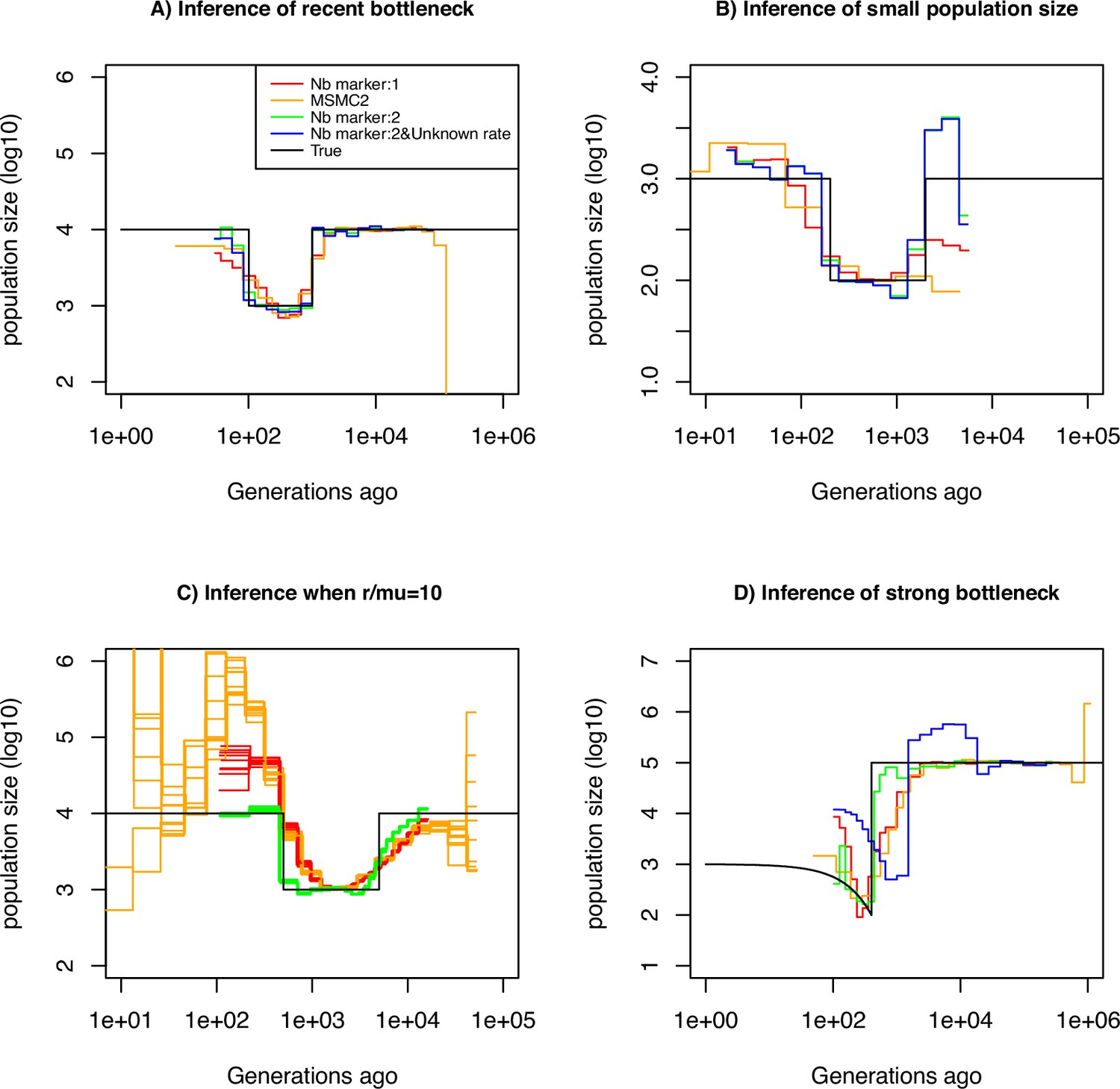
Performance of SMC approaches using different markers.
Estimated demographic history of a bottleneck (black line) by SMC approaches using two genomic markers. In orange and red are the estimates by MSMC2 and eSMC2 based on only marker 1. Estimates from SMCtheo integrating both markers are in green (with known ) and in blue with unknown . The demographic scenarios are (A) 10-fold recent bottleneck with an ancestral population size , (B) 10-fold recent bottleneck with an ancestral population size , (C) 10-fold bottleneck with an ancestral population size , and (D) a very severe (1000 fold) and very recent bottleneck with incomplete size recovery. In A, B, and D, we assume (with , per generation per bp) and in C, (with , , and per generation per bp). In all cases (A, B, C and D) 10 sequences (5 diploid indivudals) of 100 Mb were used as input.
Figure 2—figure supplement 1

Performance of of SMCtheo using two theoretical markers when marker 2 is very rare.
Estimated demographic history of a recent bottleneck using theoretical genomic markers and 10 sequences of 100 Mb: with only marker 1 (red and orange) and with two markers (green and blue). Marker 2 is found at 0.1% of the sites. The parameters are per generation per bp, and per generation per bp.
Figure 2—figure supplement 2

Performance of the SMCtheo using theoretical markers by maximizing the true Likelihood function.
Estimated demographic history by SMCtheo on theoretical genomic markers using 6 scaffolds each of 20 Mb with sample size 10: with one marker in red, and two markers in orange. We use here the likelihood function (LH) to estimate model parameters from SMCtheo. The parameters are per generation per bp, and , per generation per bp.
Figure 3

Schematic representation of site and region epimutations.
Schematic representation of a sequence undergoing epimutation at (A) the cytosine site level and (B) at the region level. A methylated cytosine in CG context is indicated in black, and an unmethylated cytosine in white.
Figure 4 with 1 supplement
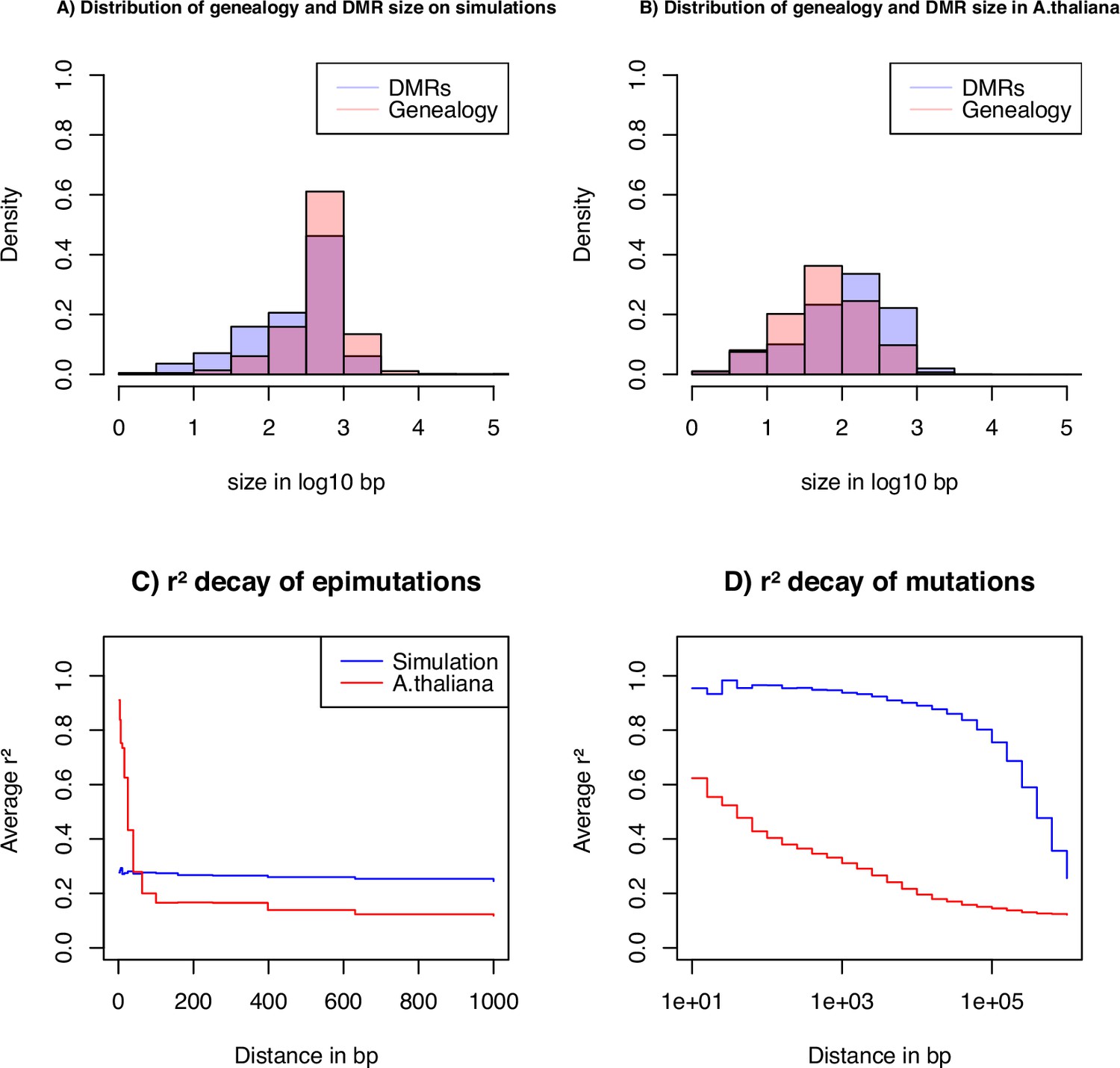
Key statistics for epimutations and mutations.
(A) Histogram of the length between two recombination events (genomic span of a genealogy) and DMRs size in bp of the simulated data. (B) Histogram of genealogy span and DMRs size in bp from the A. thaliana data (10 German accessions). (C) Linkage disequilibrium decay of epimutations in our samples of A. thaliana (red) and simulated data (blue). (D) Linkage disequilibrium decay of mutations in our A. thaliana samples (red) and simulated data (blue). The simulations reproduce the outcome of a recent bottleneck with sample size diploid of 100 Mb, the rates per generation per bp are , , , , and per 1 kb region and .
Figure 4—figure supplement 1
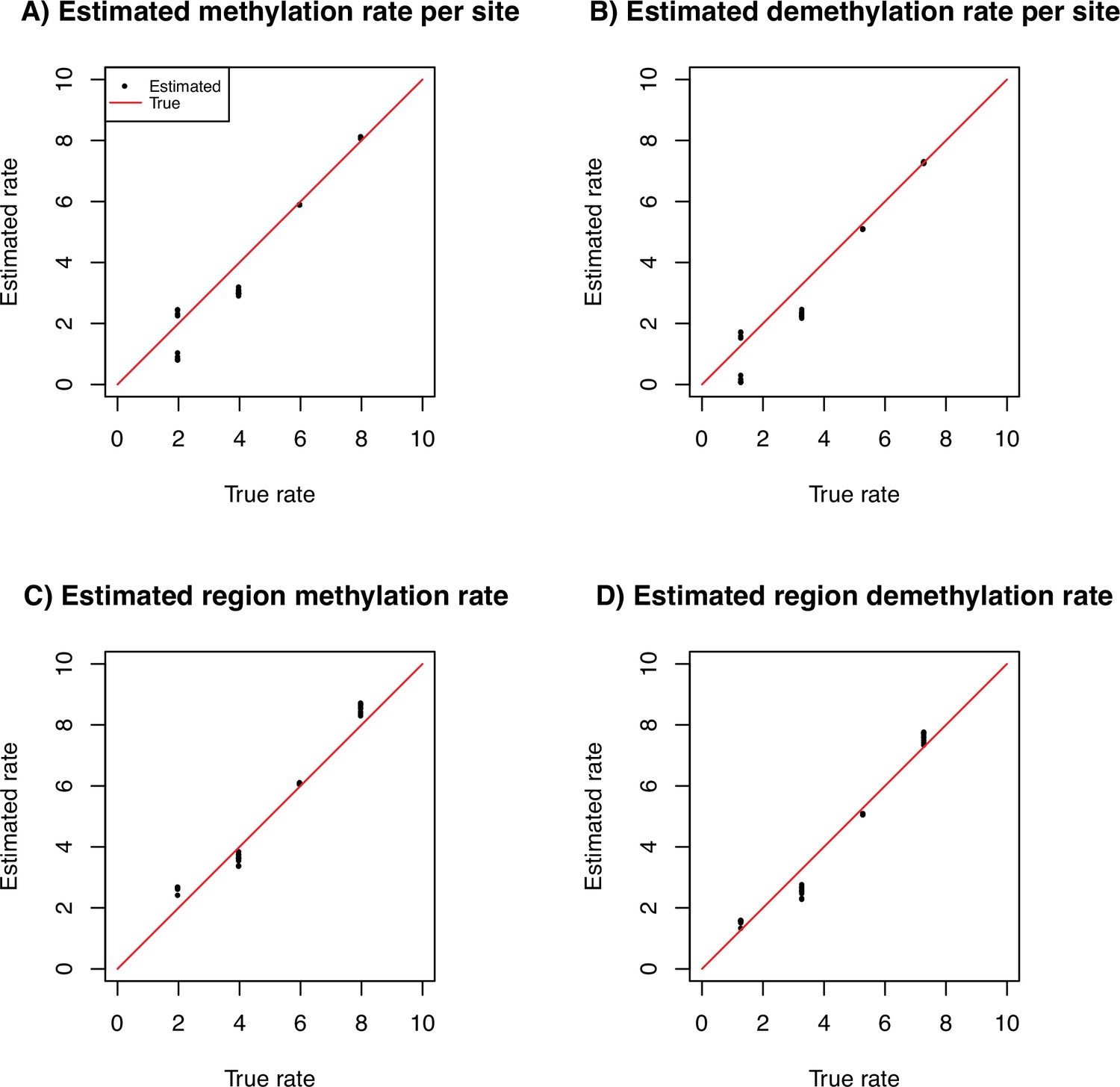
Average estimates of the site and region methylation and demethylation rates for simulated data.
The true rate is indicated as x-axis and the estimated rate on the y-axis (log10 scale). We use 10 repetitions with 10 sequences of 100 Mb with per generation per bp under a constant population size fixed to 10,000 individuals.
Figure 5 with 5 supplements
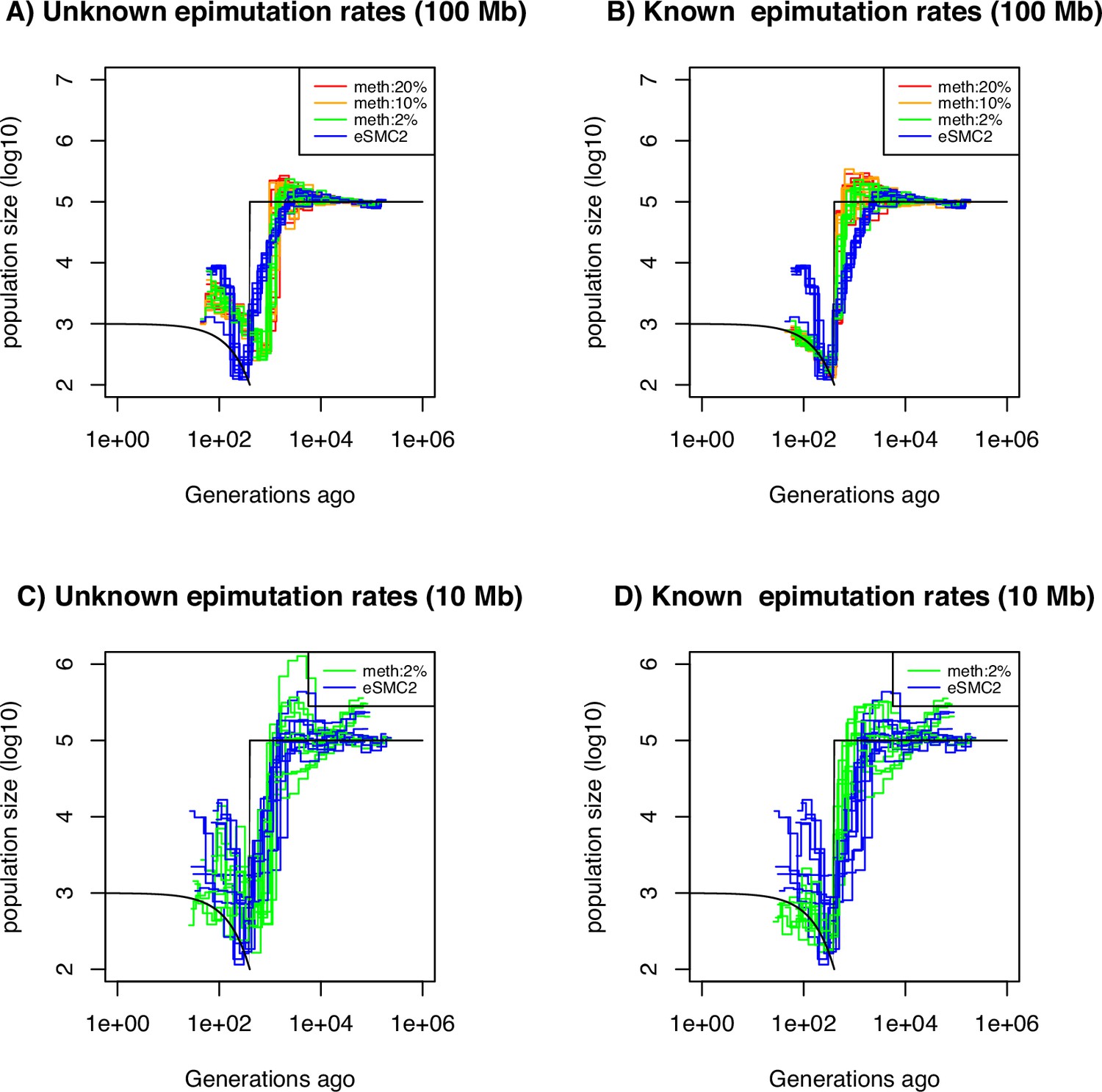
Performance of SMC approaches using site epimutations (SMPs) and mutations (SNPs) under a bottleneck scenario.
Estimated demographic history by eSMC2 (blue) and SMCm assuming the epimutation rate is known (B and D) or not (A and C) where the percentage of CG sites with methylated information varies between 20% (red), 10% (orange) and 2% (green) using 10 sequences of 100 Mb in A and B (with 10 repetitions) and 10 sequences of 10 Mb in C and D (three repetitions displayed) under a recent severe bottleneck (black). The parameters are: per generation per bp, mutation rate , methylation rate to and demethylation rate to per generation per bp.
Figure 5—figure supplement 1
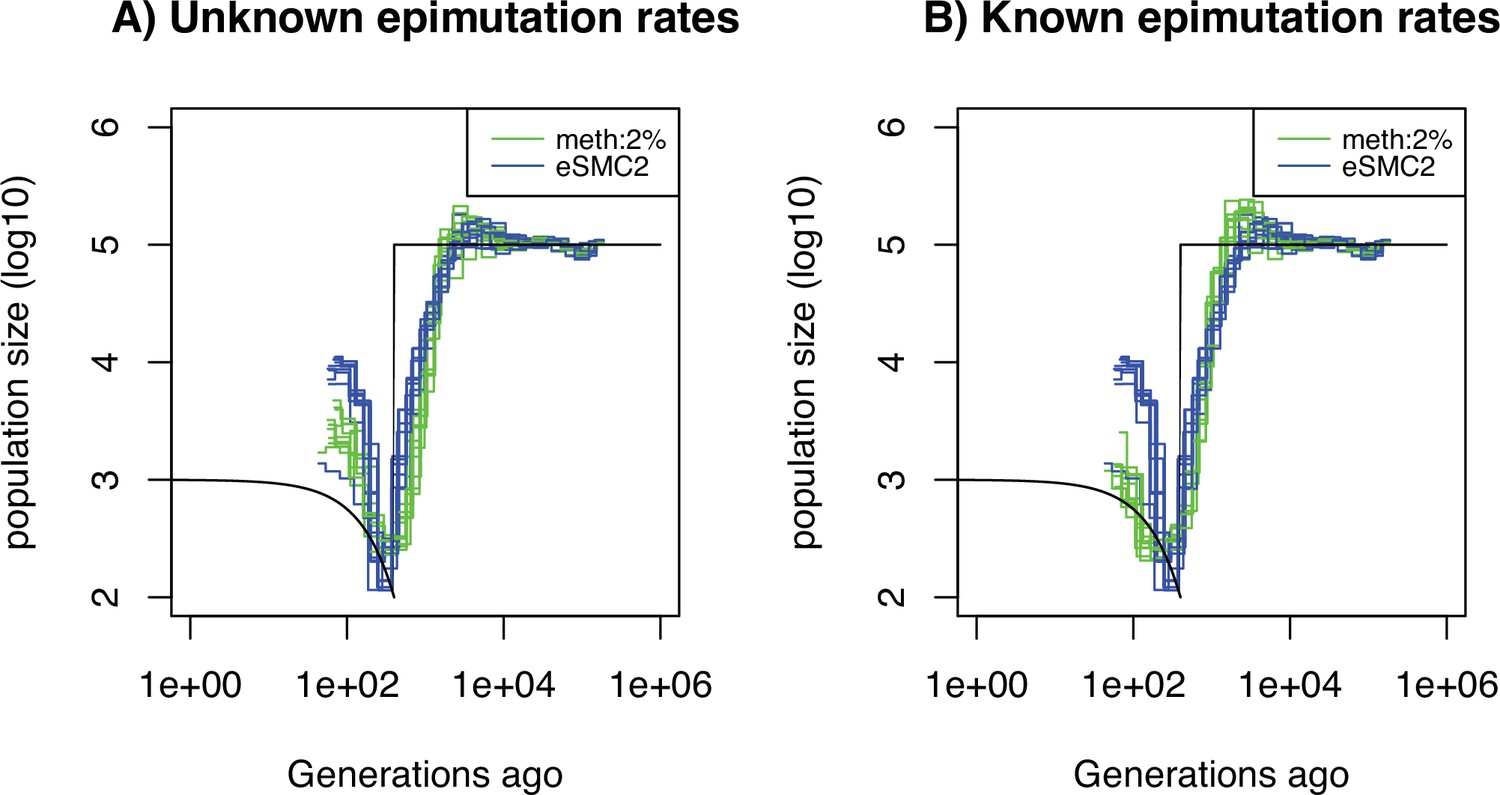
Performance of SMCm for methylation with only DMR regions of length 1kbp.
Estimated demographic history by eSMC2 (blue) and SMCm in presence of region epimutations only and of length 1kbp (green) using 10 sequences of 100 Mb under a recent bottleneck (black). The parameters are per generation per bp, , and the region methylation and demethylation rate per generation per bp.
Figure 5—figure supplement 2
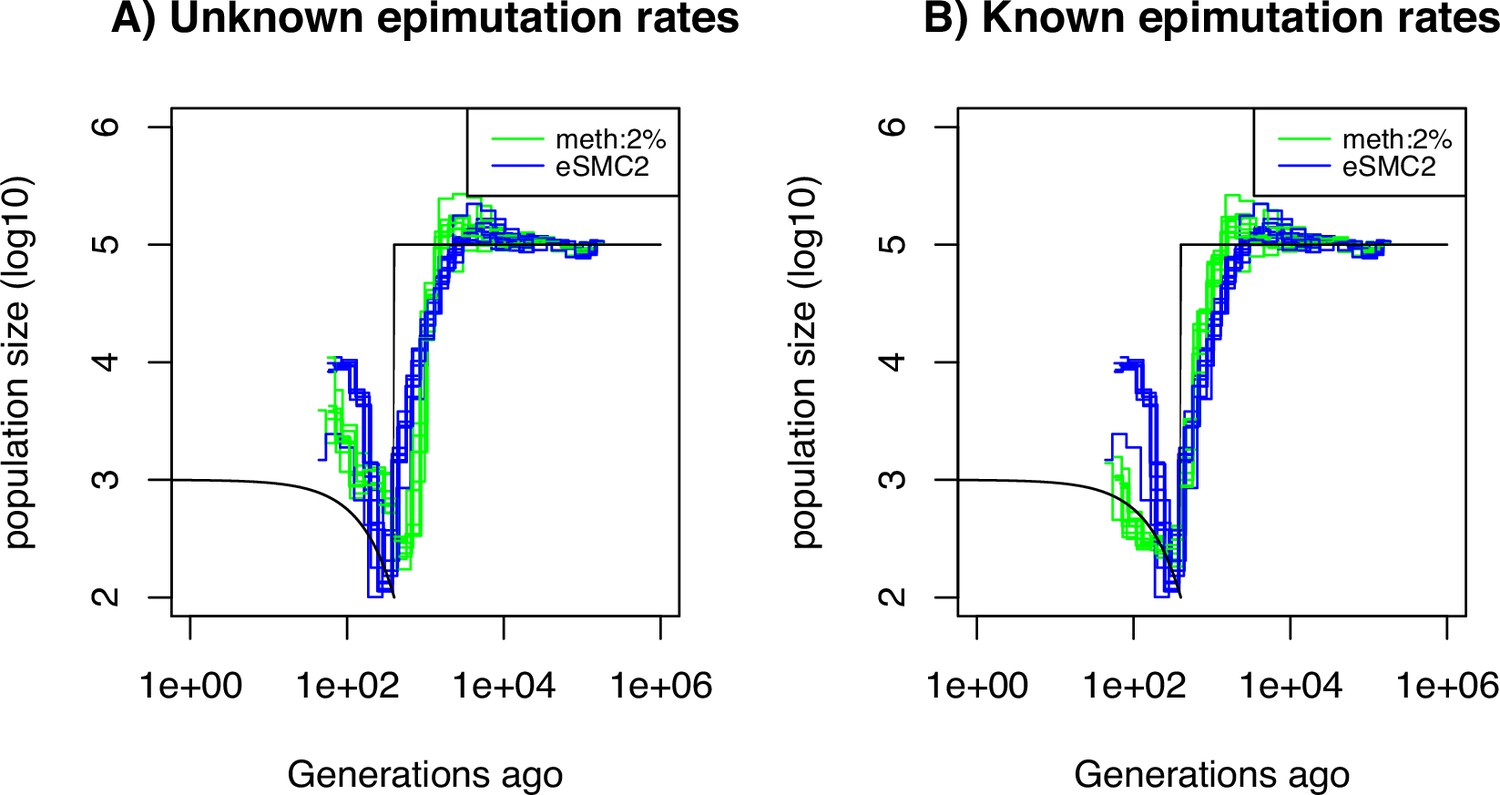
Performance of SMCm for methylation with only DMR regions of length 150 bp.
Estimated demographic history by eSMC2 (blue) and SMCm in presence of region epimutations only and of length 150 bp (green) using 10 sequences of 100 Mb under a recent bottleneck (black). The parameters are per generation per bp, , and the region methylation and demethylation rate per generation per bp.
Figure 5—figure supplement 3
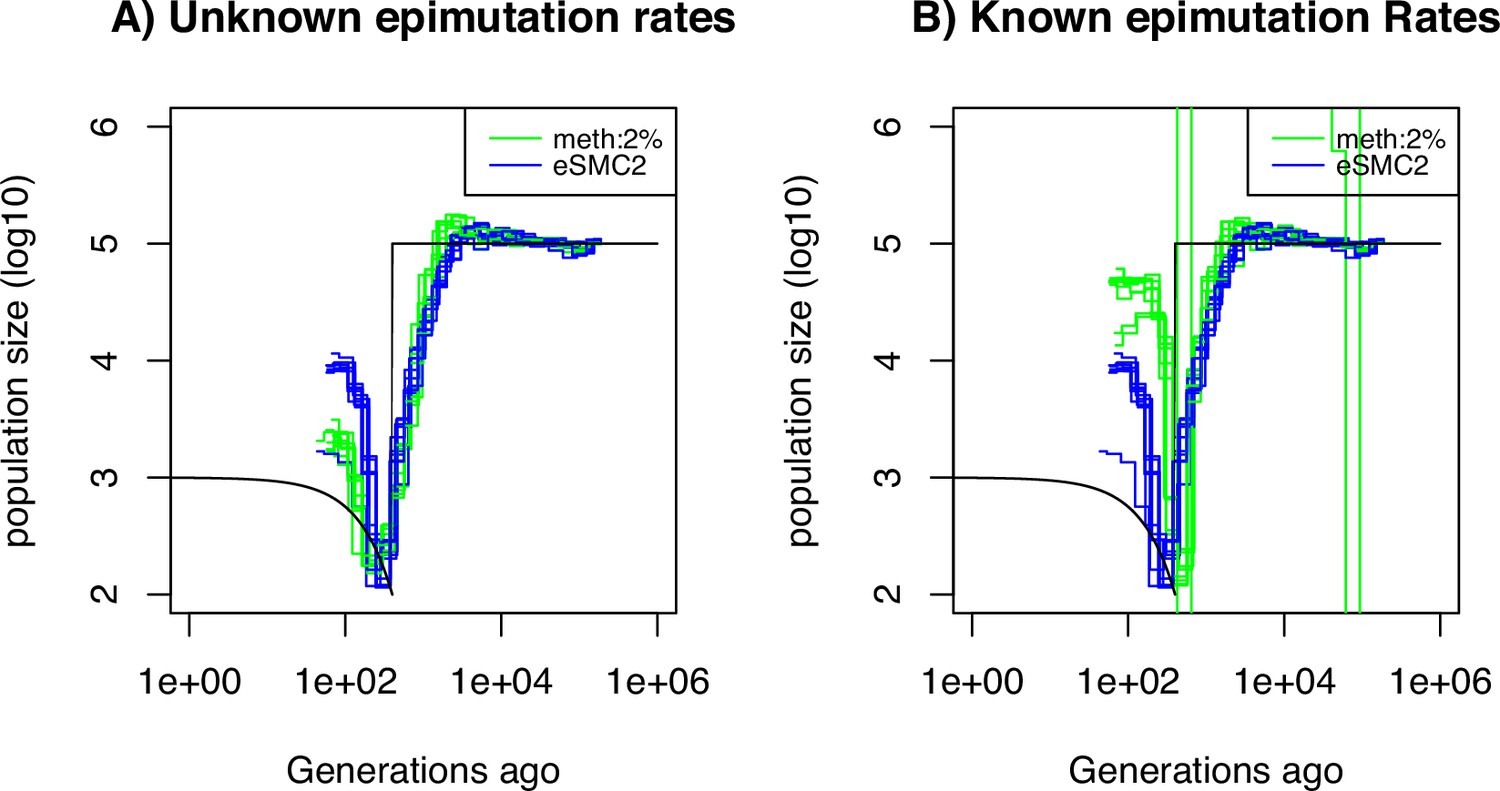
Performance of SMCm for methylation with site and region epimutations.
Estimated demographic history by eSMC2 (blue) and SMCm in presence of site and region epimutations (green) using 10 sequences of 100 Mb under a recent bottleneck (black). The recombination is set to per generation per bp, the mutation rate is set to , site methylation rate to , site demethylation rate to per generation per bp, region methylation rate to and region demethylation rate to per generation per bp.
Figure 5—figure supplement 4
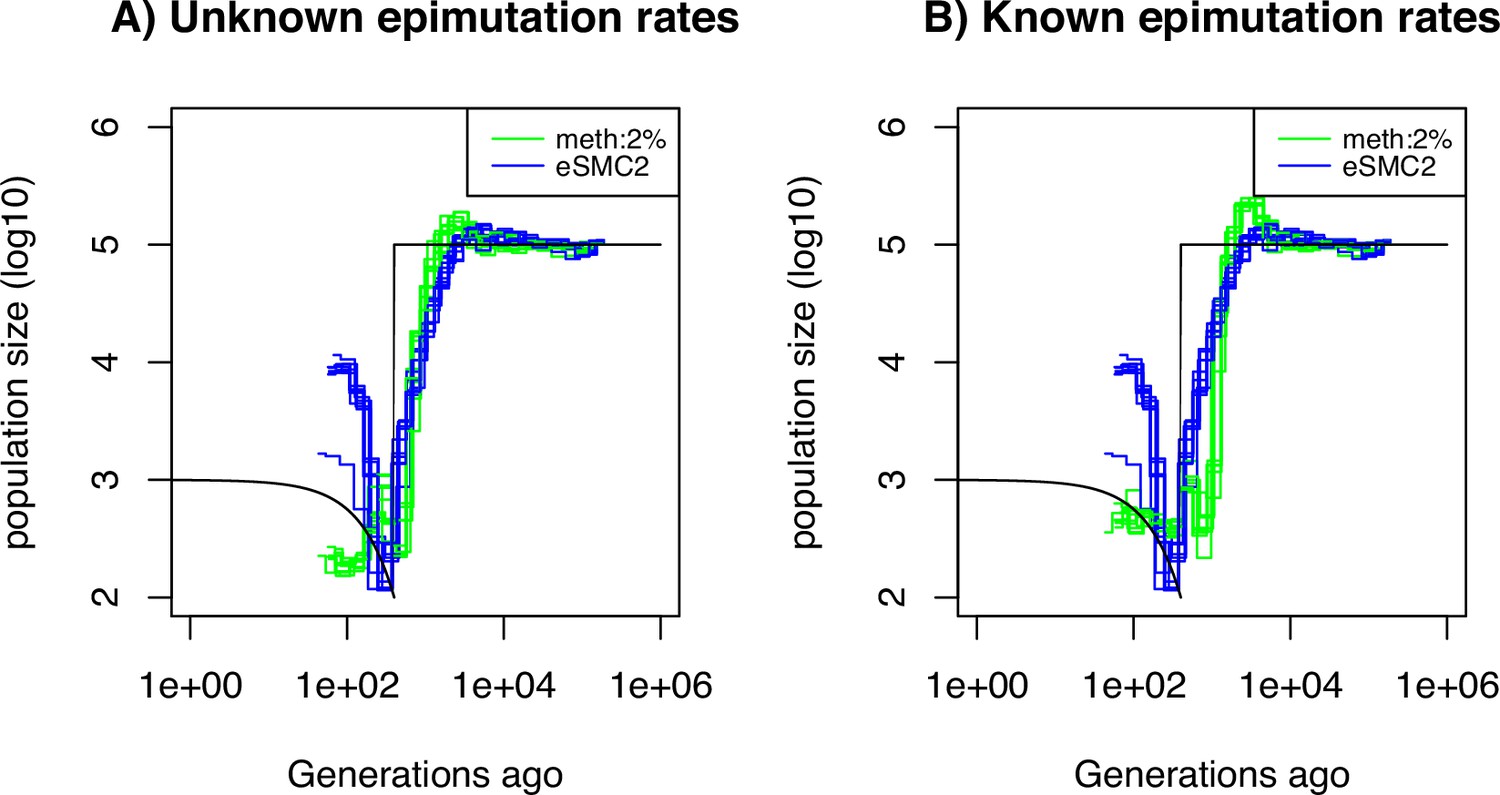
Performance of SMCm for methylation, accounting only for SMPs.
Estimated demographic history by eSMC2 (blue) and SMCm in presence of site and region epimutations but only accounting for site epimutations SMPs (green) using 10 sequences of 100 Mb under a recent bottleneck (black). The recombination is set to per generation per bp, the mutation rate is set to , site methylation rate to , site demethylation rate to per generation per bp, region methylation rate to and region demethylation rate to per generation per bp.
Figure 5—figure supplement 5
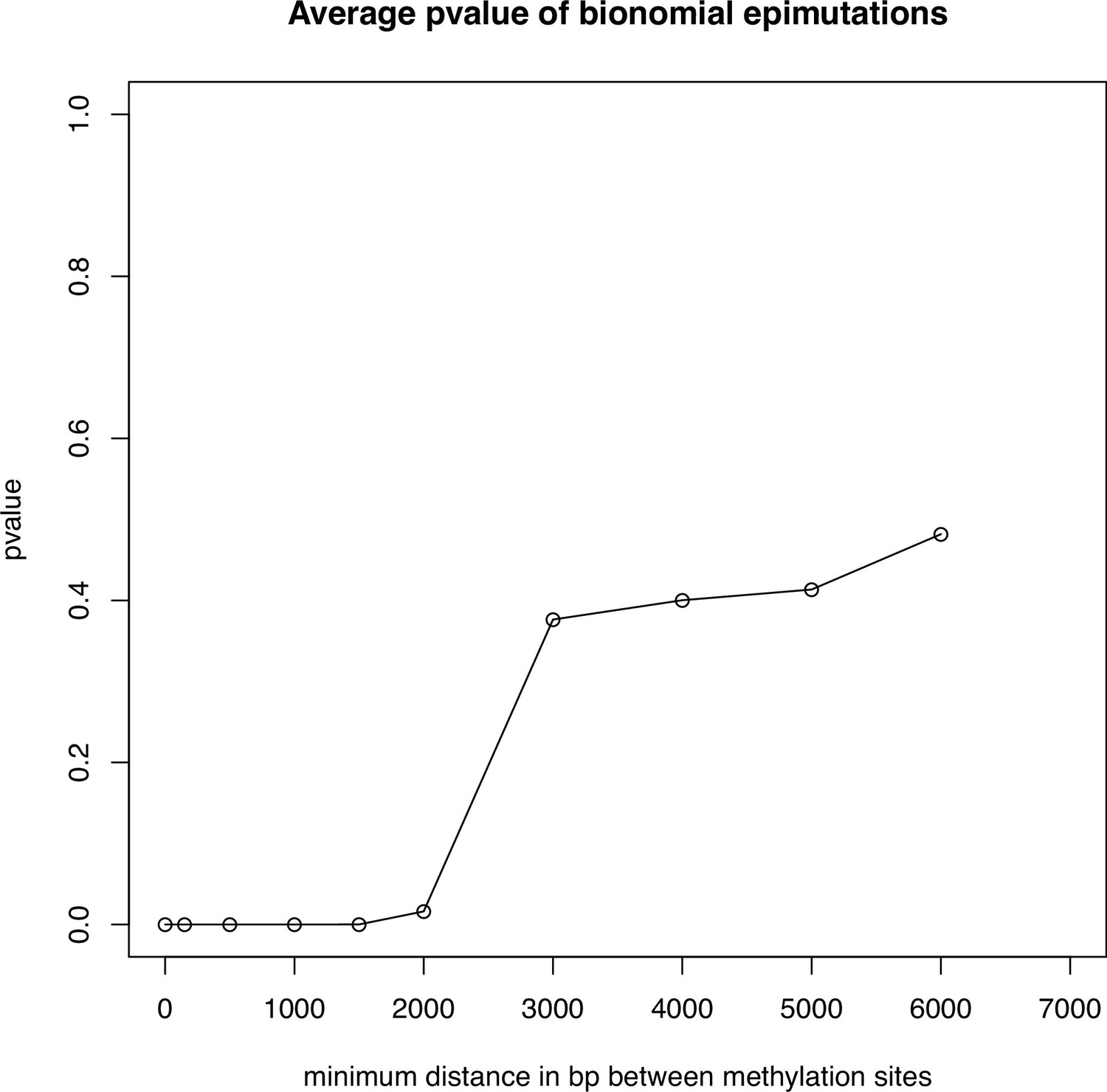
Average pvalue of the binomial test for epimutations.
Average p-value across 10 genomes of the binomial test on epimutations seperated by a minimum distance in bp (x axis) on our eight methylome scaffolds of A. thaliana.
Figure 6 with 6 supplements

Integrating epimutations and mutations on German accessions of A. thaliana.
Estimated demographic history of the German population by eSMC2 (only SNPs, purple) and SMCm when keeping polymorphic methylation sites (SMPs) only: green with epimutation rates estimated by SMCm, blue with epimutation rates fixed to empirical values. The region epimutation effect is ignored. The parameters are , , and when assumed known, the site methylation rate is and demethylation rate is .
Figure 6—figure supplement 1

Demographic estimation using all methylation sites from German accessions of A. thaliana.
Estimated demographic history of the German population by eSMC2 (purple) and SMCm under different assumptions. In green the SMCm results with epimutation rates estimated and regions epimutation (DMRs) ignored. In blue are the estimates with epimutation rates fixed and regions epimutation (DMRs) ignored. In red are the results with epimutation rates sites and regions estimated by SMCm, and in orange with both regions and site epimutation fixed to empirical values. The recombination is set to per generation per bp, the mutation rate is set to , site methylation rate to and site demethylation rate to per generation per bp (when fixed). When fixed, the region methylation and demethylation rates are set, respectively, to and .
Figure 6—figure supplement 2
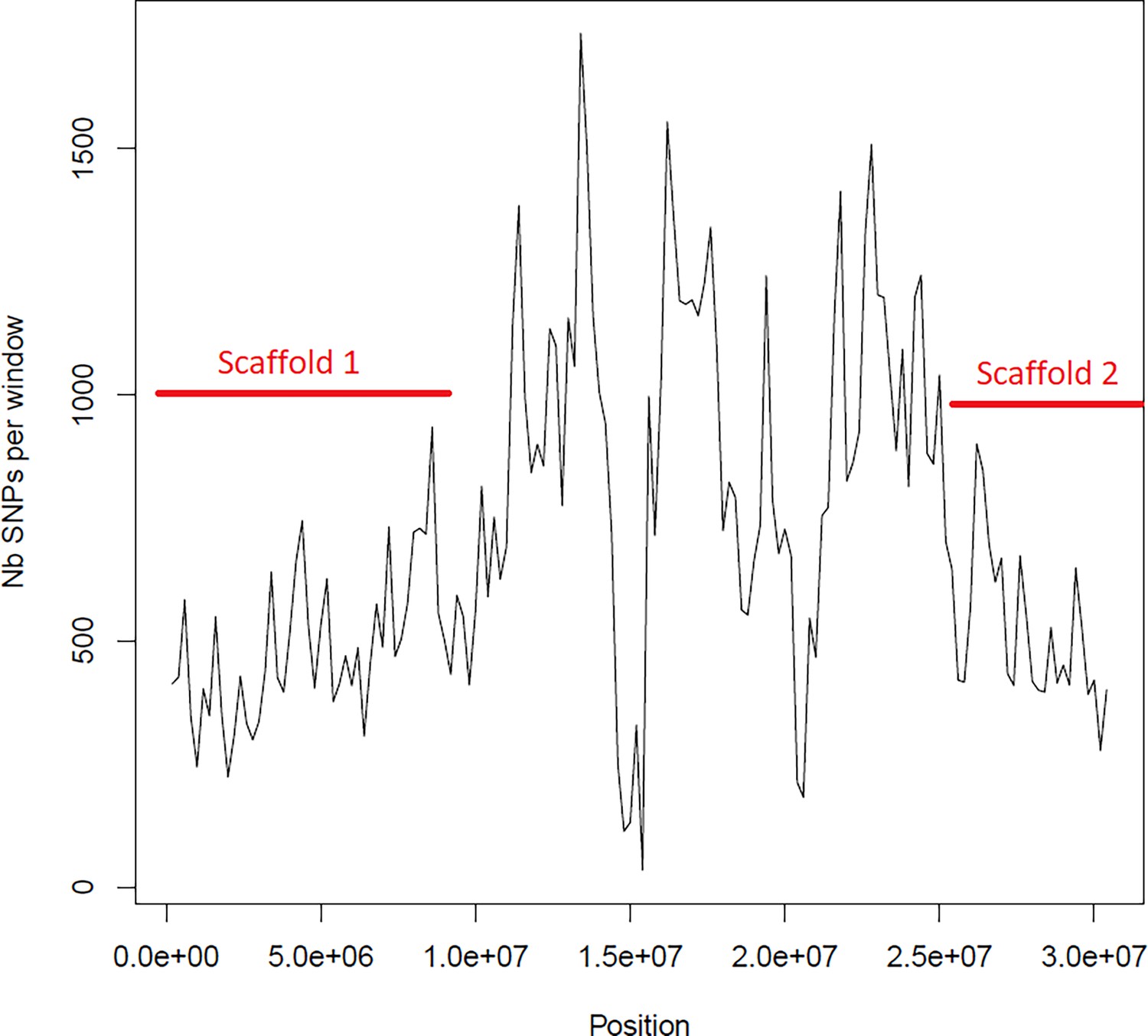
Average number of segregating site per window of 100kp on chromosome 1.
Estimated Average number of segregating site per window of 100kp on chromosome 1 on 10 individuals of the German accessions (black).
Figure 6—figure supplement 3
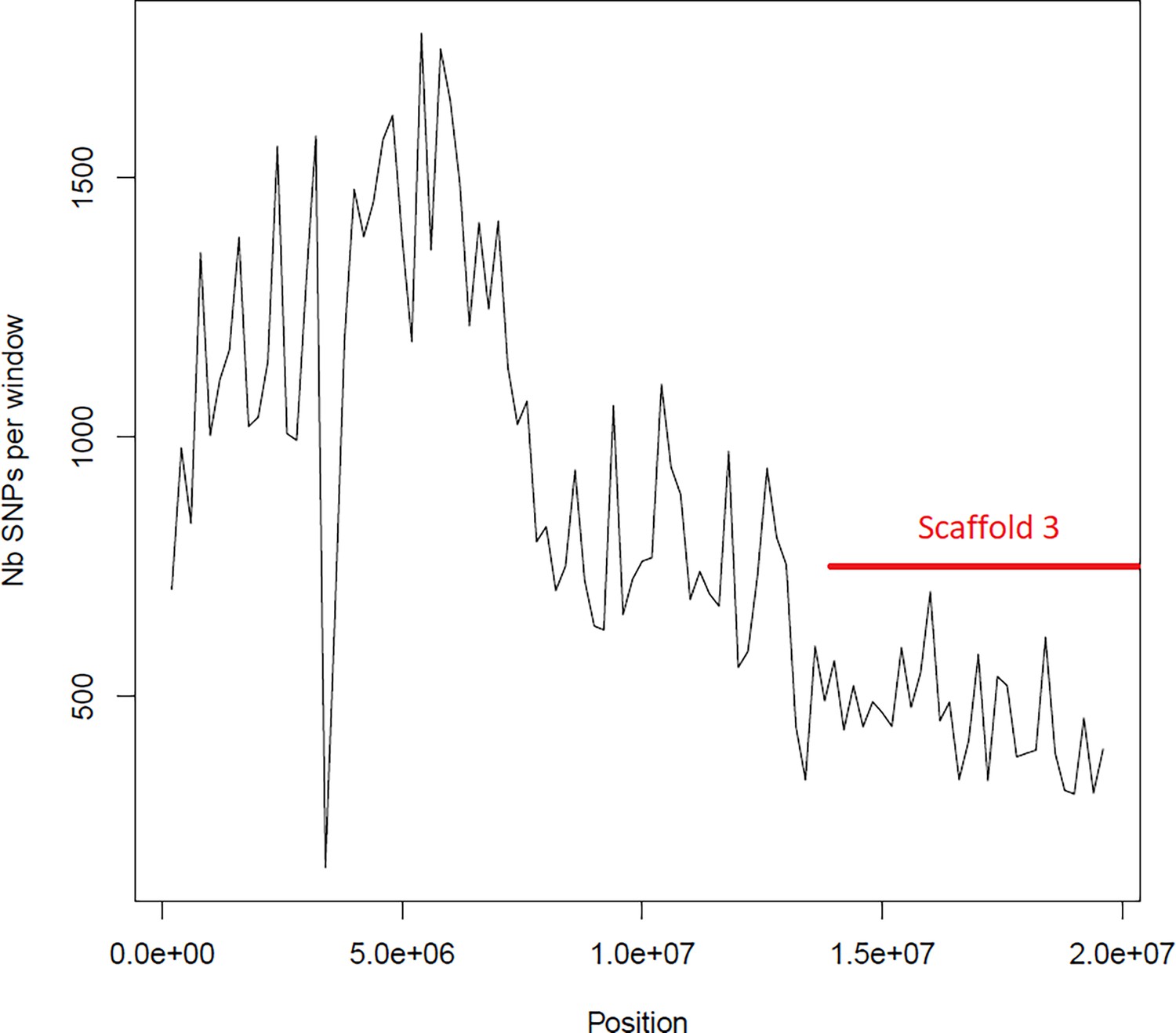
Average number of segregating site per window of 100kp on chromosome 2.
Estimated Average number of segregating site per window of 100kp on chromosome 2 on 10 individuals of the German accessions (black).
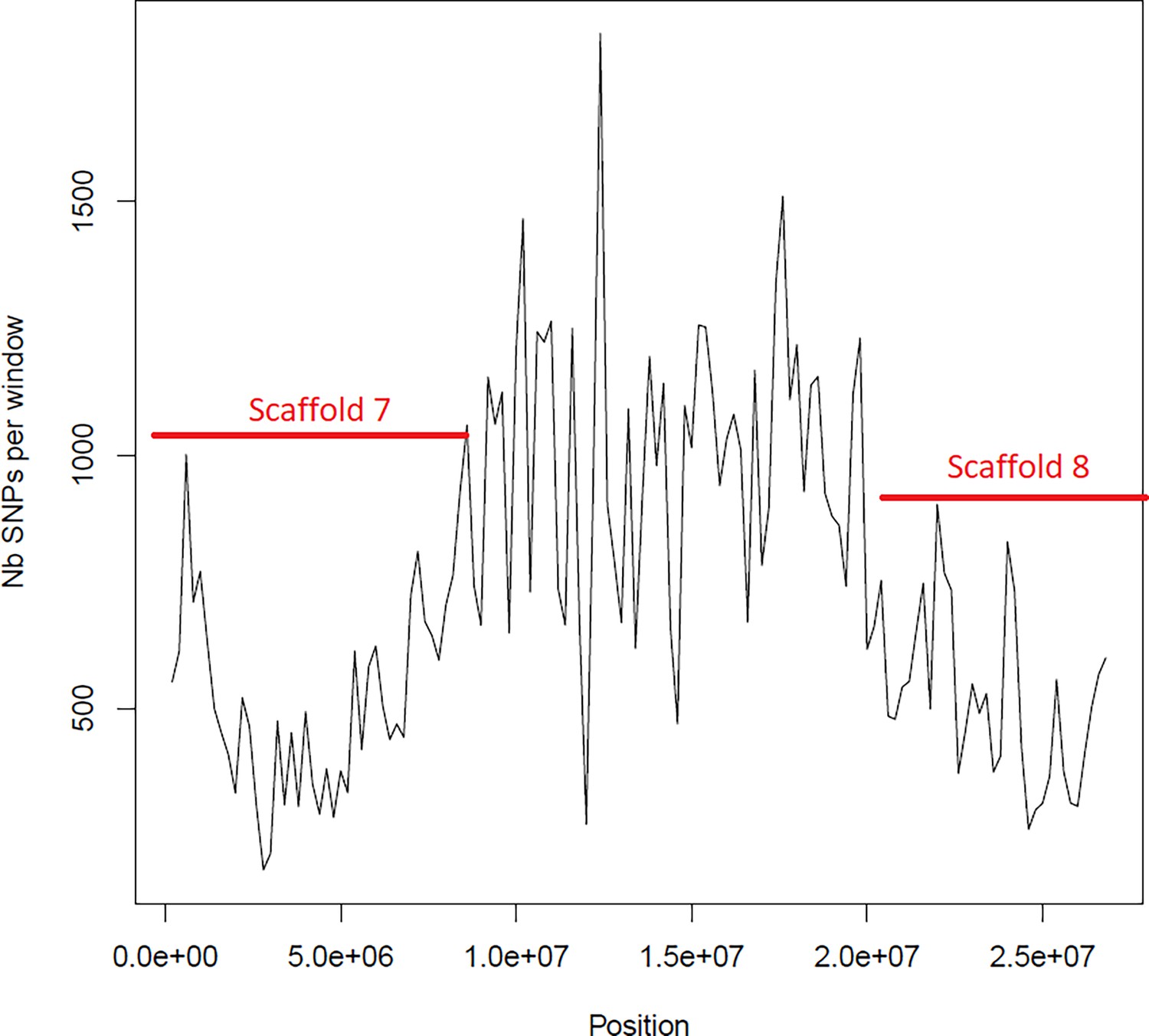
Figure 6—figure supplement 4
Average number of segregating site per window of 100kp on chromosome 3.
Estimated Average number of segregating site per window of 100kp on chromosome 3 on 10 individuals of the German accessions (black).
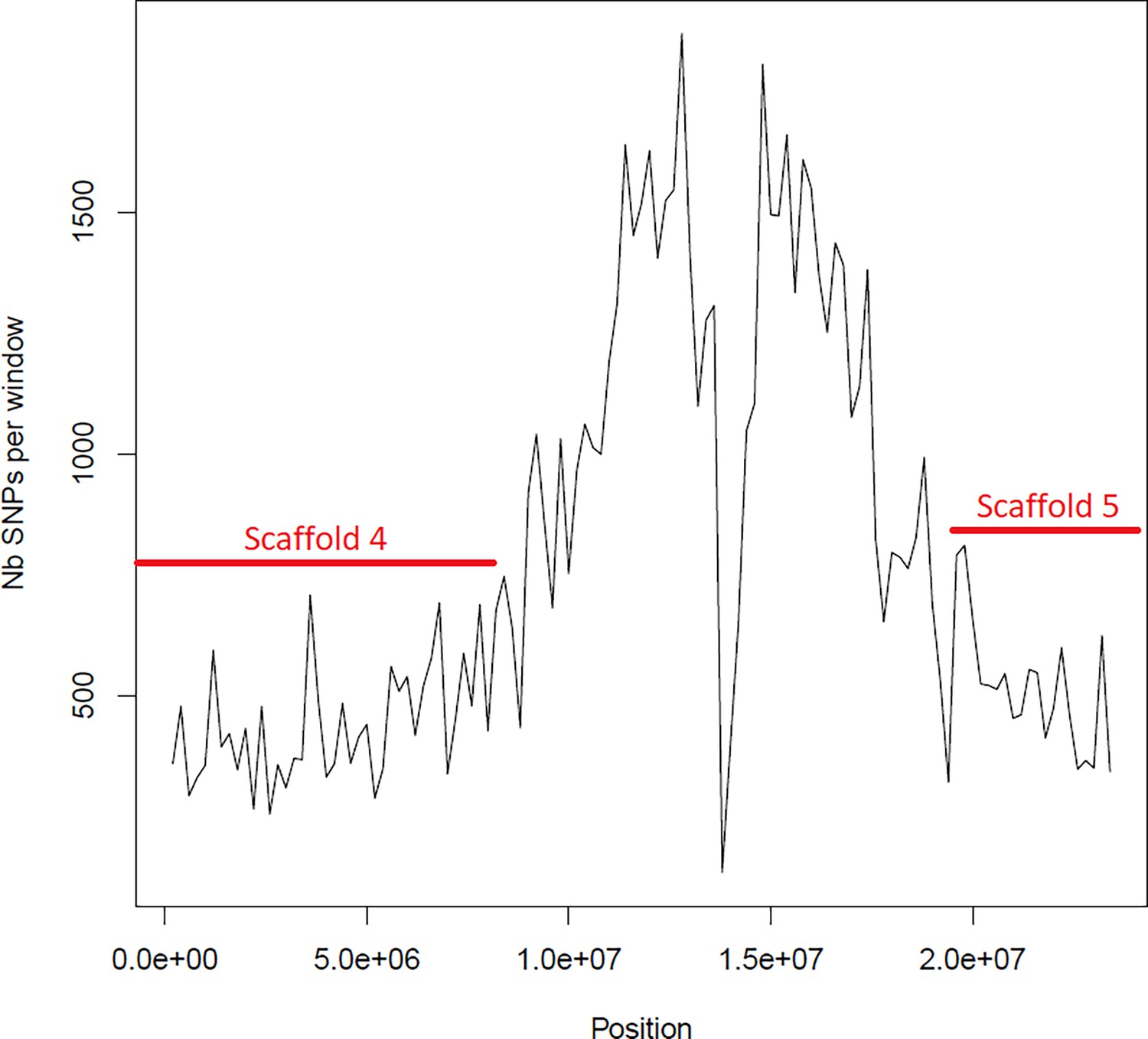
Figure 6—figure supplement 5
Average number of segregating site per window of 100kp on chromosome 4.
Estimated Average number of segregating site per window of 100kp on chromosome 4 on 10 individuals of the German accessions (black).
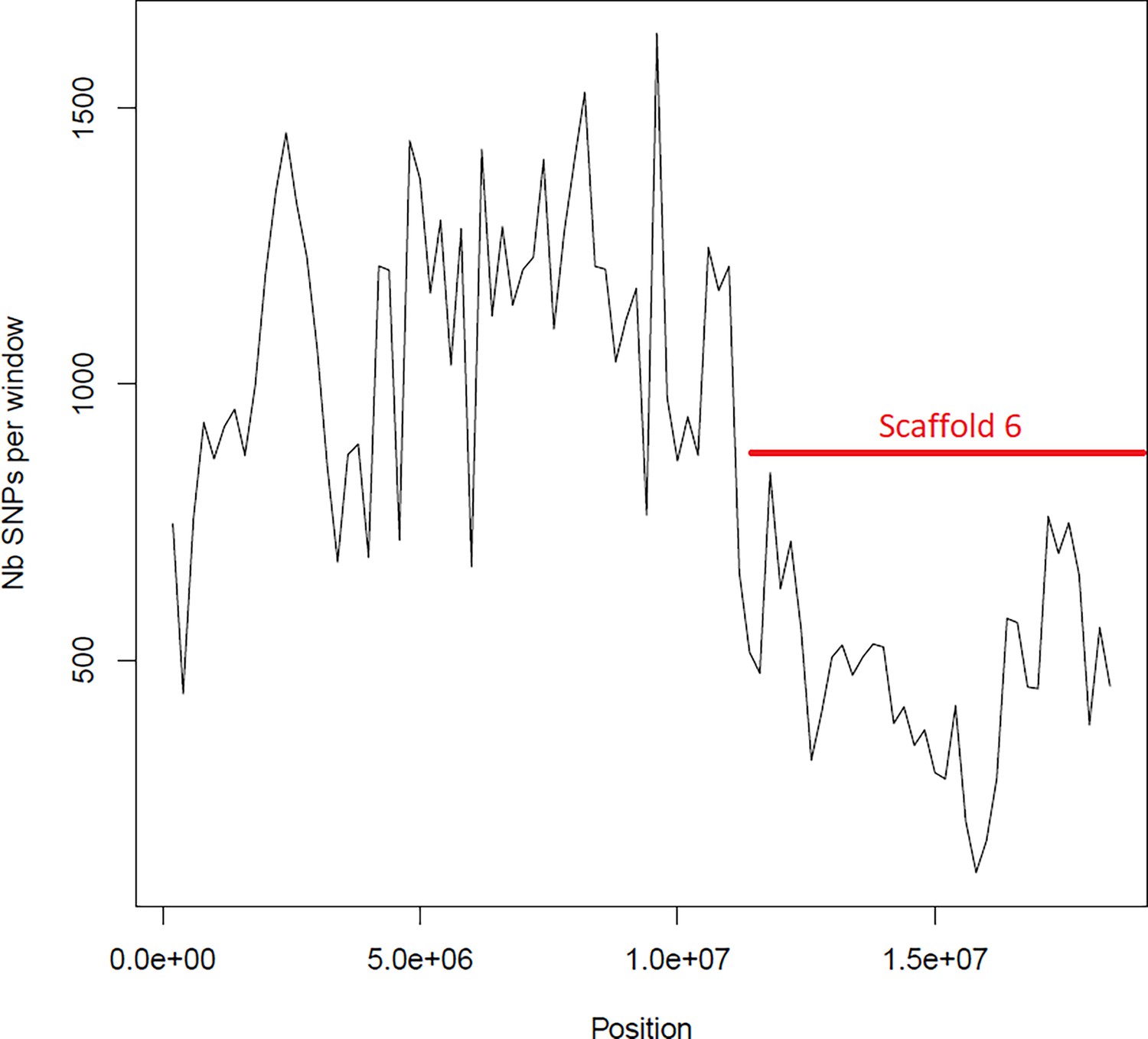
Figure 6—figure supplement 6
Average number of segregating site per window of 100kp on chromosome 5.
Estimated Average number of segregating site per window of 100kp on chromosome 5 on 10 individuals of the German accessions (black).
Tables
Table 1
Average estimated mutation rate of the second theoretical genomic marker.
Average estimated values of the mutation rate of marker 2 (), knowing that of marker 1. We use 10 sequences (5 diploid individuals) of 100 Mb ( per generation per bp) under a constant population size fixed at . The coefficient of variation over 10 repetitions is indicated in parentheses.
| True value | Estimated value of |
|---|---|
| 10−8 | 9.9×10−9 (0.02) |
| 10−6 | 1.0×10−6 (0.008) |
| 10−4 | 1.4×10−4 (0.01) |
| 10−2 | 3.05×10−3 (0.41) |
Table 2
Estimates of recombination rates with one or both markers.
For SMCtheo, BW stands for the use of the Baum-Welch algorithm to infer parameters, and LH for the use of the likelihood. We use 10 sequences of 100 Mb with , and per generation per bp in a population with a past bottleneck event. The coefficient of variation over 10 repetitions is indicated in brackets.
| Method | True recombination rate | Average estimated recombination rate |
|---|---|---|
| MSMC2 (BW) | 10−7 | 0.23×10−7 (0.017) |
| 1 Marker: BW | 10−7 | 0.25×10−7 (0.012) |
| 2 Marker: BW | 10−7 | 0.90×10−7 (0.004) |
| 1 Marker: LH | 10−7 | 0.84×10−7 (0.036) |
| 2 Marker: LH | 10−7 | 0.94×10−7 (0.01) |
Additional files
-
Supplementary file 1
Supplementary Tables.
(a) Average mean root square error (MRSE) of demographic inference in Figure 2, Figure 2—figure supplement 1 and Figure 2—figure supplement 2. Average mean root square error (in log10) of demographic inference in Figure 2A–D, Figure 2—figure supplements 1 and 2 shows the three approaches (eSMC2, SMCtheo with unknown rates, SMCtheo with known rates and MSMC2). The coefficient of variation is indicated in parentheses (b) Percentage of repetitions rejecting the hypothesis at P=0.05 of binomial distribution of epimutations over 100 repetitions using two sequences of 100 Mb with recombination and mutation rate set to per generation per bp under a constant population size fixed to 10,000. (c) Average estimated rate of the site methylation and demethylation rates from simulations. True versus average estimated values of the site methylation and demethylation rates over ten repetitions. We use two sequences of 100 Mb with per generation per bp under a constant population size fixed to 10,000. The coefficient of variation is indicated in brackets. (d) Average estimated rate of the region methylation and demethylation rates from simulations. True versus average estimated values of the region methylation and demethylation rates over ten repetitions. We use two sequences of 100 Mb with per generation per bp under a constant population size fixed to 10,000. The coefficient of variation is indicated in brackets. (e) Average estimated rate of both site and region methylation and demethylation rates from simulations. Average estimated values of the site and region methylation and demethylation rates over ten repetitions using 2 sequences of 100 Mb with recombination and mutation rate set to per generation per bp under a constant population size fixed to 10,000. The coefficient of variation is indicated in brackets. (f) Average mean root square error of demographic inference in Figure 5. Average mean root square error (in log10) of demographic inference in Figure 5 by the two approaches eSMC2, SMCm with unknown epimutations rates (A and C), and SMCm with known epimutation rates (B and D). Note the second row indicates the MRSE in recent times (younger than 400 generations ago). The coefficient of variation is indicated in parentheses (g) Average mean root square error of coalescent time along the genome inference. Average mean root square error of inferred coalescent time (in generation unit) along the genome over ten repetitions by the three approaches (eSMC2, SMCm with unknown epimutation rates and SMCm with known epimutation rates) under the same scenario from Figure 5. Inference was performed on two haploid sequences of 10 Mb with , per generation per bp. Methylation and demethylation rates were respectively fixed to and per generation per bp. The selfing rate was fixed to 90%. The coefficient of variation is indicated in parentheses. (h) Average mean root square error of demographic inference in Figure 5—figure supplements 1 and 2. Average mean root square error (in log10) of demographic inference in Figure 5—figure supplements 1 and 2 by the three approaches (eSMC2, SMCm with unknown epimutations rates and SMCm with known epimutation rates). Note that the second row indicates the MRSE in recent times (younger than 400 generations ago). The coefficient of variation is indicated in parentheses (i) Average mean root square error of demographic inference in Figure 5—figure supplement 3. Average mean root square error (in log10) of demographic inference in Figure 5—figure supplement 3 by the three approaches (eSMC2, SMCm with unknown epimutations rates, SMCm with known epimutation rates). Note that the second row indicates the MRSE in recent times (younger than 400 generations ago). The coefficient of variation is indicated in parentheses (j) Average mean root square error of demographic inference in Figure 5—figure supplement 4. Average mean root square error (in log10) of demographic inference in Figure 5—figure supplement 4 by the three approaches (eSMC2, SMCm with unknown epimutations rates, SMCm with known epimutation rates). Note that the second row indicates the MRSE in recent times (younger than 400 generations ago). The coefficient of variation is indicated in parentheses. (k) Average estimated rate of the site methylation and demethylation rates in A. thaliana. Average estimated values of the site methylation and demethylation rates by SMCm using genomes and methylomes from 10 German accessions of A. thaliana. We use eight scaffolds each of 10 sequences with recombination and mutation rate respectively set to and per generation per bp with selfing set to 90%. The polymorphic SMPs CG sites estimations corresponds to the green line in Figure 6. All CG sites estimations and CG site separated by 3,000 bp corresponds to the data of the green line in Figure 6—figure supplement 1. (l) Average estimated rate of the site and region methylation and demethylation rates in A. thaliana. Average estimated values of the site and region methylation and demethylation rates by SMCm using genomes and methylomes from 10 German accessions of A. thaliana. These estimations are produced during the inference of the red line in Figure 6—figure supplement 1. We use eight scaffolds each of 10 sequences with recombination and mutation rate respectively set to and per generation per bp with selfing set to 90%.
- https://cdn.elifesciences.org/articles/89470/elife-89470-supp1-v1.pdf
-
MDAR checklist
- https://cdn.elifesciences.org/articles/89470/elife-89470-mdarchecklist1-v1.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Improved inference of population histories by integrating genomic and epigenomic data
eLife 12:RP89470.
https://doi.org/10.7554/eLife.89470.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}