Natural variation in salt-induced changes in root:shoot ratio reveals SR3G as a negative regulator of root suberization and salt resilience in Arabidopsis
- Boyce Thompson Institute, United States
- School of Life Sciences, Lanzhou University, China
- Center for Desert Agriculture, King Abdullah University of Science and Technology, Saudi Arabia
- USDA-ARS, United States
- Visualization Core Lab, King Abdullah University of Science and Technology, Saudi Arabia
- University of California, Davis, United States
- Department of Mathematics, King Fahd University of Petroleum and Minerals, Saudi Arabia
- Julius-von-Sachs-Institute and Center for Computational and Theoretical Biology, Julius Maximilian University, Germany
- King Abdullah University of Science and Technology, Saudi Arabia
- Wageningen University & Research, Netherlands
Figures
Figure 1 with 7 supplements
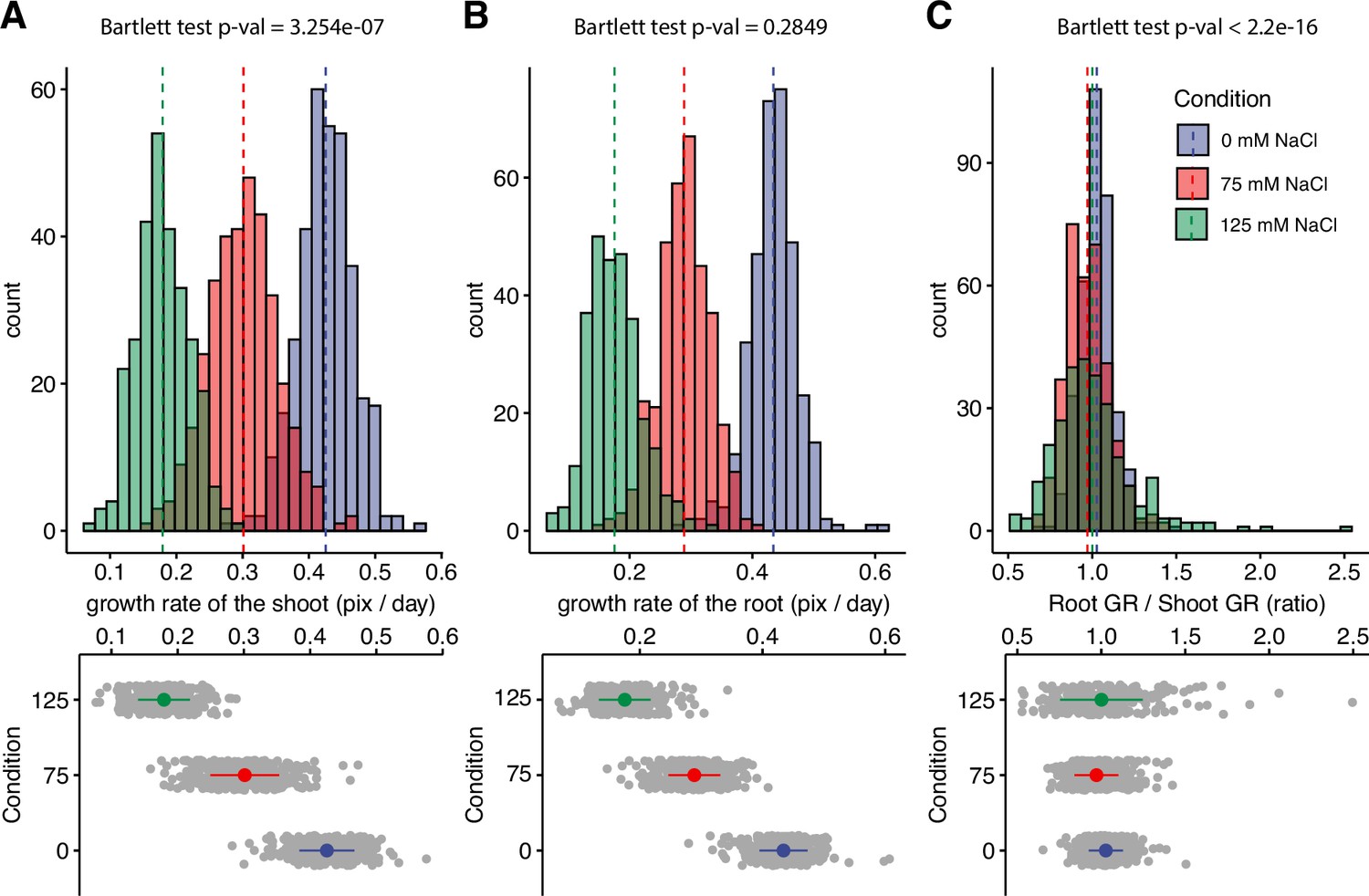
Salt stress is causing discoordination of root and shoot growth.
Arabidopsis HapMap population was screened for salt stress-induced changes in root:shoot ratio. The increase in the projected area of shoot and root (Figure 1—figure supplement 2) was used to estimate (A) shoot and (B) root exponential growth rate. (C) The root:shoot growth rate ratio was calculated per genotype. The histograms represent the number of accessions across three studied conditions (0, 75, and 125 mM NaCl), whereas the population average is indicated using the dashed line. Additionally, the distribution of the genotypes within each treatment was visualized using the error plots (lower panel), where population average and standard error is indicated using a colored point and a line, respectively. Individual gray points represent individual genotypes.
Figure 1—figure supplement 1
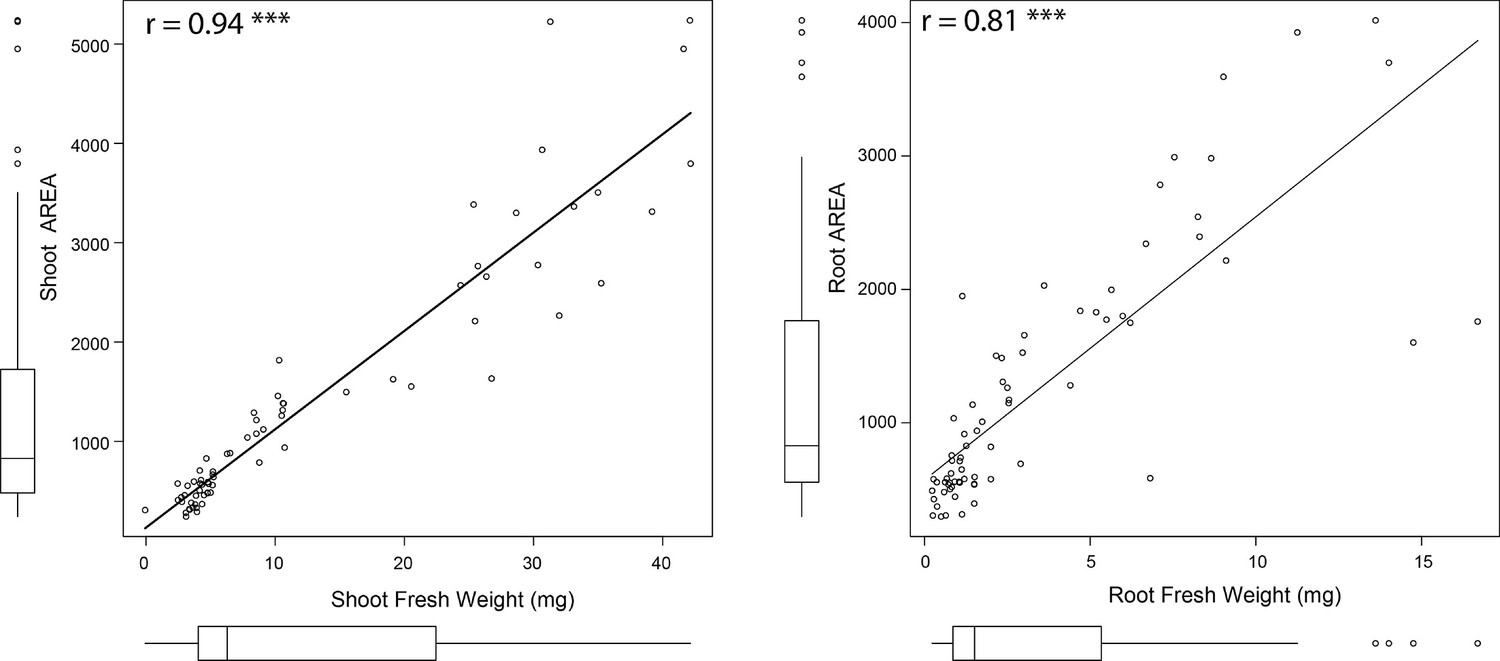
Evaluation of the tool’s precision for estimating seedling size.
The tool used to quantify root and shoot size from the agar plates was tested on the Arabidopsis Col-0 seedlings exposed to various concentrations of salt stress (0, 75, and 125 mM NaCl). The fresh weight of root and shoot of individual plants were recorded at the last day of experiment (8 d after transfer to treatment plates, 12 d after germination), and the correlation analysis between individual organ’s fresh weight and area was performed in R. The Correlation co-efficient and the p-value for individual correlations were calculated in R using stats package.
Figure 1—figure supplement 2
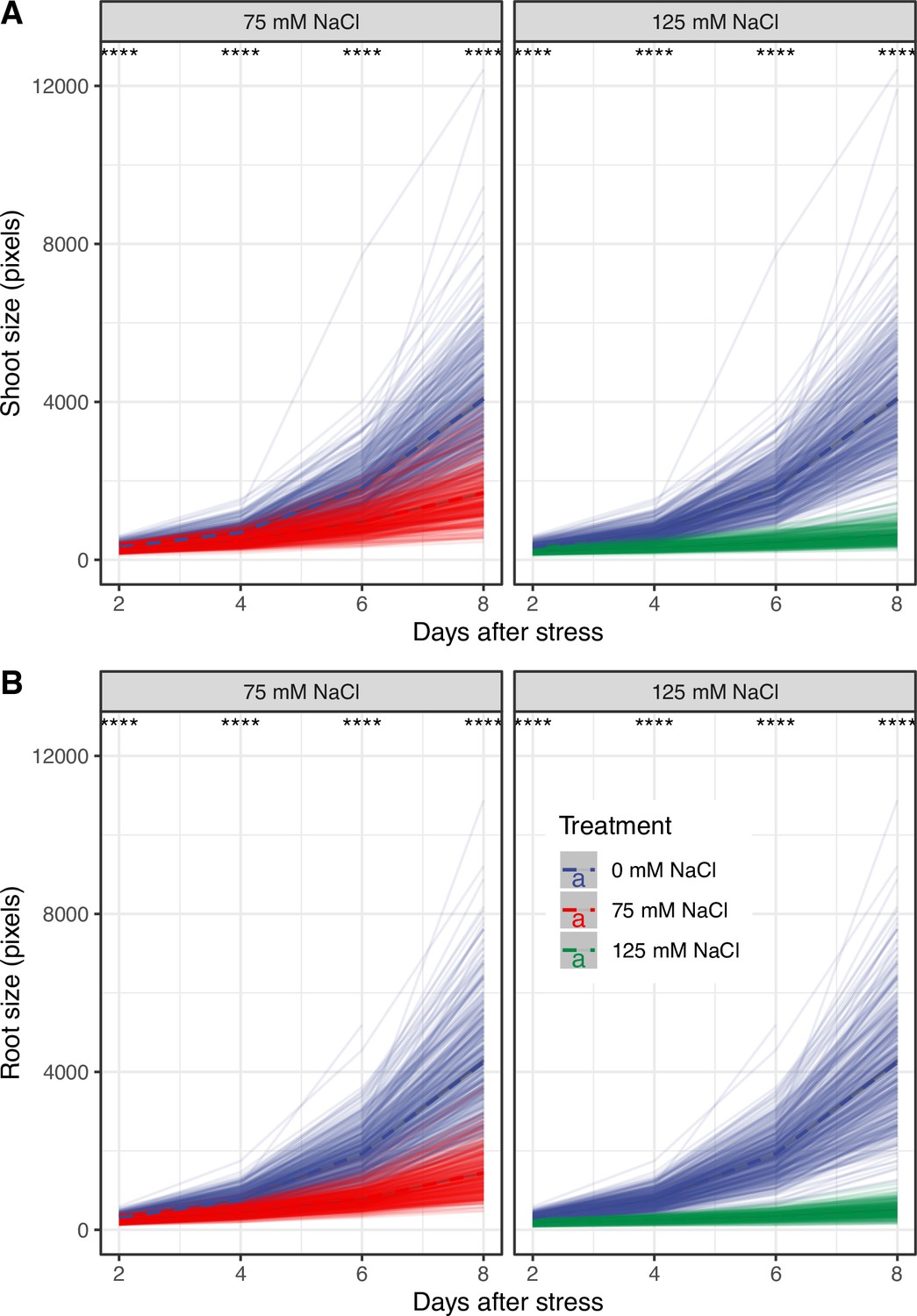
Salt stress reduced the increase in root and shoot area over time across HapMap accession.
Four-days-old seedlings exposed to 0, 75, and 125 mM NaCl grown on agar plates were scanned every second day and analyzed for root and shoot area using a custom tool. The change in genotype-specific (A) shoot and (B) root area was calculated in R using stats package. The thick dashed lines represent population-wide average in root and shoot area over the time of the experiment, while individual transparent lines indicate change in root and shoot area for individual accession. The differences between control and individual salt stress treatments were evaluated using ANOVA. The *, **, ***, and **** represent p-values below 0.05, 0.01, 0.001, and 0.0001, respectively.
Figure 1—figure supplement 3
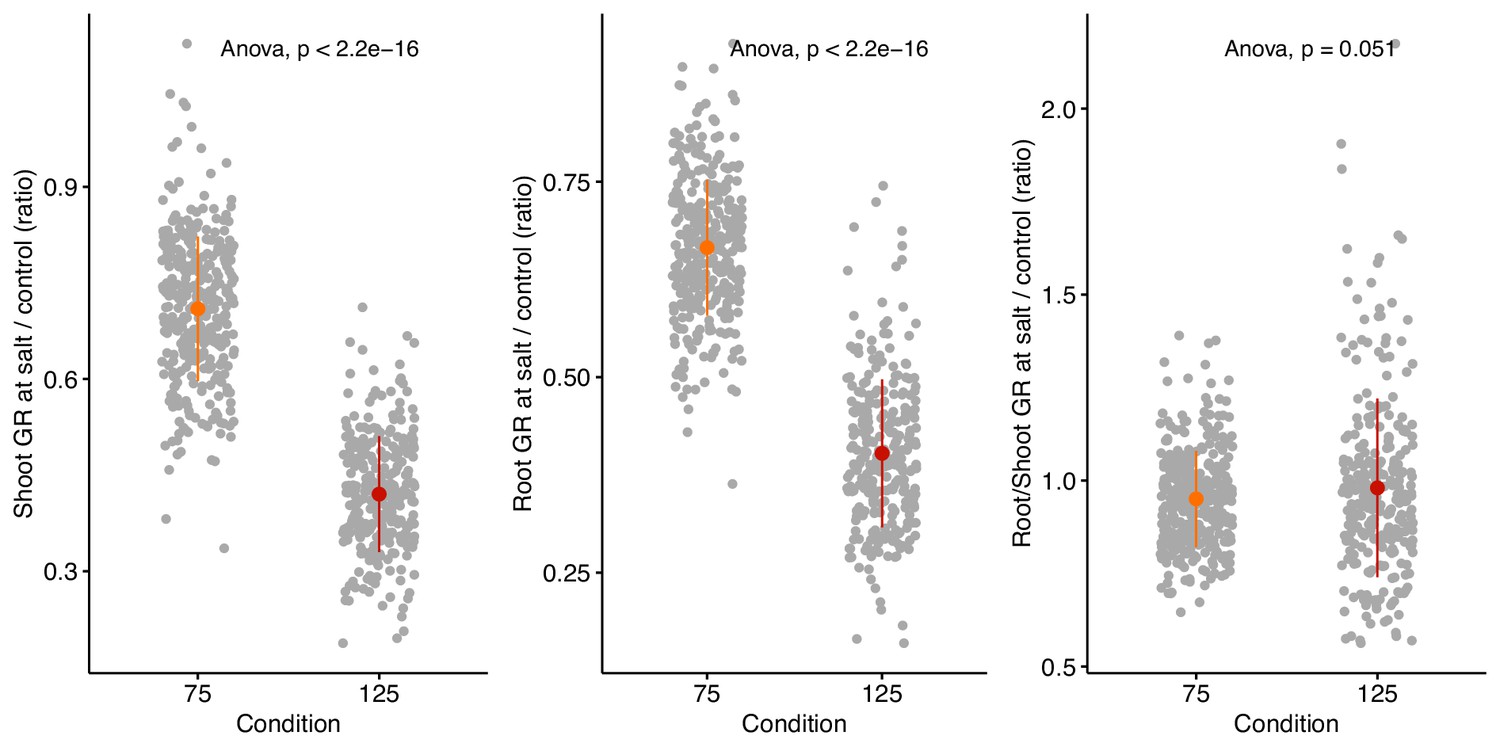
Salt stress induced changes in relative growth of shoot, root, and root:shoot ratio.
The stress tolerance index was calculated for shoot, root and root:shoot ratio by dividing genotype-specific value recorded at salt stress over the genotypic mean of the trait recorded under non-stress conditions. Individual points represent individual genotypes. The population mean for 75 and 125 mM NaCl treated plants is represented by the colored point, whereas the population-wide standard error by whiskers.
Figure 1—figure supplement 4
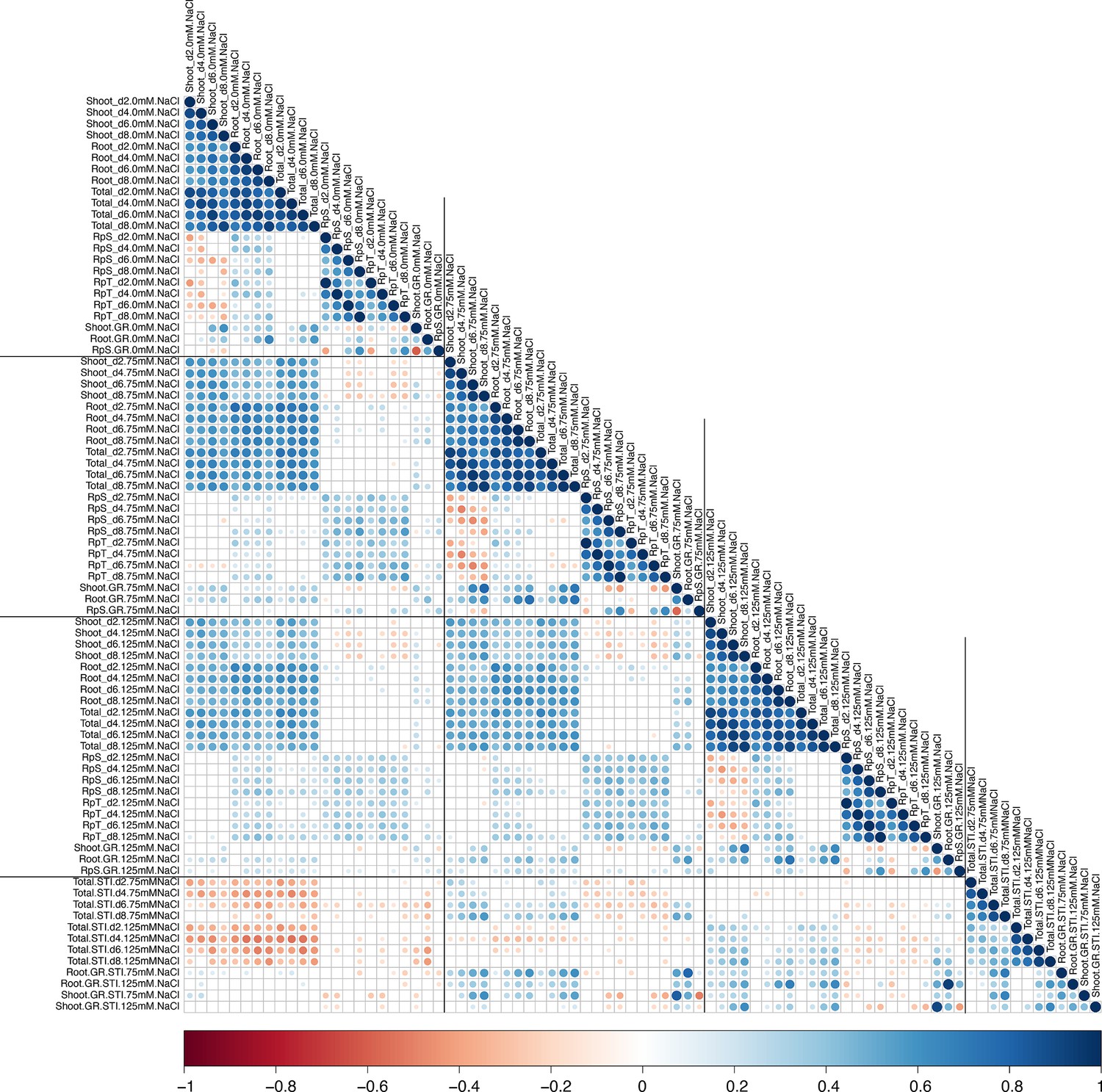
The correlation between all measured traits and calculated stress tolerance indices (STI).
The correlations between all measured and calculated traits was performed using the Pearson’s correlation coefficient. All non-significant correlations were replaced by blank circles. The size and color of individual circles indicates the strength and significance of individual correlation between two traits. Blue and red shades indicate positive and negative correlations, respectively.
Figure 1—figure supplement 5
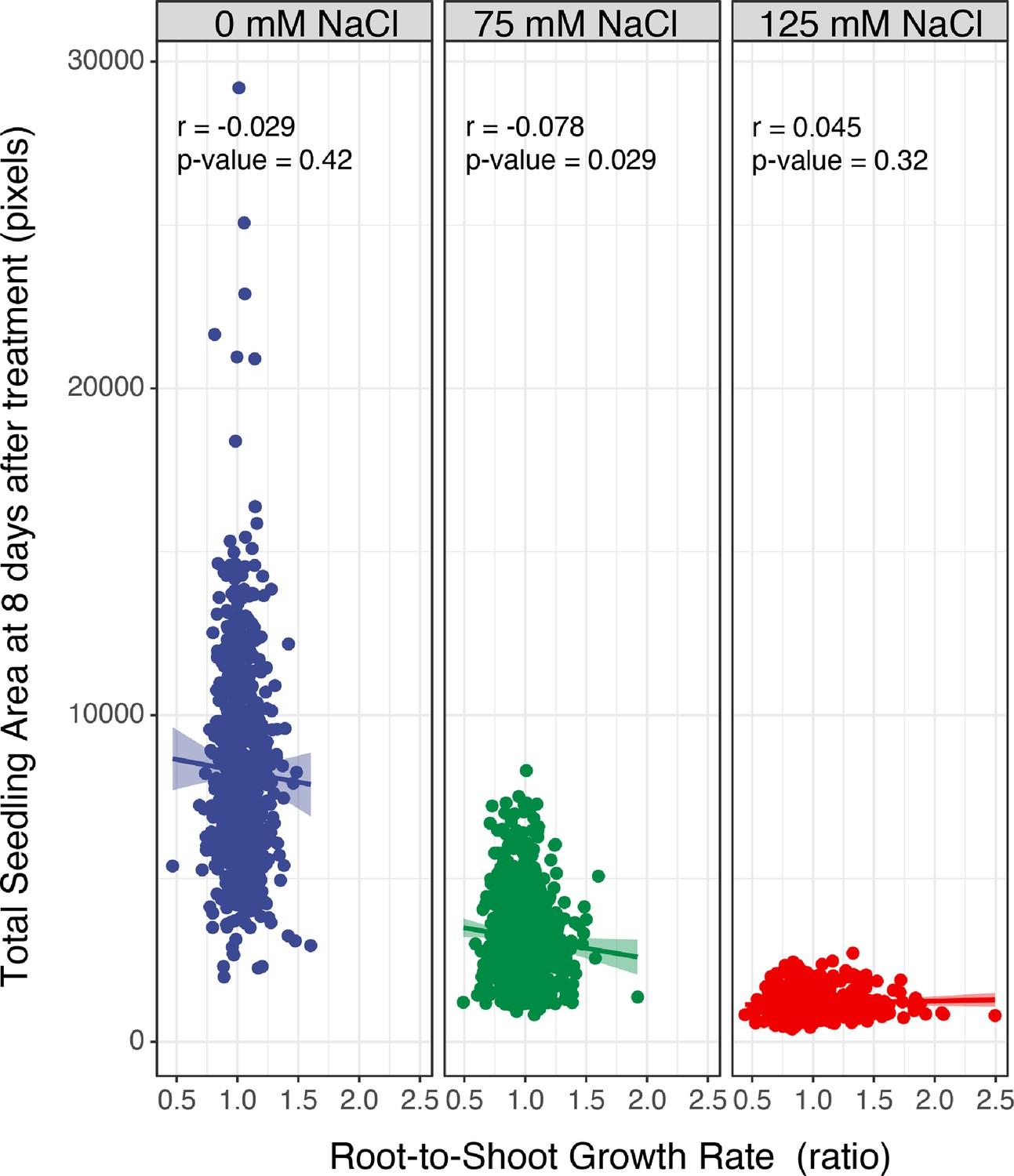
The correlation between total seedling area and salt-induced changes in the root-to-shoot ratio.
The correlation between the total seedling area quantified using the custom tool at the last day of experiment (8 d after transfer and 12 d after germination), and the ratio of root-and-shoot growth rate was examined across Arabidopsis HapMap population and various concentrations of salt stress (0, 75, and 125 mM NaCl). The correlation analysis was performed in R, and the Correlation co-efficient (r) and the p-values for individual correlations were calculated in R using stats package.
Figure 1—figure supplement 6
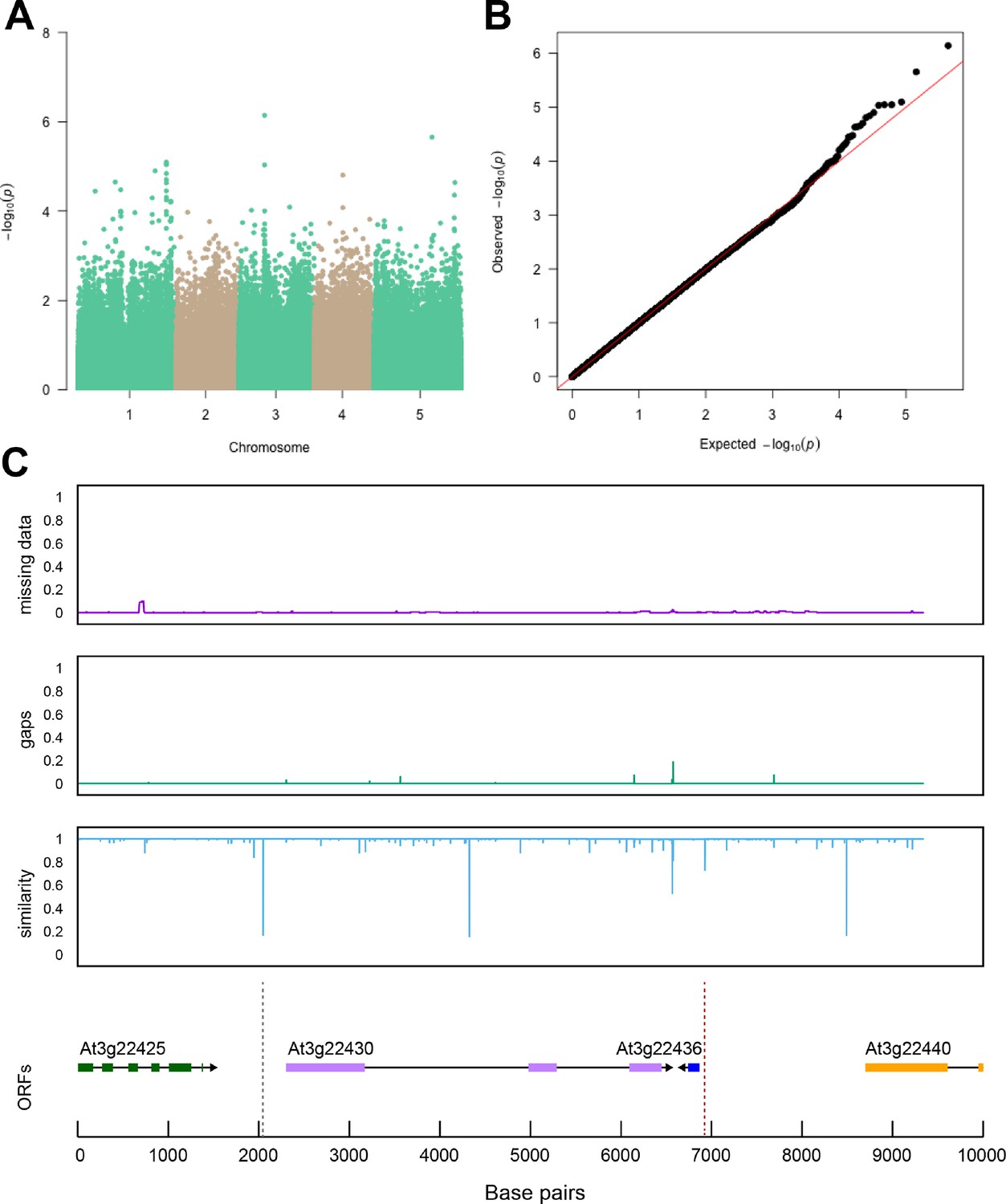
Region around NTL8 is associated with salt stress-induced changes in root-per-shoot growth.
The salt-induced changes in root-to-shoot and shoot-per-total seedling area ratios were used as an input for genome-wide association study (GWAS). (A) Manhattan plot represents the associations of root-per-shoot growth factors recorded at 75 mM NaCl with 250 k SNPs while (B) represents the QQ plot for this association study. (C) Significant associations were found with the SNPs forming a locus on chromosome 2 in and around AT2G27300, encoding a NAC-domain containing transcription factor (NTL8). The natural variation in the linkage-disequilibrium (LD) region was studied in 162 accessions sequenced by 1001 Genomes Project. The upper panel represents portion of the missing data, upper middle panel represents deletions relative to Col-0, while lower middle panel represents the sequence similarity compared to Col-0. The bottom panel represents the open reading frames (ORFs) within the LD, and the location of associated SNPs is indicated with the dashed lines. Red dashed lines represent associations above the Bonferroni threshold in 250 k SNP mapping, while gray lines represent associations with -log10(p-value)>5 in the 4 M SNP mapping.
Figure 1—figure supplement 7
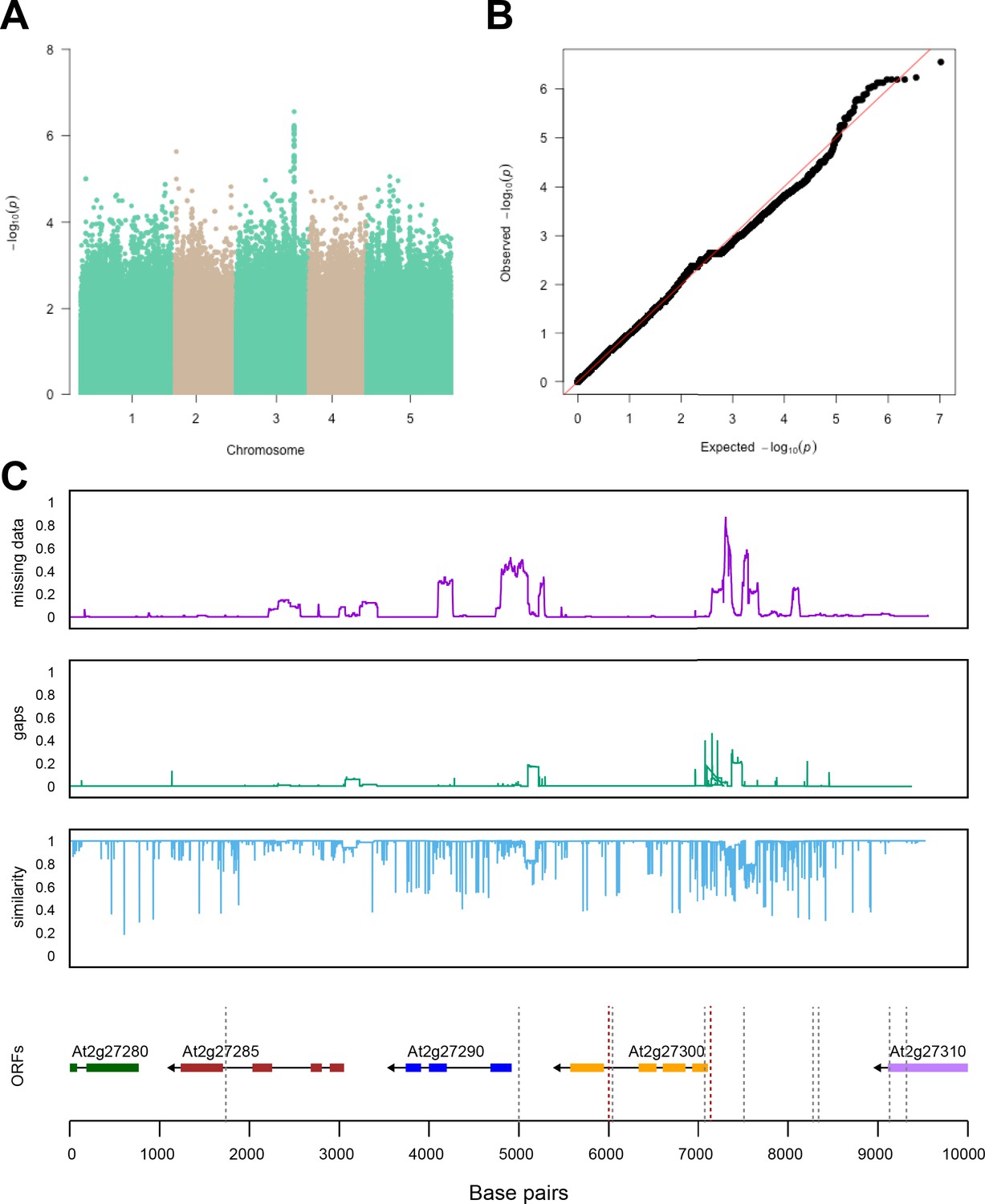
Region around unknown gene is associated with salt stress-induced changes in relative changes in shoot-per-total seedling area.
The salt-induced changes in root-to-shoot and shoot-per-total seedling area ratios were used as an input for genome-wide association study (GWAS). (A) Manhattan plot represents the associations of relative change (125 mM/0 mM) in shoot-per-total seedling area at 4 d after transfer to 125 mM NaCl, while (B) represents the QQ plot for this association study. (C) Significant associations were found with the SNPs forming a locus on chromosome 3 in and around AT3G22430, encoding an unknown gene encoding a domain of unknown function. The natural variation in the linkage-disequilibrium (LD) region was studied in 162 accessions sequenced by 1001 Genomes Project. The upper panel represents portion of the missing data, upper middle panel represents deletions relative to Col-0, while lower middle panel represents the sequence similarity compared to Col-0. The bottom panel represents the open reading frames (ORFs) within the LD, and the location of associated SNPs is indicated with the dashed lines. Red dashed lines represent associations above Bonferroni threshold in 250 k SNP mapping, while gray lines represent associations with -log10(p-value)>5 in the 4 M SNP mapping.
Figure 2 with 5 supplements
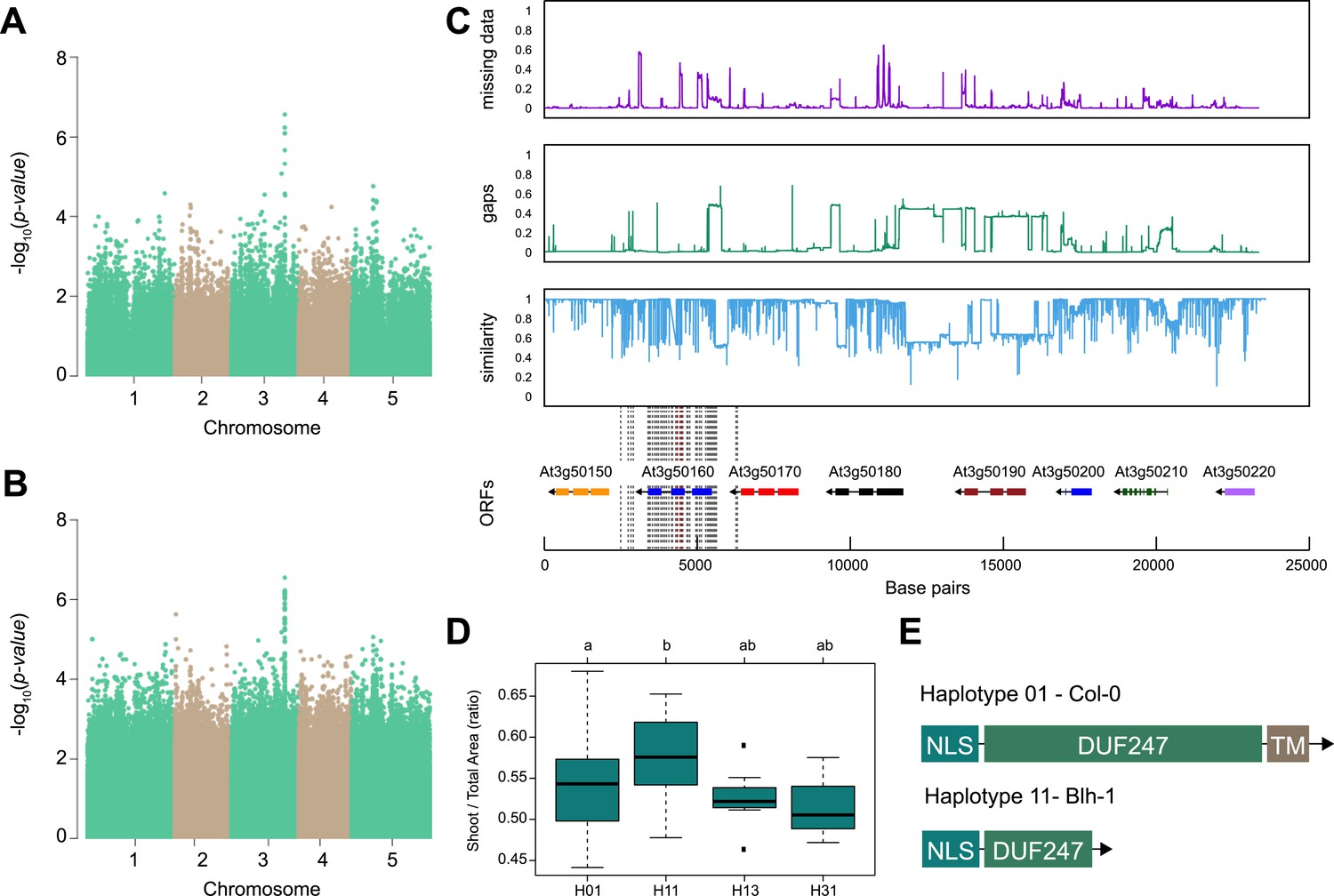
SR3G is associated with salt stress-induced changes in shoot-per-total seedling area ratio.
The salt-induced changes in root-to-shoot and shoot-per-total seedling area ratios were used as an input for genome-wide association study (GWAS). Manhattan plots representing the associations of shoot-per-total seedling area recorded 6 d after transfer to 125 mM NaCl and (A) 250 k SNPs and (B) 4 M SNPs. (C) Significant associations were found with the SNPs forming a locus on chromosome 3 in and around SR3G (AT3G50160), encoding Domain of Unknown Function 247 (DUF247). The natural variation in the linkage-disequilibrium (LD) region was studied in 162 accessions sequenced by 1001 Genomes Project. The upper panel represents portion of the missing data, upper middle panel represents deletions relative to Col-0, while lower middle panel represents the sequence similarity compared to Col-0. The bottom panel represents the open reading frames (ORFs) within the LD, and the location of associated SNPs is indicated with the dashed lines. Red dashed lines represent associations above Bonferroni threshold in 250 k SNP mapping, while gray lines represent associations with -log10(p-value)>5 in the 4 M SNP mapping. (D) The haplotype analysis performed using SNPs located within the coding region of the SR3G revealed significant differences in shoot-per-total seedling area recorded 6 d after transfer to 125 mM NaCl between Haplotype groups 1 (represented by 46 accessions, including Col-0) and 11 (represented by 11 accessions, including Blh-1). The significant differences between individual haplotype groups were tested using ANOVA with Tukey HSD to identify significantly different groups. (E) Upon further sequencing of SR3G in accessions from Haplotype 1 and 11, revealed two 200 bp insertions within SR3G exons in four tested accessions belonging to haplotype 11 group. These 200 bp insertions resulted in a premature STOP-codon within the DUF247 domain (after Gly-215).
Figure 2—figure supplement 1
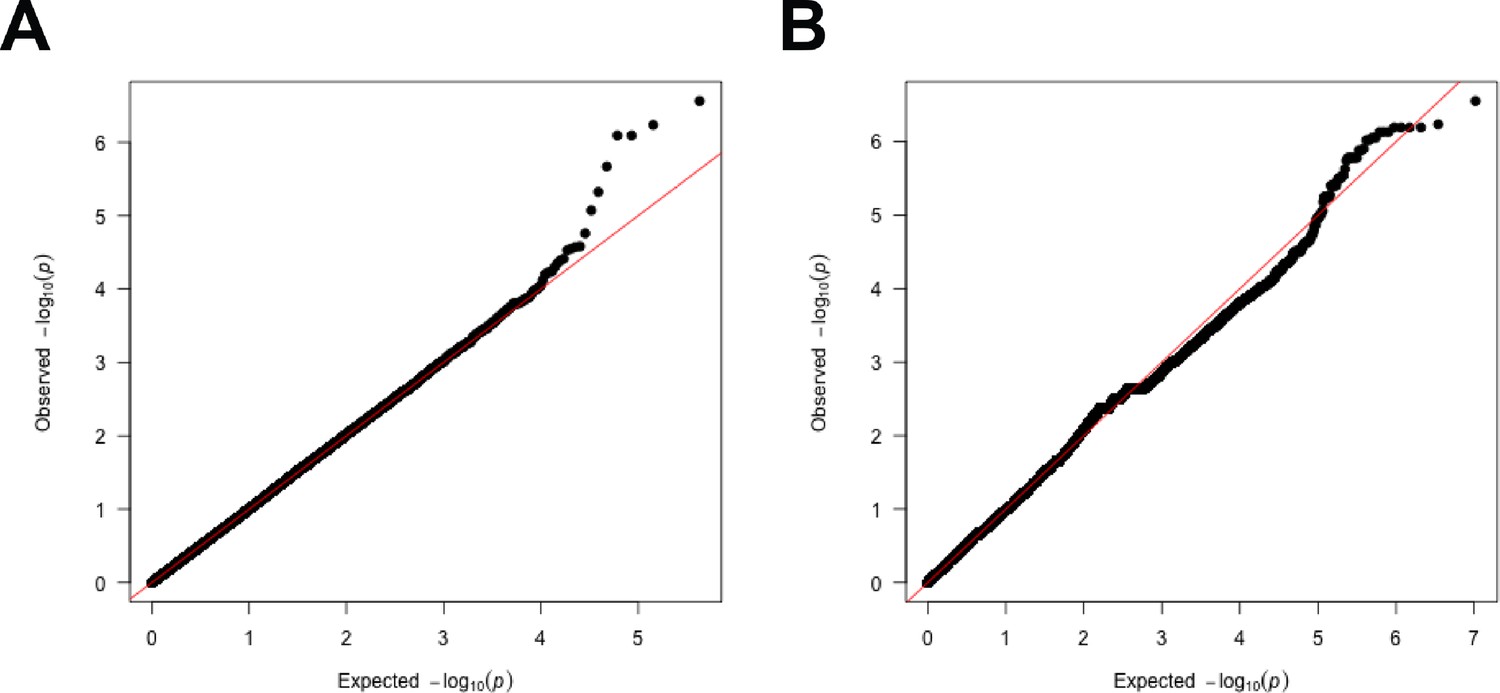
The QQ-plots for genome-wide association study (GWAS) models with shoot-per-total seedling area.
The QQ-plots for association between shoot-per-total seedling area recorded 6 d after transfer to 125 mM NaCl and (A) 250 k SNPs and (B) 4 M SNPs. The red line indicates the expected vs. observed ratio of association, while black points indicate the observed associations.
Figure 2—figure supplement 2
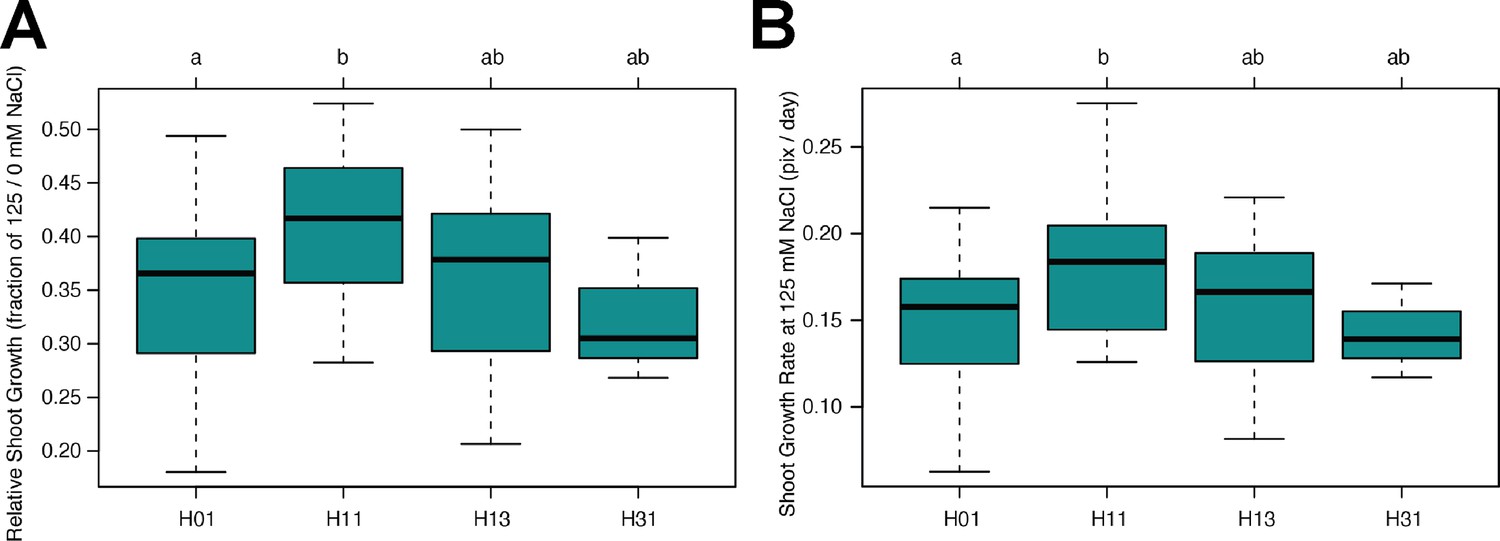
Haplotype analysis of AT3G50160 (SR3G).
The haplotype analysis performed using SNPs located within the coding region of the SR3G (AT3G50160) revealed significant differences in (A) Relative shoot growth (as fraction of shoot growth at 125 mM NaCl and 0 mM NaCl) and (B) Shoot growth at 125 mM NaCl. The significant differences between individual haplotype groups (each haplotype represented by at least three accessions), were tested using ANOVA with Tukey HSD to identify significantly different groups. Significant differences were identified between Haplotype groups 1 (represented by 46 accessions, including Col-0) and 11 (represented by 11 accessions, including Blh-1).
Figure 2—figure supplement 3
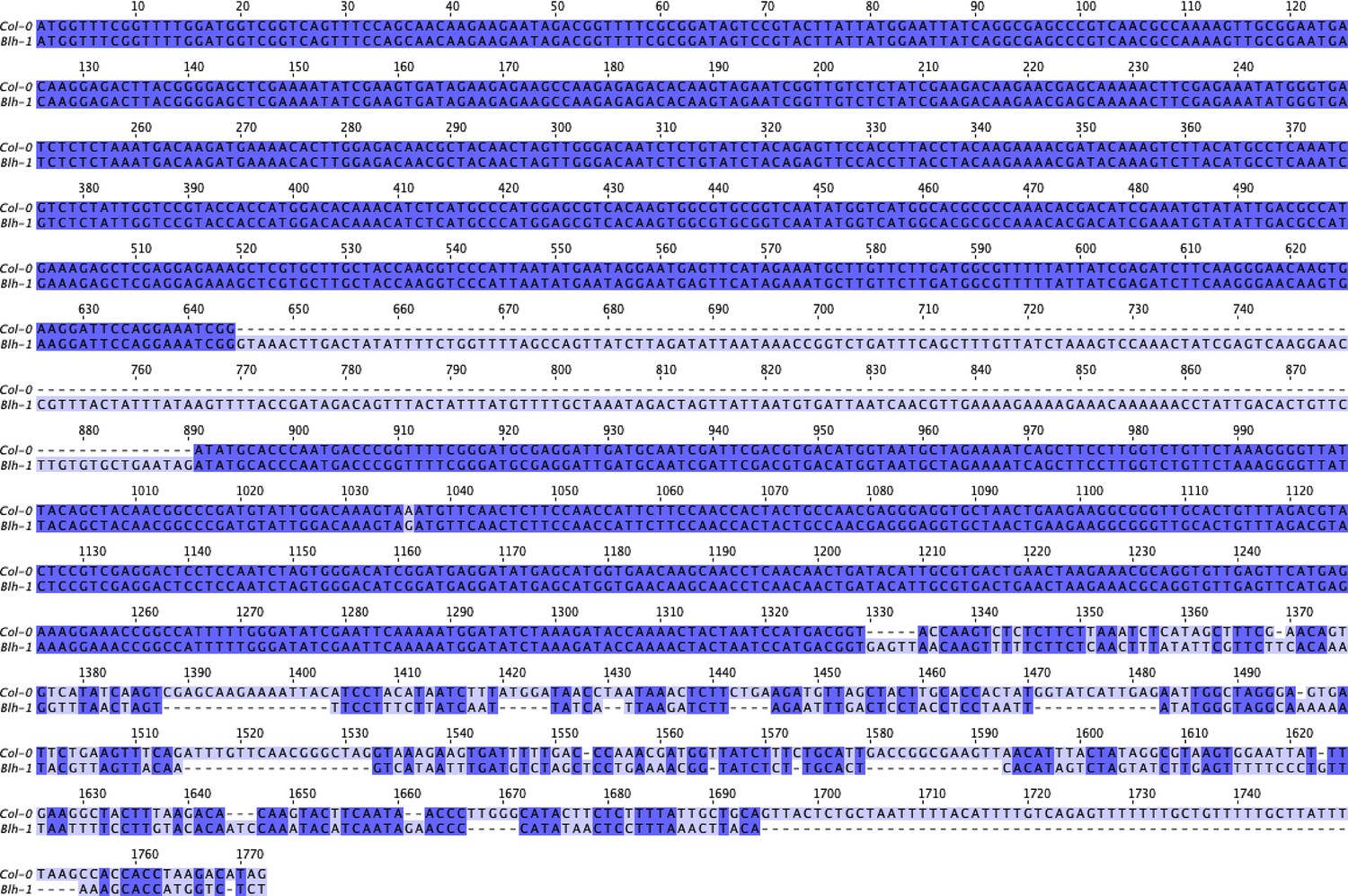
Nucleotide sequence alignment of SR3G CDS between Col-0 and Blh-1 alleles.
The nucleotide sequences from SR3G CDS were obtained from Col-0 and Blh-1 cDNA, and compared to Col-0 cDNA sequence on TAIR. The cDNA sequences were subsequently aligned in JalView using ProbconsWS alignment. The intensity of purple hue indicates the nucleotide identity, while dash marks indicate insertion in Blh-1 compared to Col-0 allele.
Figure 2—figure supplement 4

Amino acid sequence alignment of SR3G CDS between Col-0 and Blh-1 alleles.
The amino acid sequences from SR3G CDS were obtained by translating Col-0 and Blh-1 cDNA into amino acid sequence in JalView using Standard settings. The amino acid sequences were subsequently aligned in JalView using ProbconsWS alignment. The sequence length markers for are indicated above the aligned sequence. The intensity of purple hue indicates the nucleotide identity, while dash marks indicate insertion in one of the alleles. * indicate STOP codons.
Figure 2—figure supplement 5

Sequence alignment between Col-0 and Blh-1 alleles for SR3G promotor.
The nucleotide sequences from SR3G promotor were obtained from Col-0 and Blh-1 genomic DNA, and compared to Col-0 sequence on TAIR. The gDNA sequences were subsequently aligned in JalView using ProbconsWS alignment. The intensity of purple hue is indicating the nucleotide identity, while dash marks indicate insertions.
Figure 3
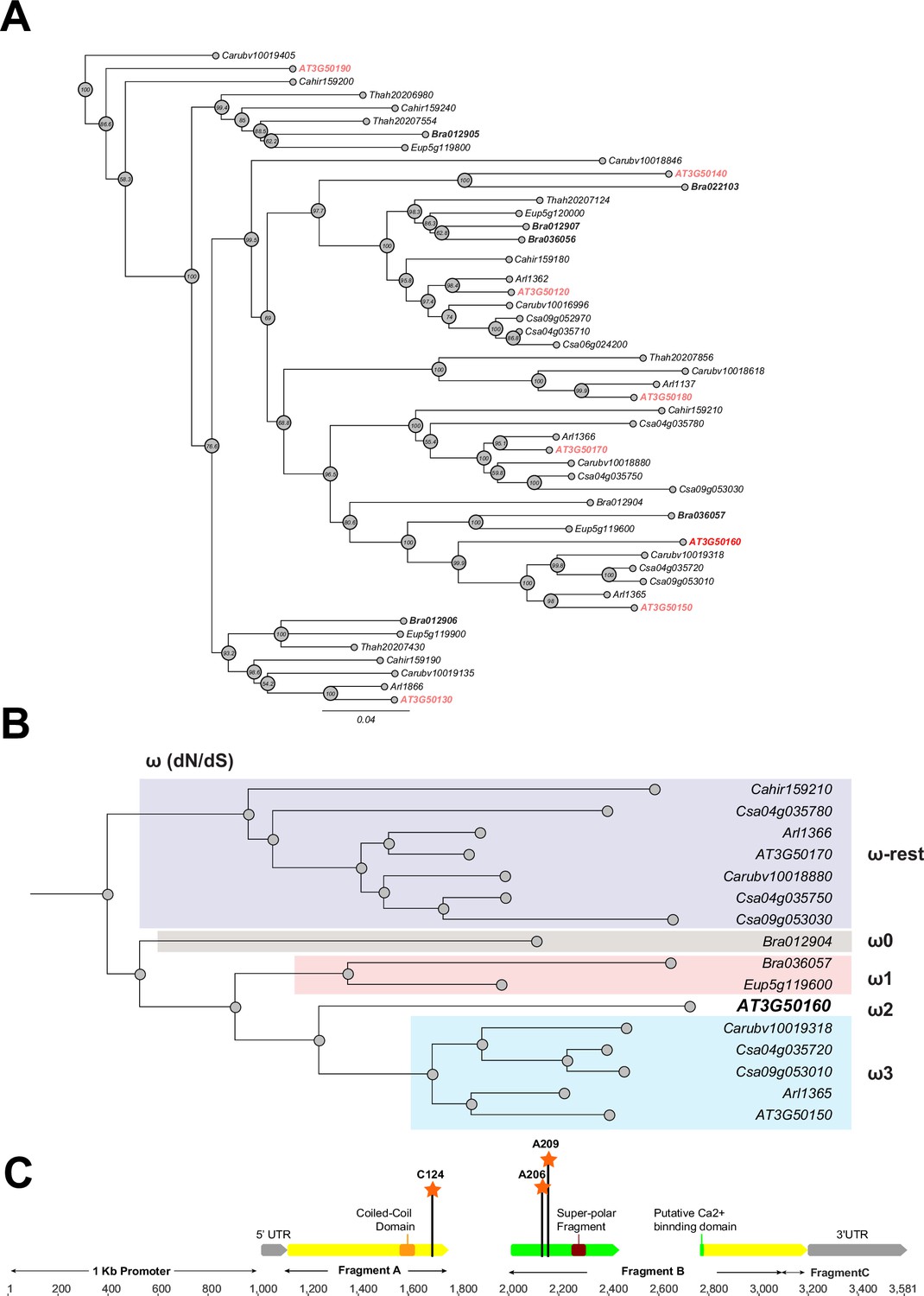
Phylogeny and positive selection analysis of SR3G and its orthologs from other species.
The orthologous genes of SR3G (AT3G50160) from other seven species were identified using GoGeBlast (E-value <1.00E-10). (A) The protein sequences of the 50 homologous genes were aligned by MUSCLE and then an unrooted phylogenetic tree was reconstructed by a Maximum -likelihood (ML)-derived style by RAxML (Bootstrap number: 100). (B) A positive selection analysis was conducted by branch-site model A test: Specifically, we hypothesized the differential or constant dN/dS (ω) substitution rate among the closely distant branches leading to DUF247 (AT3G50160) (See Method and materials for details). These branches were marked as ω1 (light brown branch), ω2 (DUF247-specific branch), ω3 (light blue branch), and ω-rest (light purple branch). (C) The gene structure of DUF247 (AT3G50160) was illustrated by wide arrows (gray arrow: 5 and 3’ UTR, yellow arrow: coding-region). The regions used for cloning (Fragments A, B, and C) were marked by narrow arrows along with ‘Coil-Coil Domain’ and ‘Super-polar Fragment’ marked on Fragments A and B, respectively. Lastly, the Bayes Empirical Bayes (BEB) was performed to test the probability of sites along with ω>1 over the DUF247 (AT3G50160). Three sites were generated with a posterior probability >0.90: 124 C (p=0.992), 206 A (p=0.951), and 209 A (p=0.988). These three sites were marked with an orange asterisk and assumed to have been undergoing positive selection.
Figure 4 with 2 supplements
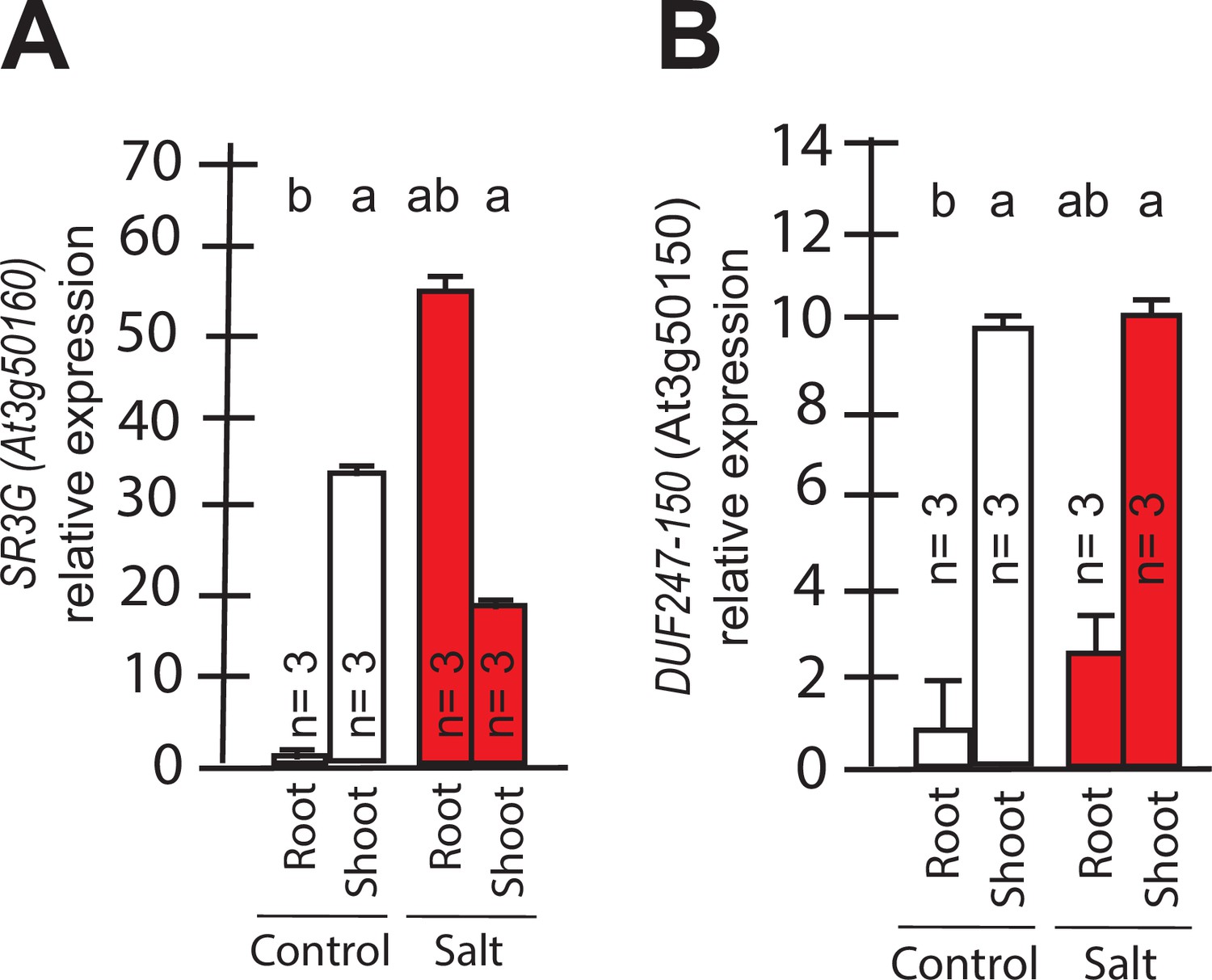
Transcript abundances for the two closely related DUF247s.
Expression of (A) SR3G (AT3G50160) (B) and its closest homolog DUF247-150 (AT3G50150) were measured in Col-0 seedlings grown with and without salt stress. RT-qPCR analyses were conducted using seedlings grown on 1/2 x MS for 4 d and then followed by transferring to the treatment plates with or without 75 mM NaCl for 1 wk. Mean values are shown ±SE, with number of replicates (n) shown in each graph. AT4G04120 was used as a reference gene for normalization. Significance was determined by the Tukey–Kramer HSD test in JMP.
Figure 4—figure supplement 1
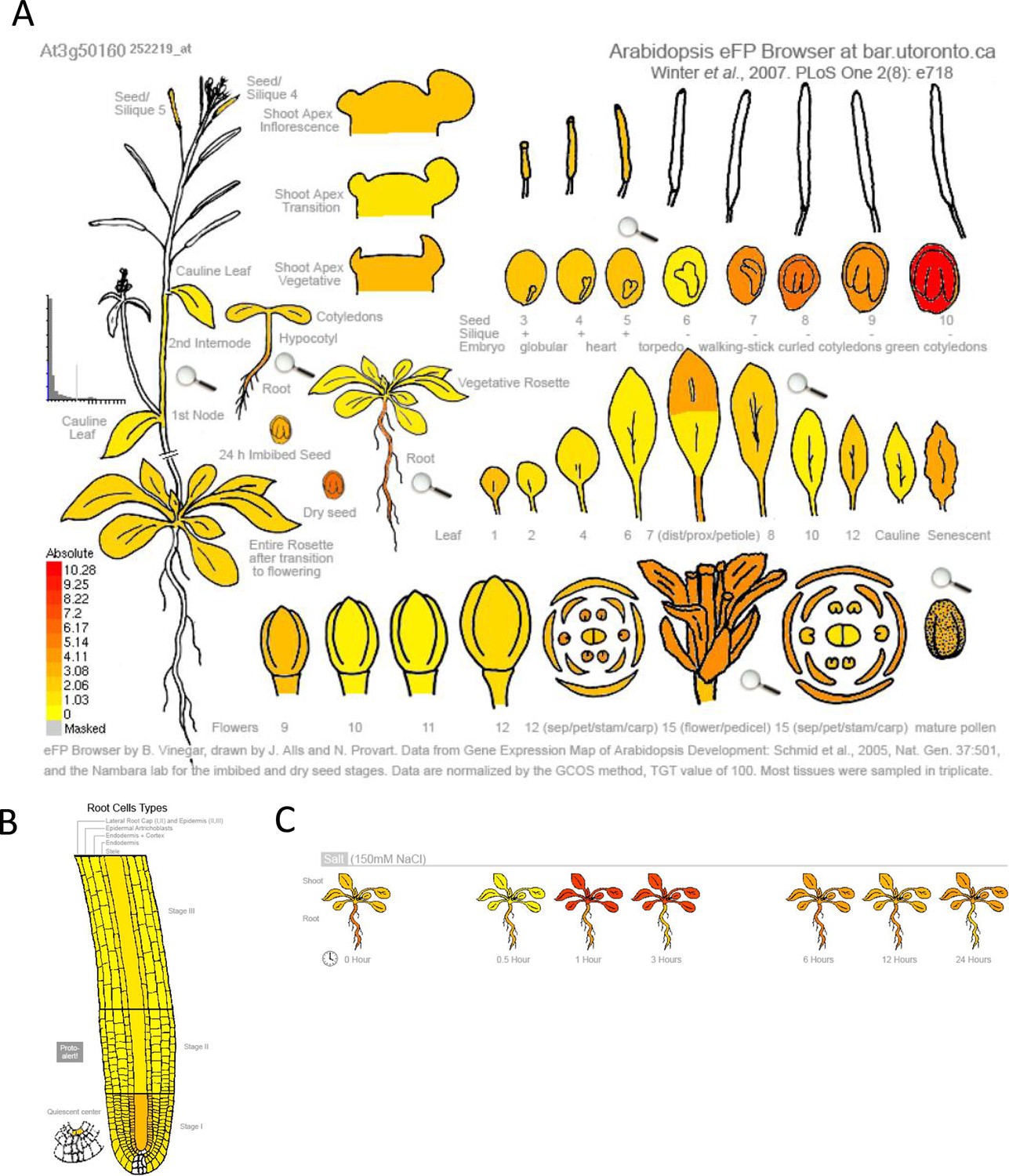
Gene expression profile of SR3G across various developmental stages and salt stress.
The absolute gene expression of SR3G is depicted across (A) various developmental stages, (B) root cell types, and (C) in response to salt stress. The images were acquired through the eFP Browser using data sources specified for the developmental map, abiotic stress, and tissue specific.
Figure 4—figure supplement 2
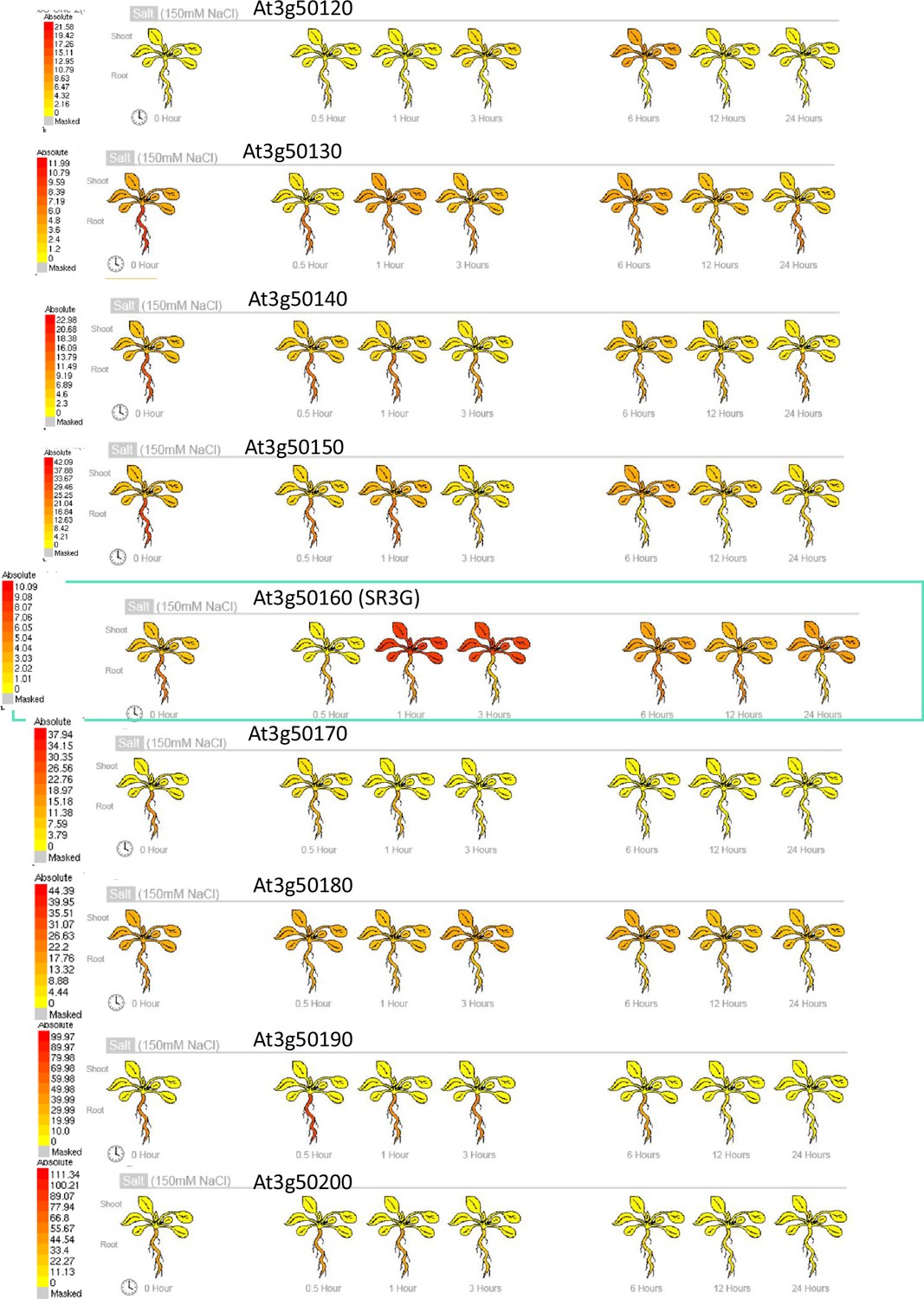
Gene expression profile of other DUF247s under salt stress.
Absolute gene expression of other DUF247s is illustrated in response to salt-induced changes. The images were acquired through the eFP Browser using data sources specified for the abiotic stress. The SR3G is shown in a green box for comparison.
Figure 5 with 1 supplement
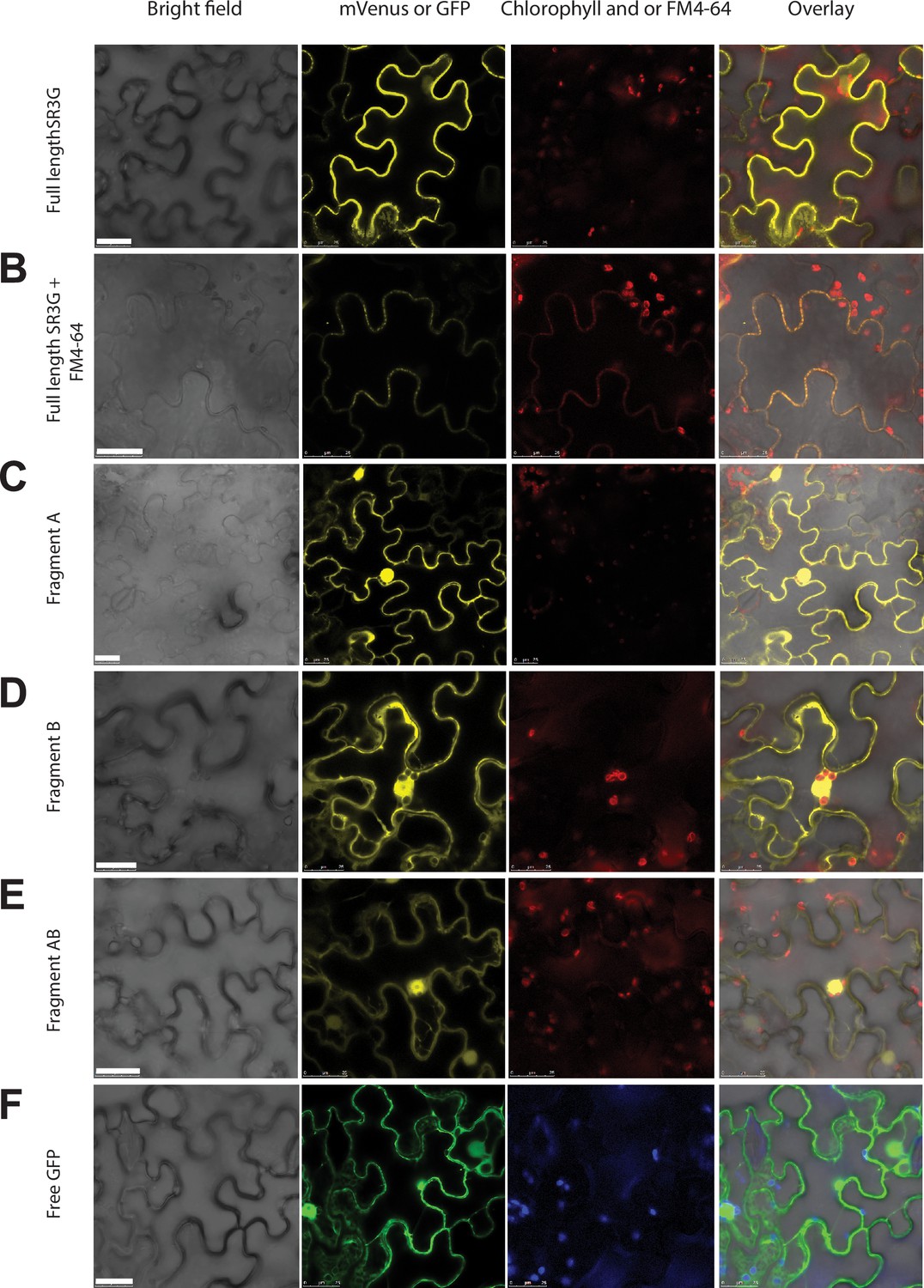
SR3G full-length protein resides in the plasma membrane while removal of its transmembrane domain results in protein relocation to nucleus and cytosol.
(A-B) SR3G full-length protein or (C-E) truncated versions fused to mVenus at the N-terminus were agro-infiltrated into the Nicotiana benthamiana leaves for transient expression. Shown are a bright-field image of transfected leaf cells and mVenus-SR3G-mediated fluorescence as well as GFP-based subcellular marker. (A) Localization of full-length SR3G alone or (B) in combination with the known plasma membrane dye, FM4-64. Localization of truncated versions of SR3G are shown for (C) SR3G-Fragment A, (D) SR3G-Fragment B, and (E) SR3G-Fragment AB. (F) Free GFP was used as a nuclear and cytosolic marker. Scale bar = 25 µm.
Figure 5—figure supplement 1

SR3G predicted protein domains.
The SR3G protein sequence (Col-0 allele) was used to identify predicted protein domains. The identified domains include: nuclear localization signal (NLS): K15-F19; coiled-coil domain: M153-K173; potential Ca2 + binding pocket: G210-F372; and transmembrane domain P469-T503. Informed by these predicted protein domains, we designed SR3G Fragments A, B, and C for subsequent localization studies and functional characterization. The pink mark at Y216 indicates a position of a premature STOP codon in Blh-1 allele.
Figure 6 with 8 supplements
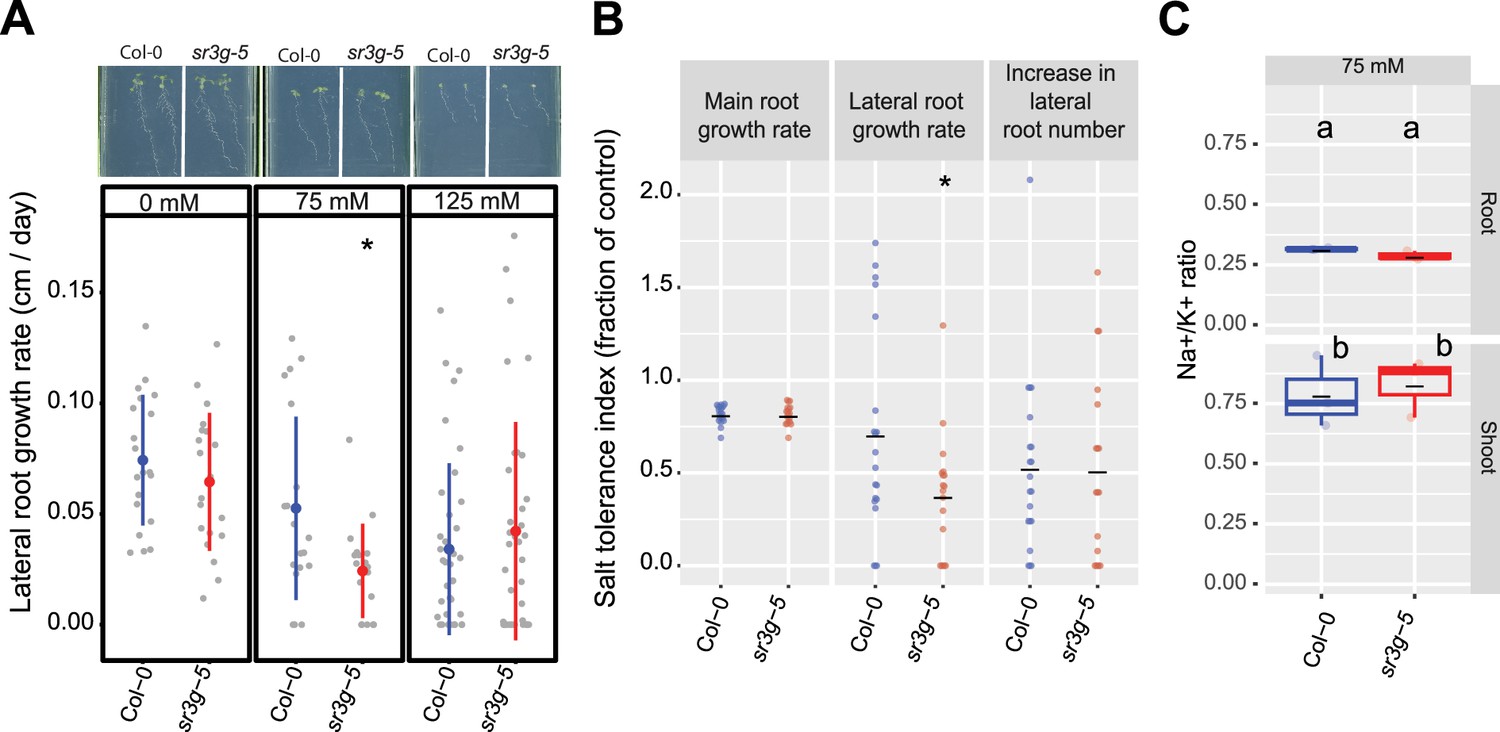
sr3g mutant displays reduced growth rate for the lateral root length.
Root system architecture analysis of Col-0 and sr3g-5 plants under various concentrations of NaCl are shown here. (A) The representative images of 13-d-old Col-0 and sr3g-5 genotypes that experienced 9 d of salt treatment at indicated concentrations as well as growth rate for lateral root. (B) Salt Tolerance Index (STI) for the main root length, average lateral root length, and lateral root number at 75 mM NaCl. The STI was calculated by dividing the growth rate measured under salt stress by the growth rate measured under control condition for each genotype. (C) Na+/K+ ratio in root and shoot of Col-0 and sr3g-5 after 2 wk on 75 mM salt are shown. Each dot in (A) and (B) represents individual replicate per genotype. Lines in (A) and (B) graphs represent median and average, respectively. In (A) and (B), the asterisks above the graphs indicate significant differences between Col-0 and the sr3g-5 mutant, as determined by the Student ‘s t-test: *p<0.05. Statistical analysis in (C) was done by comparison of the means for all pairs using Tukey–Kramer HSD test. Levels not connected by the same letter are significantly different (p<0.05). Root system architecture and ICP-AES analyses of Col-0 and sr3g-5 mutant are shown in details in Figure 6—figure supplement 2.
Figure 6—figure supplement 1
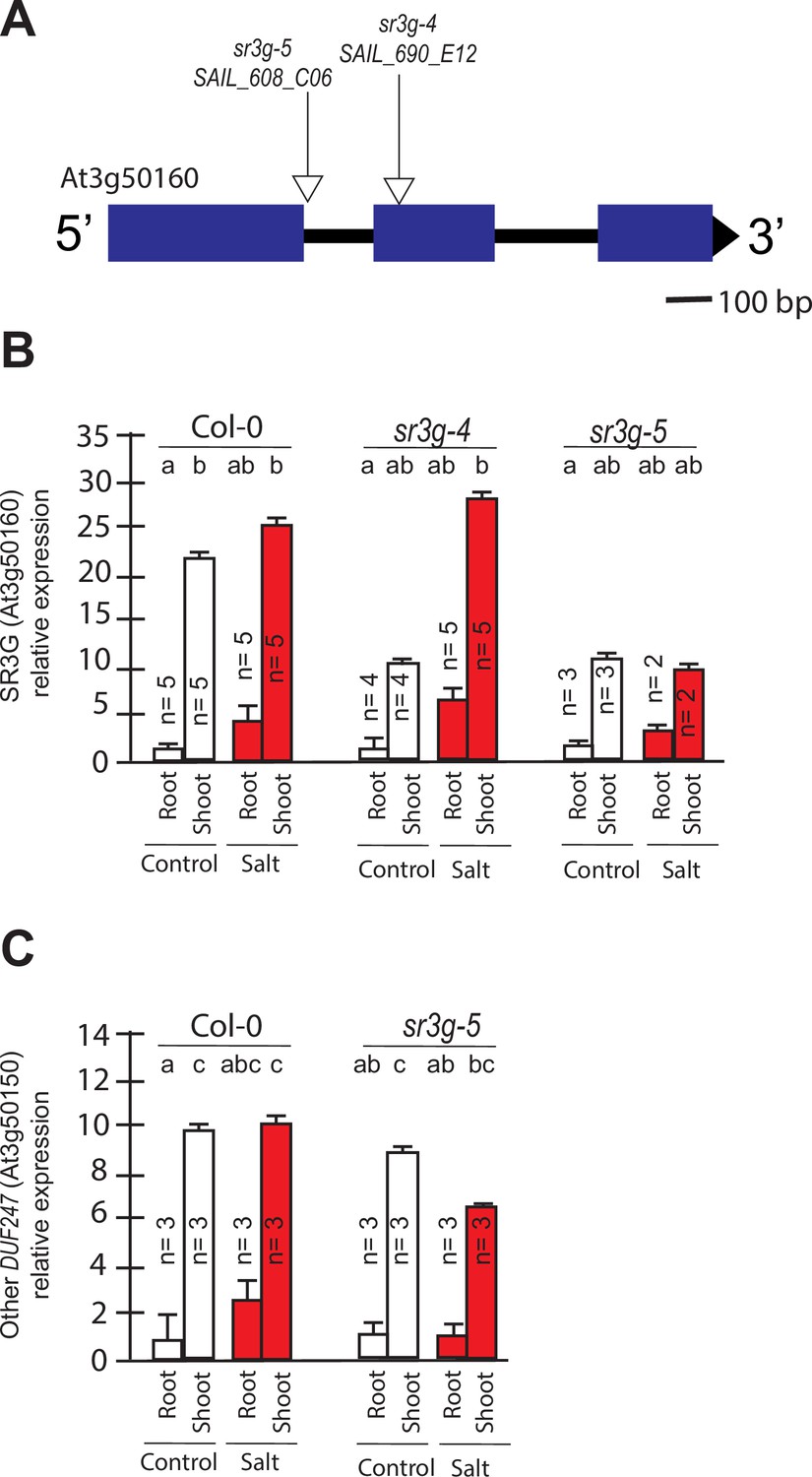
The expression of DUF247-150 remains unaltered in the sr3g mutant.
(A) Schematic diagram of main SR3G gene model and the location of T-DNA insertions. The location of T-DNA insertions is shown for sr3g-4, and –5 mutants using triangles. RT-qPCR showing expression of SR3G gene (AT3G50160) (B) and its neighboring gene, DUF247-150 (At3g50150) (C) in Col-0 and two sr3g mutants. RT-qPCR analyses were conducted using seedlings grown on 1/2 x MS for 4 d and then followed by transferring to the treatment plates with or without 75 mM NaCl for 1 wk. Mean values are shown ±SE, with the number of replicates (n) shown in each graph. AT4G04120 (transposable_element_gene) was used as a reference gene for normalization. Significance was determined by the Tukey–Kramer HSD test in JMP. Levels not connected by same letter are significantly different.
Figure 6—figure supplement 2
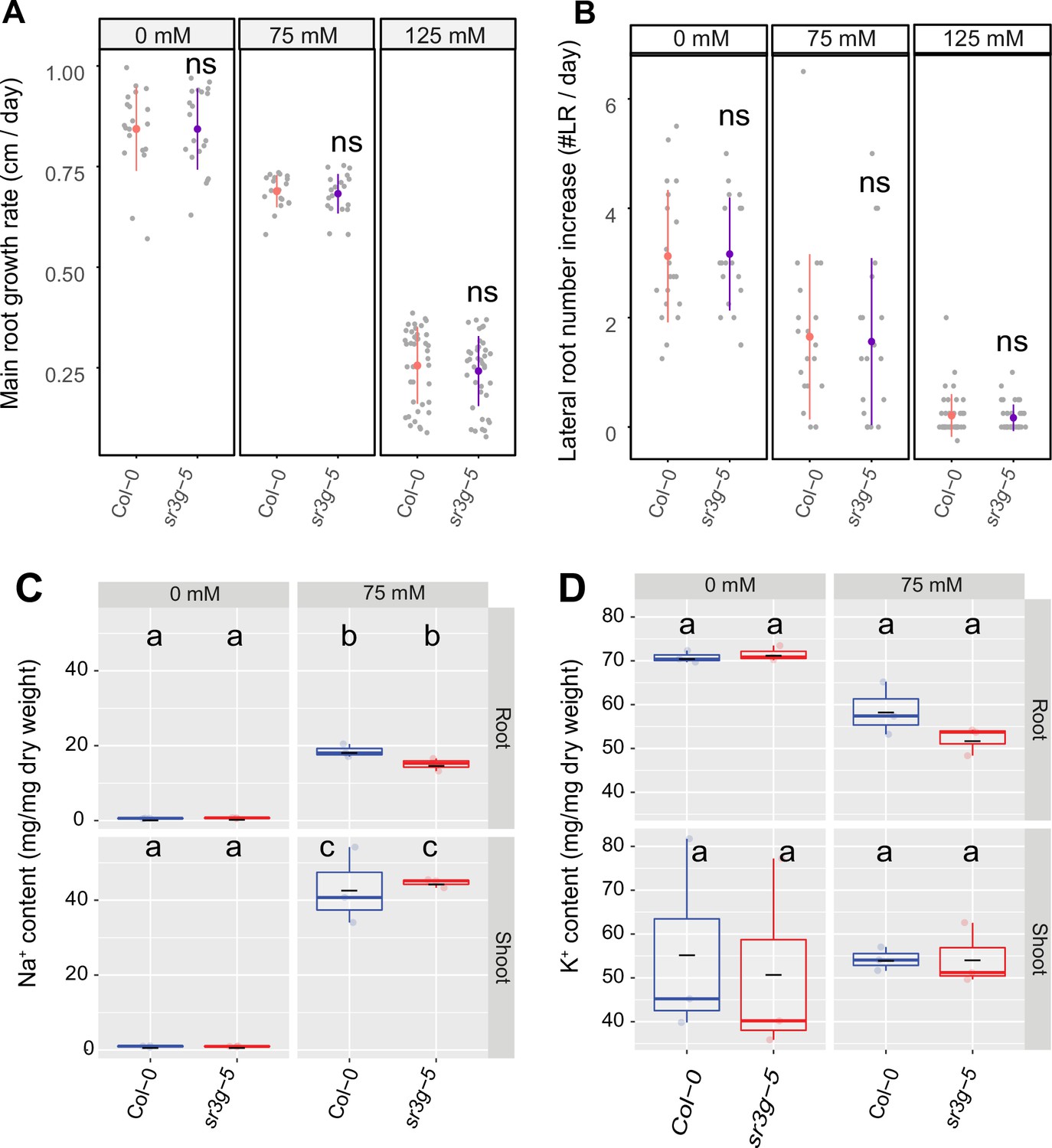
sr3g-5 mutant displays no difference in main root length and lateral root numbers compared to Col-0.
Root system architecture analysis of Col-0 and sr3g-5 plants under various concentrations of NaCl are shown here for (A) main root length and (B) increase in lateral root number. 13-d-old Col-0 and sr3g-5 genotypes experienced 9 d of salt treatment at indicated concentrations were used here. Each dot in (A) and (B) represents individual replicate per genotype. Statistical analysis was performed between Col-0 and the sr3g-5 mutant using the Student’s t-test: *p<0.05. ns stands for not significant. (C) Na+ and (D) K+ contents in root and shoot of Col-0 and sr3g-5 after 2 wk on 75 mM salt are shown. Statistical analysis in (C-D) was done by comparison of the means for all pairs using Tukey–Kramer HSD test. Levels not connected by the same letter are significantly different (p<0.05).
Figure 6—figure supplement 3
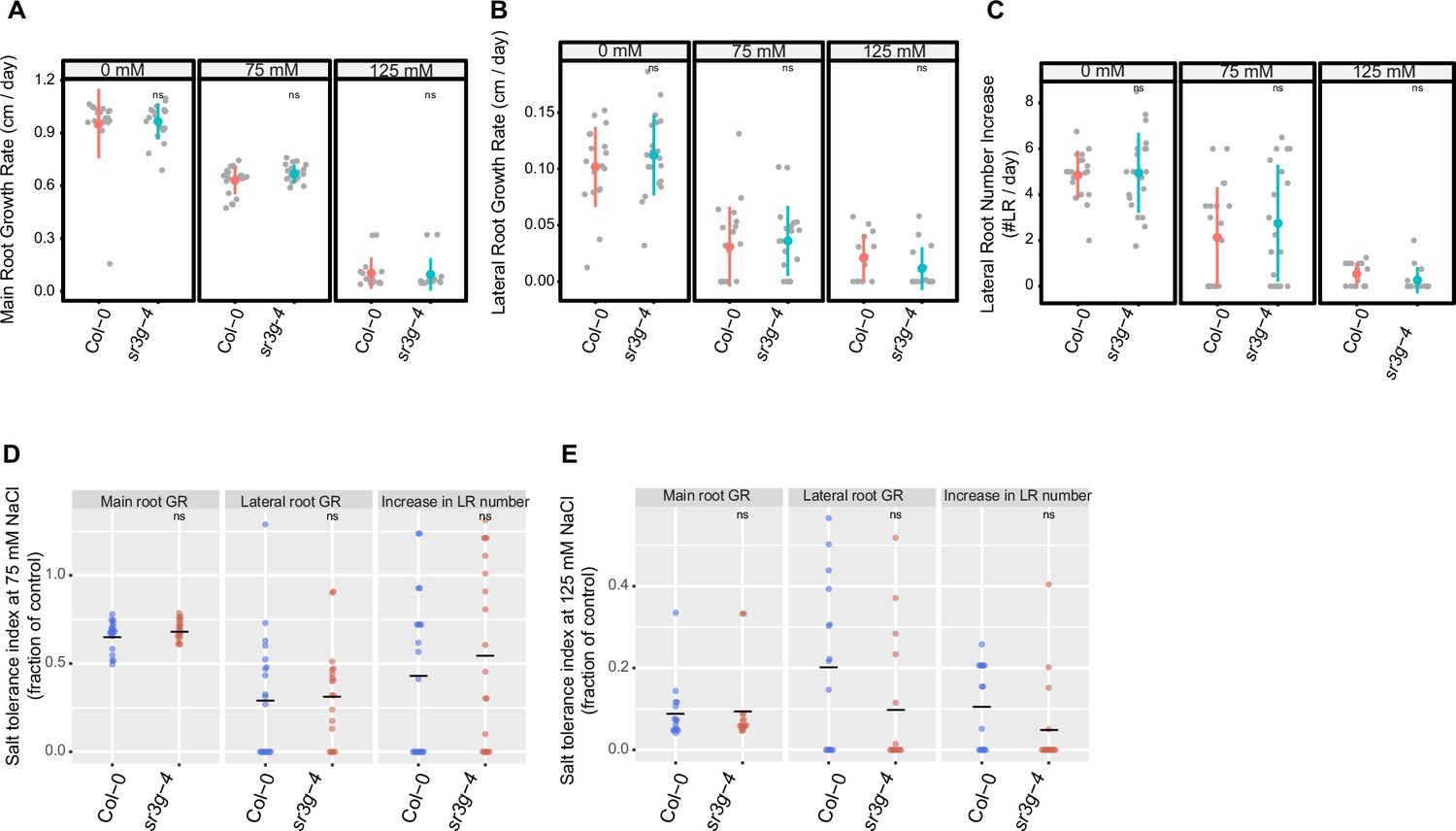
sr3g-4 mutant has no alteration in root system architectures compared to Col-0.
Root system architecture analysis of Col-0 and sr3g-4 plants under various concentrations of NaCl are shown here for (A) main root length, (B) lateral root length, and (C) lateral root number. Salt Tolerance Index (STI) for the main root length, average lateral root length, and lateral root number are shown at (D) 75 and (E) 125 mM NaCl, respectively. The STI was calculated by dividing the growth rate measured under salt stress by the growth rate measured under control condition for each genotype. Statistical analysis was performed between Col-0 and the sr3g-4 mutant using the Student‘s t-test: *p<0.05.
Figure 6—figure supplement 4
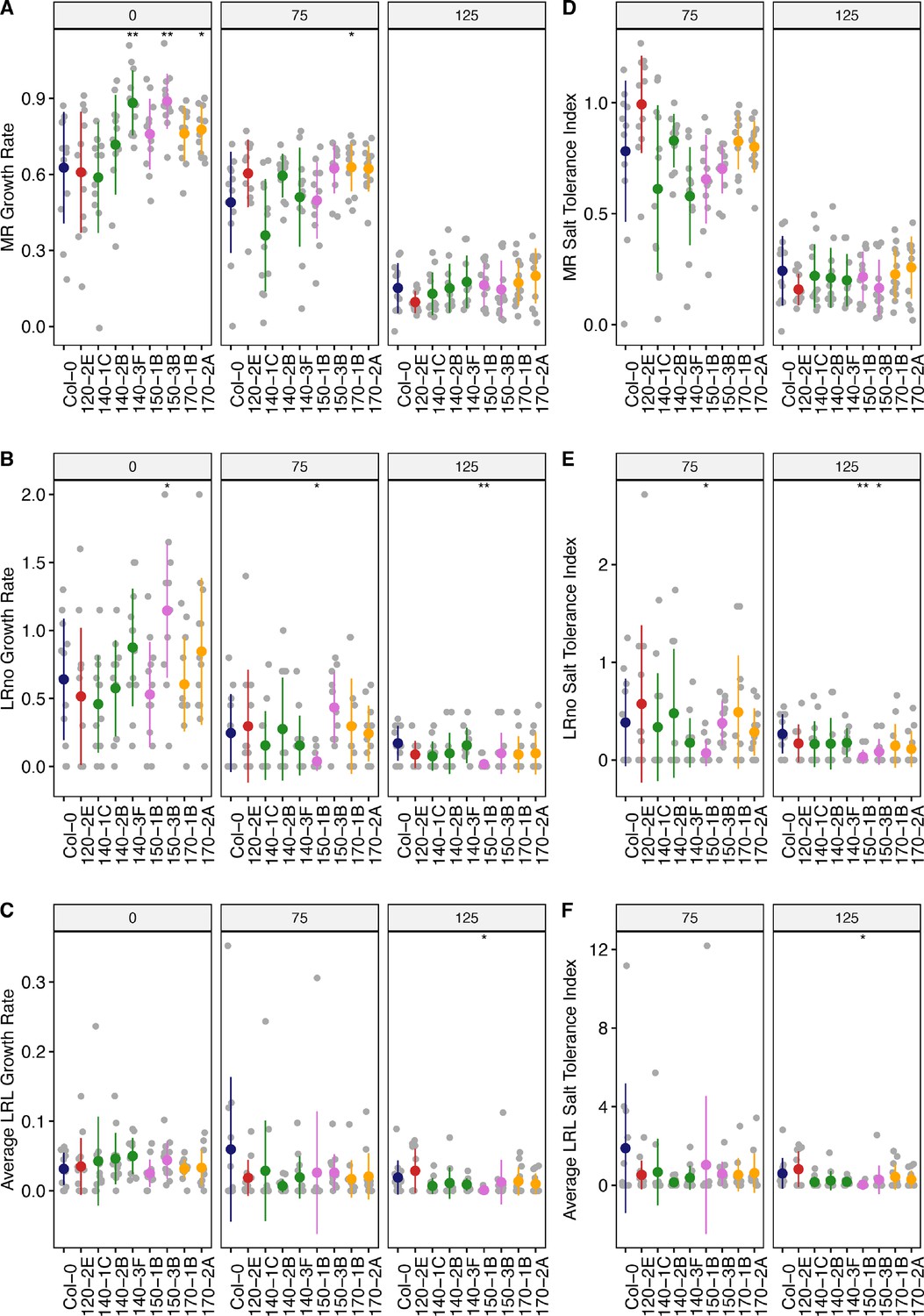
Root system architecture analysis of neighboring DUF247 mutants under the salt.
(A) Main root length, (B) lateral root number, and (C) lateral root length are shown for neighboring DUF247s of the SR3G, including At3g50120 (shown as 120, with one T-DNA representative, 120-2(SAIL_382_A09)), At3g50140 (shown as 140, with three T-DNA representatives, 140–1 (SALK_044685), 140–2 (SALK_005466), and 140–3 (SALK_122700)), At3g50150 (shown as 150, with two T-DNA representatives, 150–1 (SALK_003824) and 150–3 (SALK_071080)), and At3g50170 (shown as 170, with two T-DNA representatives, 170–1 (SALK_009186), and 170–2 (SALK_112602)) compared to Col-0 under various concentrations of NaCl as indicated. The corresponding Salt Tolerance Index (STI) was calculated for (D) main root growth, (E) increase in lateral root number, and (F) lateral root growth rate. Statistical analysis was performed between Col-0 and the other mutants using the Student’s t-test: *p<0.05. The root system architecture analysis was performed similar to what described for Figure 6.
Figure 6—figure supplement 5
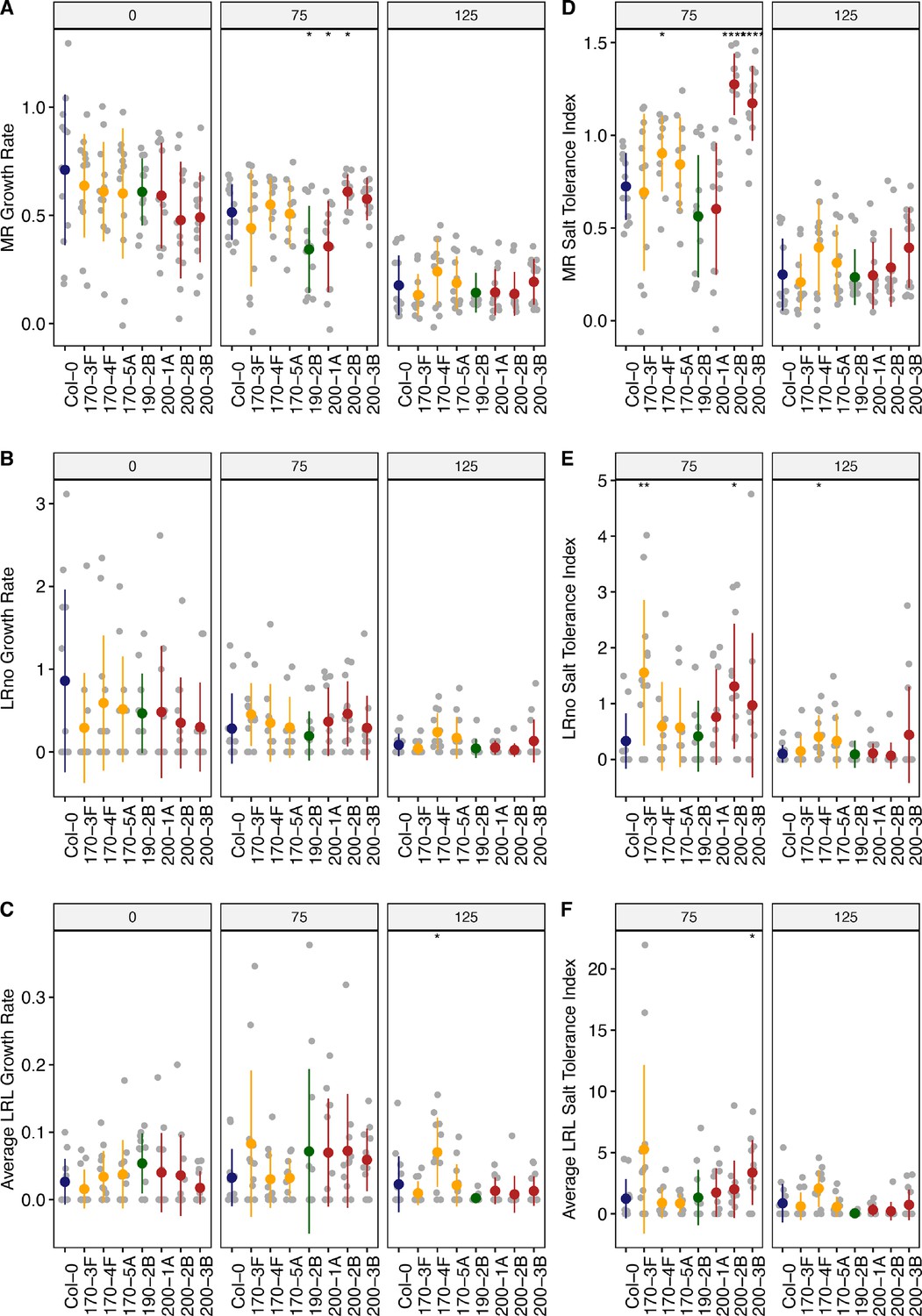
Root system architecture analysis of neighboring DUF247 mutants under the salt, continues.
(A) Main root length, (B) lateral root number, and (C) lateral root length are shown for neighboring of the SR3G, including At3g50170 (shown as 170, with three T-DNA representatives, 170–3 F (SALK_008710), 170–4 F (SALK_145999), 170–5 A (SALK_072937)), At3g50190 (shown as 190, with one T-DNA representative, 190-2B (SALK_137791)), and At3g50200 (shown as 200, with three T-DNA representatives, 200–1 A (SALK_113759), 200-2B (SALK_093701), and 200-3B (SALK_129634)) compared to Col-0 under various concentrations of NaCl as indicated. The corresponding Salt Tolerance Index (STI) was calculated for (D) main root growth, (E) increase in lateral root number, and (F) lateral root growth rate. Statistical analysis was performed between Col-0 and the other mutants using the Student’s t-test: *p<0.05. The root system architecture analysis was performed similar to what described for Figure 6.
Figure 6—figure supplement 6
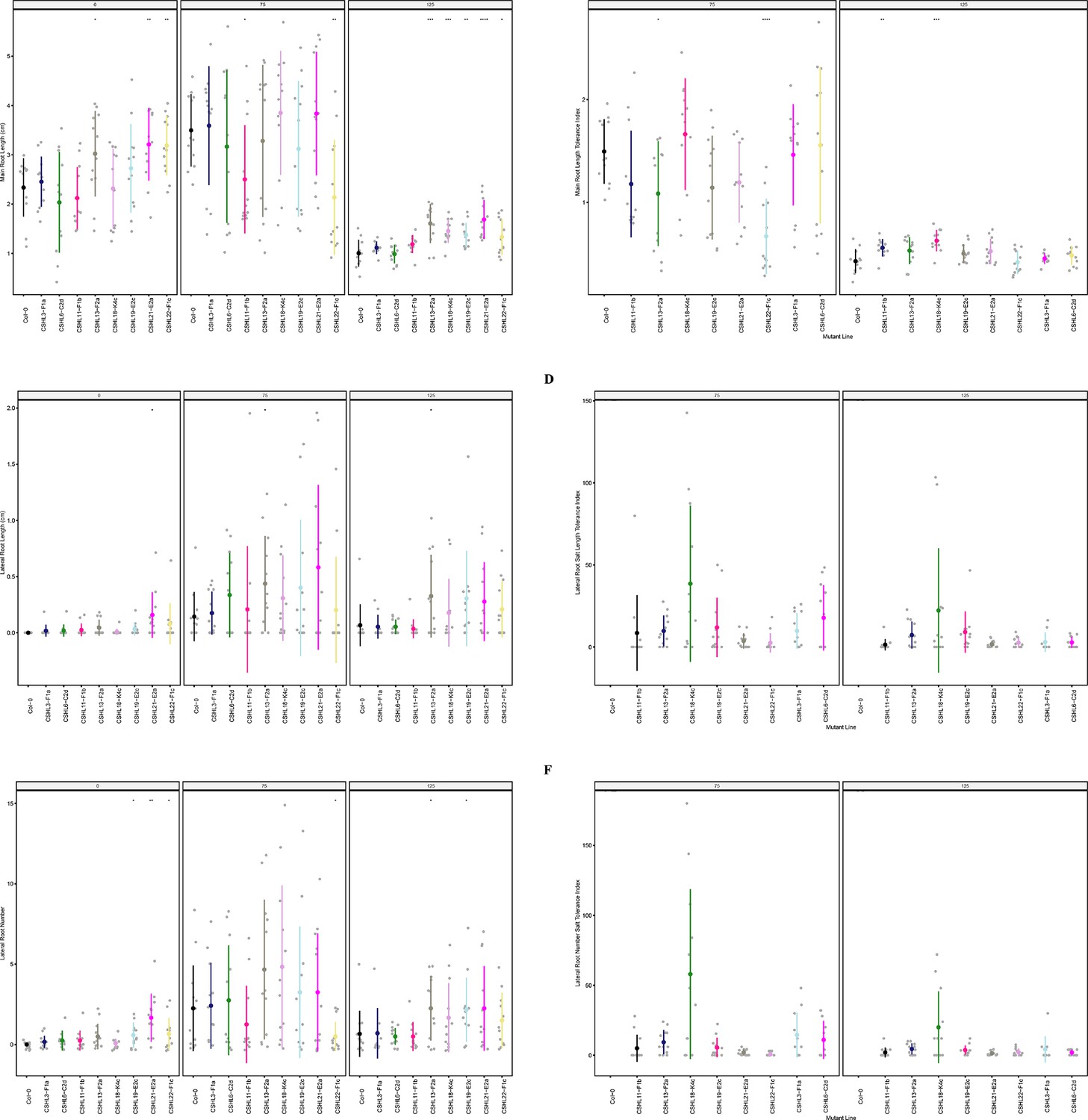
Root system architecture analysis of RNAi lines targeting neighboring DUF247s.
(A) Main root length, and (B) its stress tolerance index (Salt / Control), (C) Lateral root length, and (D) its stress tolerance index (Salt / Control), (E) Lateral root number and (F) its stress tolerance index (Salt / Control) are shown for RNAi lines targeting AT3G50140 (CSHL3-F1a), targeting AT3G50130 and AT3G50140 (CSHL6-C2d), targeting AT3G50150 and AT3G50170 (CSHL11-F1b), targeting AT3G50140 and AT3G50170 (CSHL13-F2a), targetingAT3G50150 and AT3G50190 (CSHL18-K4c), targeting AT3G50130, AT3G50140, AT3G50150, AT3G50160, AT3G50170, AT3G50180, and AT3G50190 (CSHL19-E2c), targeting AT3G50130 and AT3G50150 (CSHL21-E2a), and targeting AT3G50150 and AT3G50160 (CSHL22-F1c). Statistical analysis was performed between Col-0 and the RNAi lines using a Student’s t-test: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 6—figure supplement 7
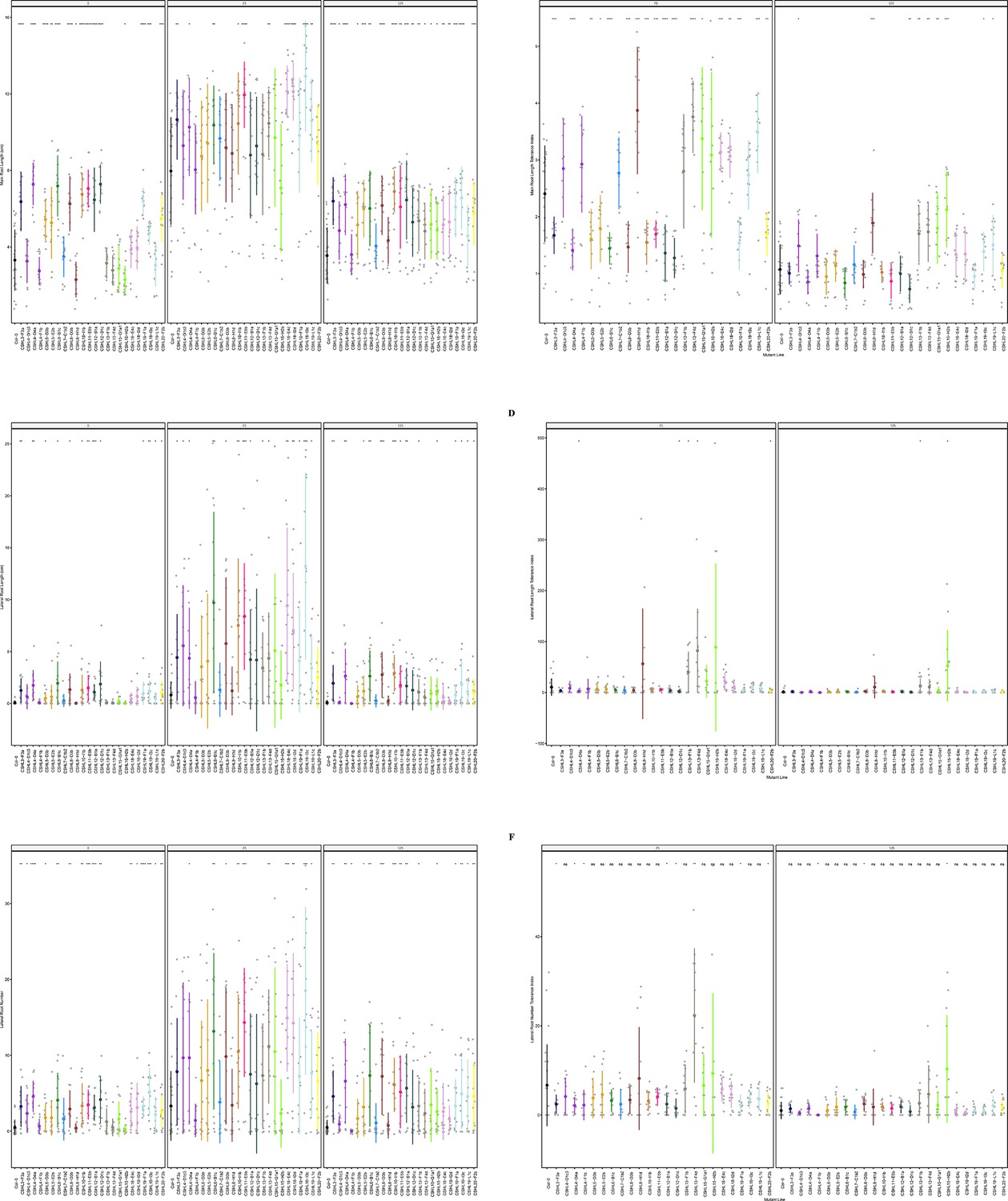
Root system architecture analysis of RNAi lines targeting neighboring DUF247s continued.
(A) Main root length, and (B) its stress tolerance index (Salt / Control), (C) Lateral root length, and (D) its stress tolerance index (Salt / Control), (E) Lateral root number and (F) its stress tolerance index (Salt / Control) are shown for RNAi lines targeting AT3G50160 (CSHL4-B2a and CSHL4G1d), targeting AT3G50140 and AT3G50150 (CSHL7-A3a), targeting AT3G50140 and AT3G50190 (CSHL8-G2b), targeting AT3G50180 (CSHL10-I1c), targeting AT3G50130, AT3G50140, AT3G50170, AT3G50180, and AT3G50190 (CSHL12-A1a), targeting AT3G50140, AT3G50170, AT3G50180, and AT3G50190 (CSHL15-E1b), targeting AT3G50130, AT3G50160, and AT3G50170 (CSHL-A2c and CSHL16-B1a), targeting AT3G50130 and AT3G50160 (CSHL20-G3b), and targeting AT3G50130 and AT3G50150 (CSHL21-L2c and CSHL21-M1d). Statistical analysis was performed between Col-0 and the RNAi lines using a Student’s t-test: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 6—figure supplement 8
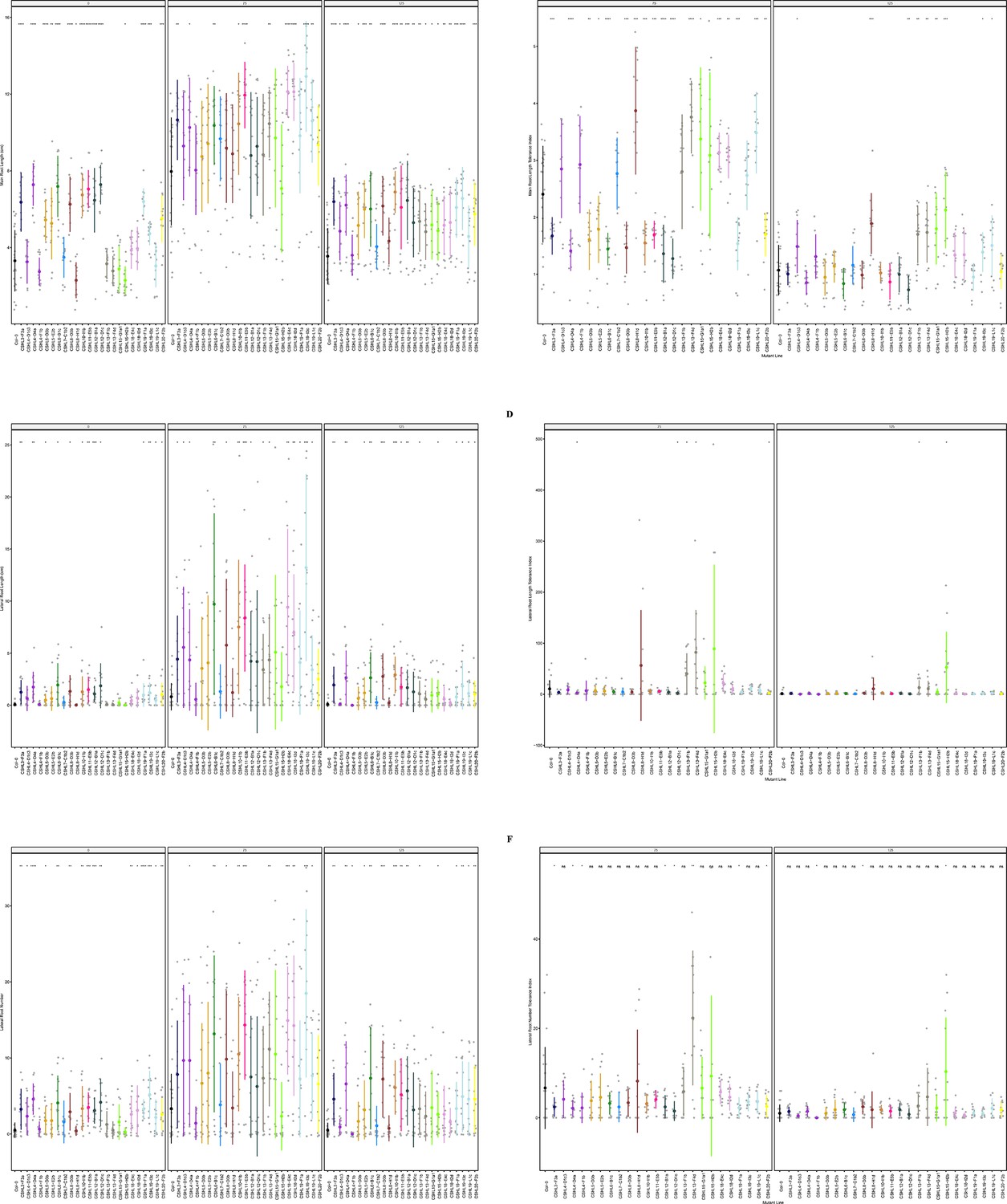
Root system architecture analysis of RNAi lines targeting neighboring DUF247s continued.
(A) Main root length, and (B) its stress tolerance index (Salt / Control), (C) Lateral root length, and (D) its stress tolerance index (Salt / Control), (E) Lateral root number and (F) its stress tolerance index (Salt / Control) are shown for RNAi lines targeting AT3G50140 (CSHL3-F3a), targeting AT3G50160 (CSHL4-D1c3, CSHL4-D4a, and CSHL4-F1b), targeting AT3G50130 and AT3G50170 (CSHL5-D3b and CSHL5-E2b), targeting AT3G50130 and AT3G50140 (CSHL6-B1c), targeting AT3G50140 and AT3G50150 (CSHL7-C1b2), targeting AT3G50140 and AT3G50190 (CSHL8-D3b and CSHL8-H1d), targeting AT3G50180 (CSHL10-I1b), targeting AT3G50150 and AT3G50170 (CSHL11-E5b), targeting AT3G50130, AT3G50140, AT3G50170, AT3G50180, and AT3G50190 (CSHL12-B1a and CSHL12-D1c), targeting AT3G50140 and AT3G50170 (CSHL13-F1b and CSHL13-F4d), targeting AT3G50140, AT3G50170, AT3G50180, and AT3G50190 (CSHL15-G1a1 and CSHL15-H2b), targeting AT3G50150 and AT3G50190 (CSHL18-E4c and CSHL18-I2d), targeting AT3G50130, AT3G50140, AT3G50150, AT3G50160, AT3G50170, AT3G50180, and AT3G50190 (CSHL19-F1a, CSHL19-I3c, and CSHL19-L1c), and targeting AT3G50130 and AT3G50160 (CSHL20-F2b). Statistical analysis was performed between Col-0 and the RNAi lines using a Student’s t-test: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 7 with 1 supplement
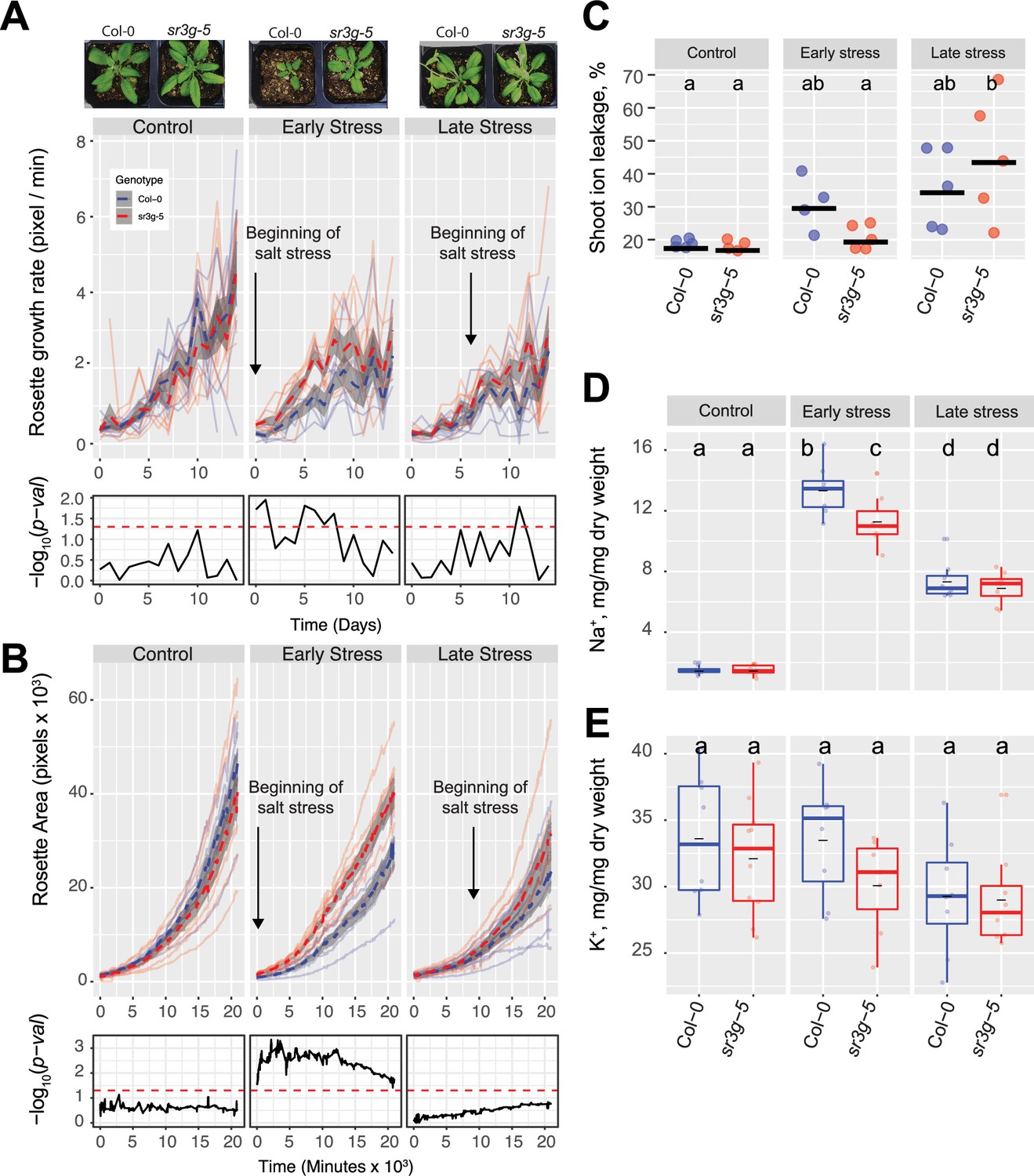
sr3g mutant has higher growth rate and larger rosette area while displaying less ion leakage and Na+ accumulation in shoot under salt stress.
The salt stress responses of 2-wk-old (referred to as ‘early salt stress’) or 3-wk-old (referred to as ‘late salt stress’) soil-grown Col-0 and sr3g-5 plants that were exposed to a final concentration of 100 mM NaCl were examined here. (A) Rosette growth rate and (B) area were monitored over a period of 2 wk, during which the ‘early salt stress’ and ‘late salt stress’ groups were exposed to 2 or 1 wk of salt stress, respectively. Each line represents the trajectory of individual plant throughout time of experiments, where red and blue lines indicate Col-0 and sr3g-5 plants, respectively. The dashed lines represent the mean values of the genotype per condition, whereas the gray band represents the confidence interval. The significant differences between genotypes, determined by one-way analysis of variance, are illustrated in a plot below each graph, with dashed red line representing a threshold corresponding to p-value of 0.05. Representative images of 4-wk-old Col-0 and sr3g-5 plants were shown in (A). (C) Ion leakage percentage, (D) Na+ and (E) K+ contents in shoot were measured in 4-wk-old plants. The asterisks above the graphs indicate significant differences between Col-0 and sr3g-5 plants, as determined by one-way analysis of variance: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Statistically different groups were determined using Tukey–Kramer HSD test. Groups that are not assigned the same letter are significantly different (p<0.05).
Figure 7—figure supplement 1
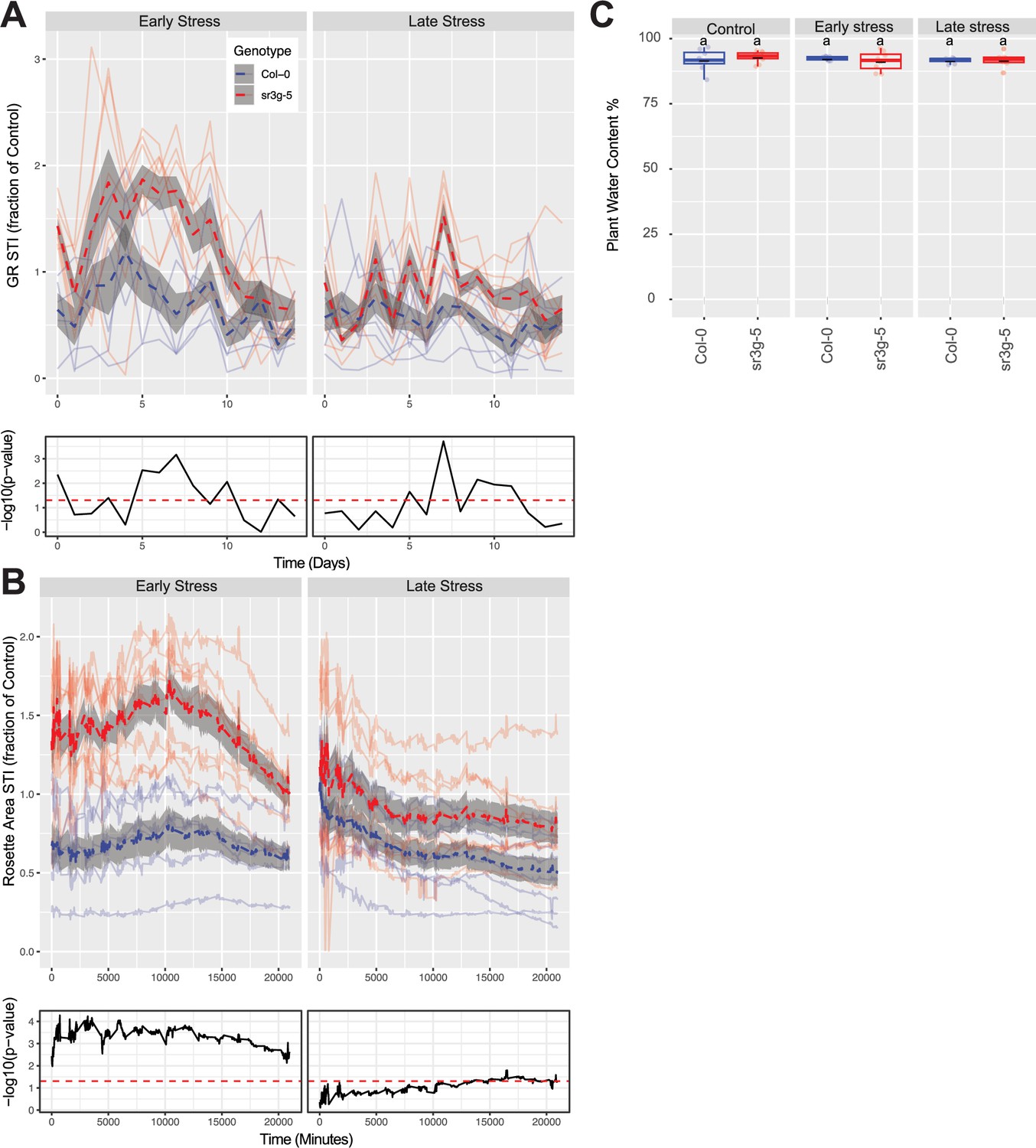
Additional phenotypes of soil grown sr3g mutant and Col-0 wild-type plants.
Salt Tolerance Index (STI) was calculated by dividing the value of each trait for each genotype by its corresponding mean value for that trait and timepoint. The STI was calculated for (A) Rosette growth rate and (B) area. Each line represents the relative trajectory of individual plant throughout time of experiments, where red and blue lines indicate Col-0 and sr3g-5 plants, respectively. The dashed lines represent the mean values of the genotype per condition, whereas the gray band represents the confidence interval. The significant differences between genotypes, determined by one-way analysis of variance, are illustrated in a plot below each graph, with dashed red line representing a threshold corresponding to p-value of 0.05. (C) The percentage of plant water content was calculated using the formula: (Fresh Weight−Dry Weight)/Fresh Weight ×100 for the 2-wk-old (referred to as ‘early salt stress’) or 3-wk-old (referred to as ‘late salt stress’) soil-grown Col-0 and sr3g-5 plants that were exposed to a final concentration of 100 mM NaCl. ‘early salt stress’ group experienced 2 wk of salt stress while the ‘late salt stress’ group experienced 1 wk. Statistical analysis was done by comparison of the means for all pairs using Tukey–Kramer HSD test. Levels not connected by the same letter are significantly different (p<0.05).
Figure 8
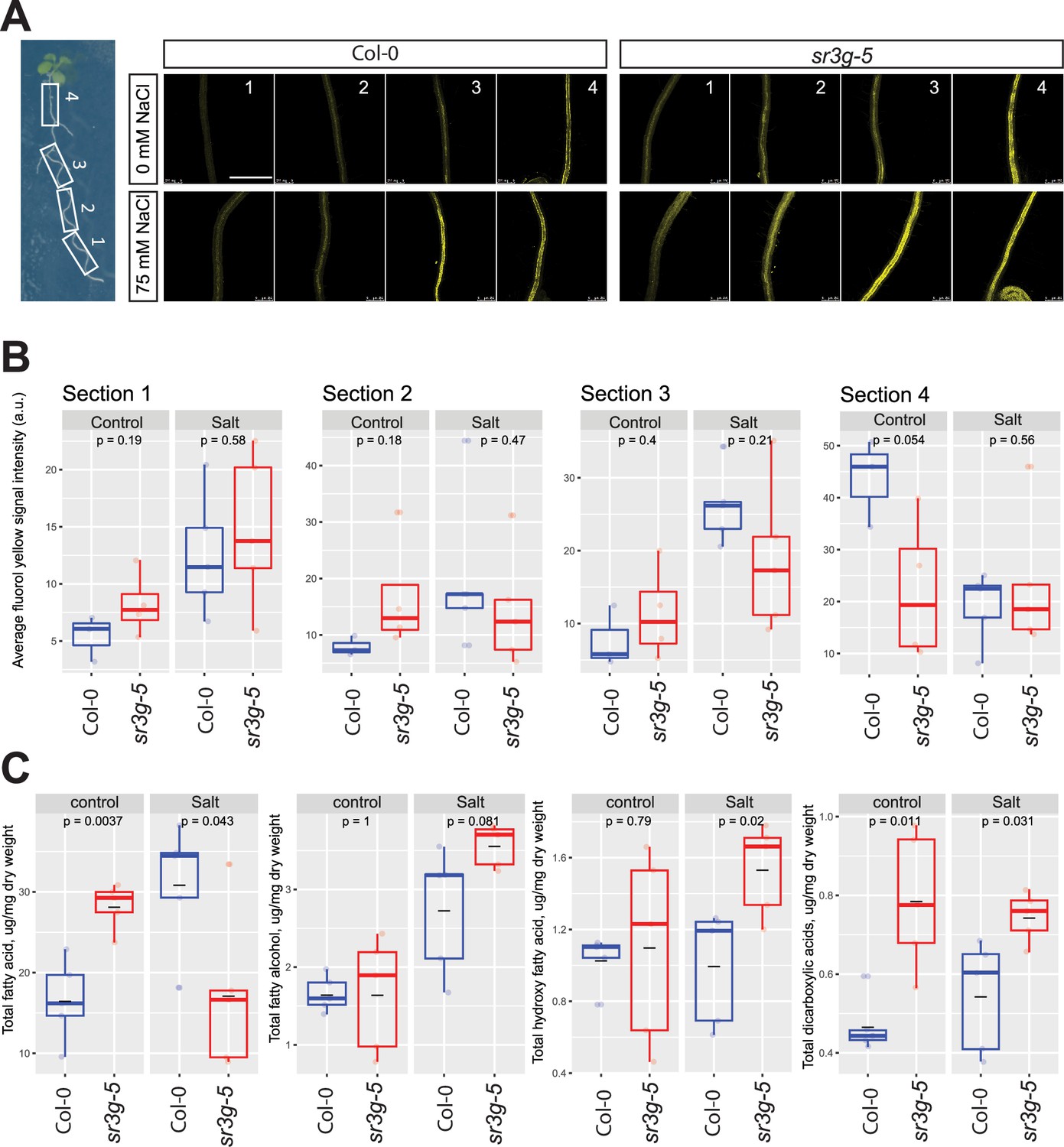
sr3g mutant root is more suberized than Col-0.
(A) Representative images of fluorol yellow (FY) staining of four root sections (# 1–4, from root tip to root base as shown by white rectangles across the seedling) were shown for Col-0 and sr3g-5 mutant with or without 75 mM NaCl. Seeds were germinated on the 1/2 MS plates for 4 d and then seedlings were transferred to the treatment plates with or without 75 mM NaCl for wo more days. (B) Quantification of fluorol yellow (FY) signal intensity for each root section. The FY quantification was done using ImageJ. Scale bar is equal to 500 µm in all. Three to five biological replicates were used for FY signal quantification, with n=3 for Col control, n=4 for sr3g control, n=5 for Col under salt stress, and n=5 for sr3g under salt stress. (C) Suberin monomers detected using Gas Chromatography–Mass Spectrometry (GC-MS) in the Col-0 and sr3g-5 mutant roots with or without 75 mM NaCl included fatty acid (FA), fatty alcohol (OH-FA), α,ω-dicarboxylic acids (DCA), and ω-hydroxy fatty acid (ωOH-FA). The significant differences between Col-0 and sr3g-5 mutant were evaluated using Student ‘s t-test. Five biological replicates per genotype per condition used for this experiment.
Figure 9
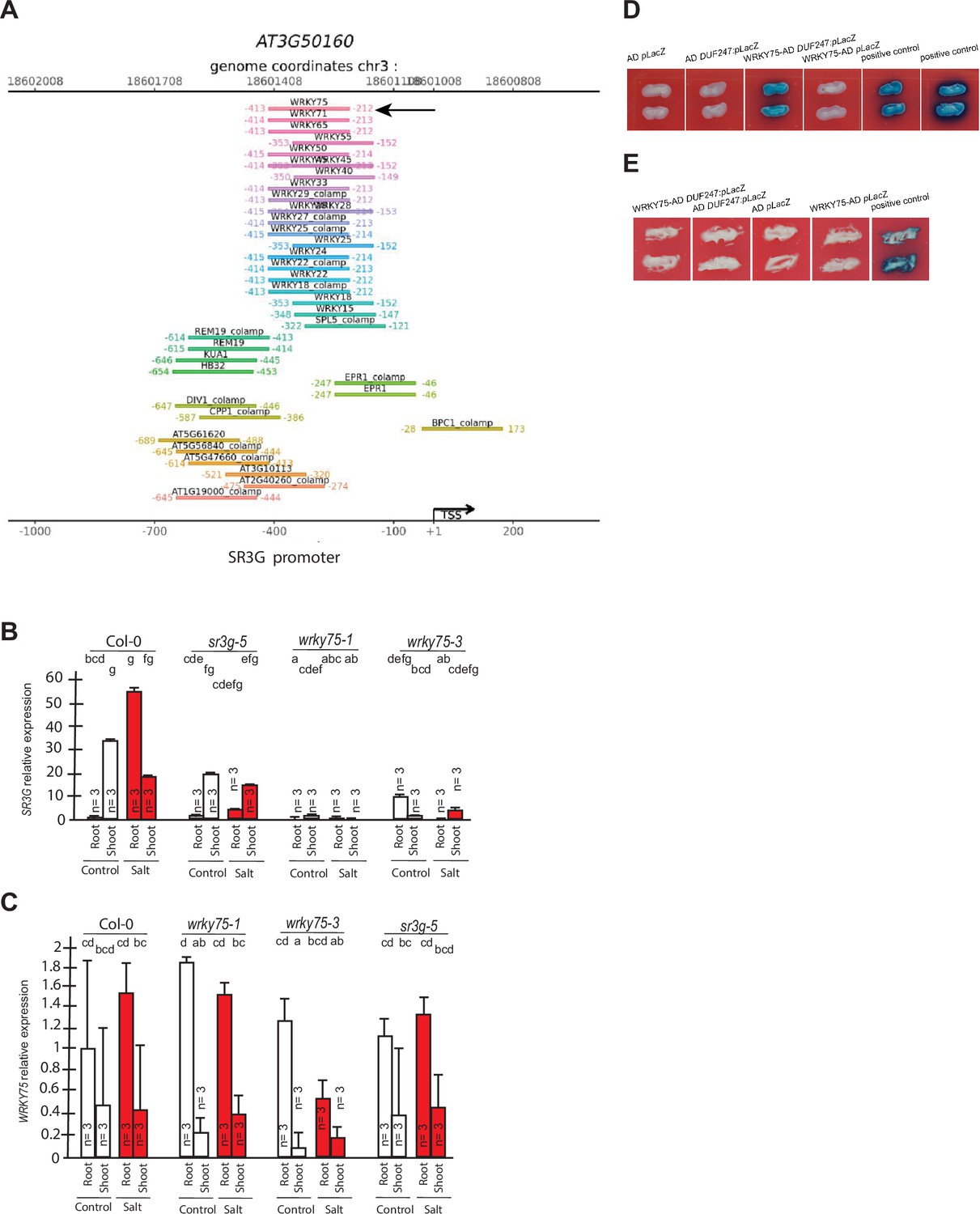
WRKY75 binds to the promoter region of the SR3G but not to its neighboring DUF247.
(A) In silico searches in CisCross (https://plamorph.sysbio.ru/ciscross/CCL_index.html) shows potential transcription factors with binding sites on the main SR3G (At3g50160) promoter region. A black-arrow points to the WRKY75 (AT5G13080). (B) RT-qPCR showing expression of SR3G (AT3G50160) and (C) WRKY75 (AT5G13080) genes, respectively, in Col-0, sr3g-5 mutant, and two different mutant alleles of the wrky75, i.e., wrky75-1 and wrky75-3. RT-qPCR analyses were conducted using seedlings grown on 1/2 x MS for 4 d and then followed by transferring to the treatment plates with or without 75 mM NaCl for 1 wk. Mean values are shown ±SE, with three biological replicates used in each condition and genotype. AT4G04120 (transposable_element_gene) was used as a reference gene for normalization. Significance was determined by the Tukey–Kramer HSD test in JMP. Levels not connected by same letter are significantly different. (D) Yeast one-hybrid (Y1H) assay showing WRKY75 (438 bp) binds to the promoter region of the main SR3G (AT3G50160, 953 bp) (E) but not to its neighboring promoter, i.e., DUF247-150 (At3g50150, 631 bp). pB42AD (AD) and pLacZ were used as effector and reporter construct, respectively. Effector and reporter constructs were co-transformed into yeast strain EGY48. Transformants were selected and grown on SD/-Trp-Ura medium. The interactions were observed on SD/-Trp/-Ura+X Gal medium. Empty vector expressing AD domain and pLacZ were used as negative control. The two positive controls are NIGT1.4-GolS2 and NIGT1.4-GAE1. The oligo sequences used for Y1H and luciferase assay were provided in Supplementary file 4.
Figure 10 with 2 supplements
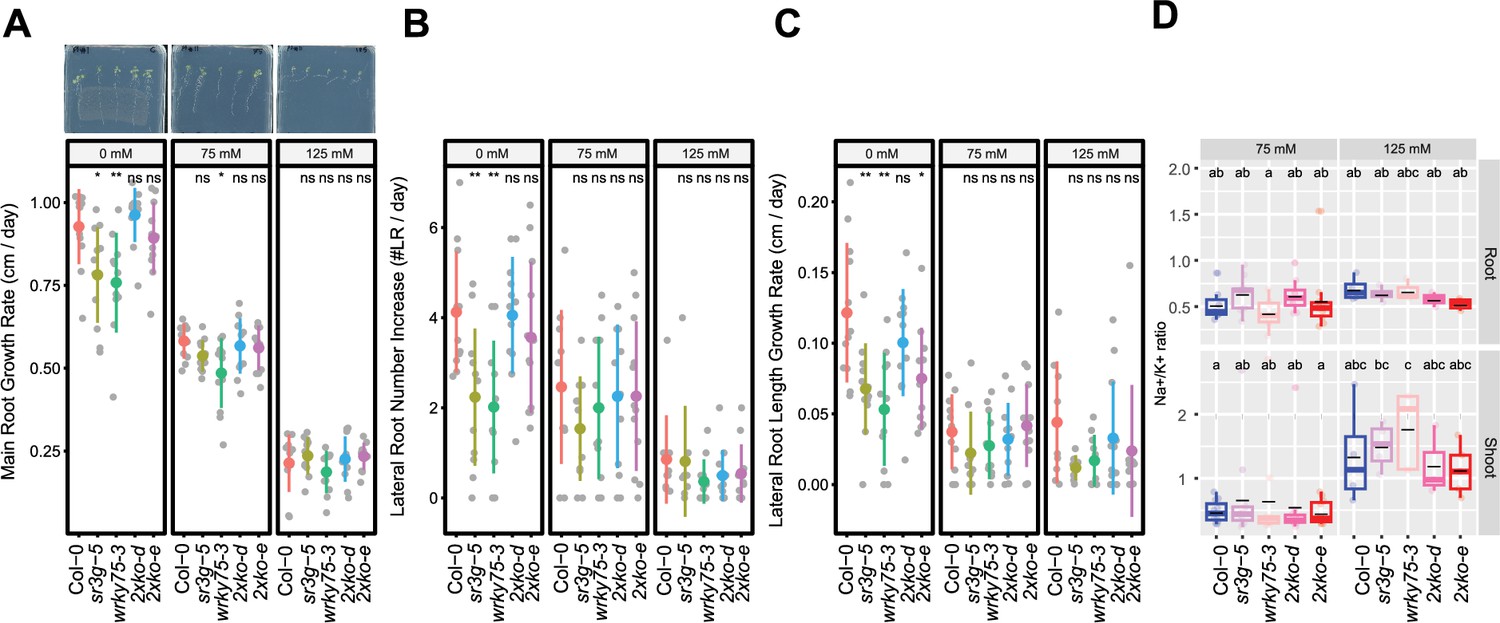
wrky75/sr3g double mutants roots exhibit reduced sensitivity to salt stress and accumulate lower levels of Na+ in their shoots.
(A-C) Root system architecture analysis for the Col-0, sr3g-5, wrky75-3, and two double mutants under various concentrations of NaCl are shown here. (A) Main root growth rate, (B) Lateral root number increase, and (C) Lateral root length increase are shown for the indicated genotypes at various salt concentrations. (D) Na+/k+ ratio in root and shoot of indicated genotypes after 2 wk on treatment plates. Each dot represents individual replicate per genotype. The asterisks above the graphs (A-C) indicate significant differences between Col-0 and the genotype, as determined by the Student’s t-test: *p<0.05 and **p<0.01. ns denotes no statistically significant. Statistical analysis in (D) was done by comparison of the means for all pairs using Tukey–Kramer HSD test. Levels not connected by the same letter are significantly different (p<0.05).
Figure 10—figure supplement 1
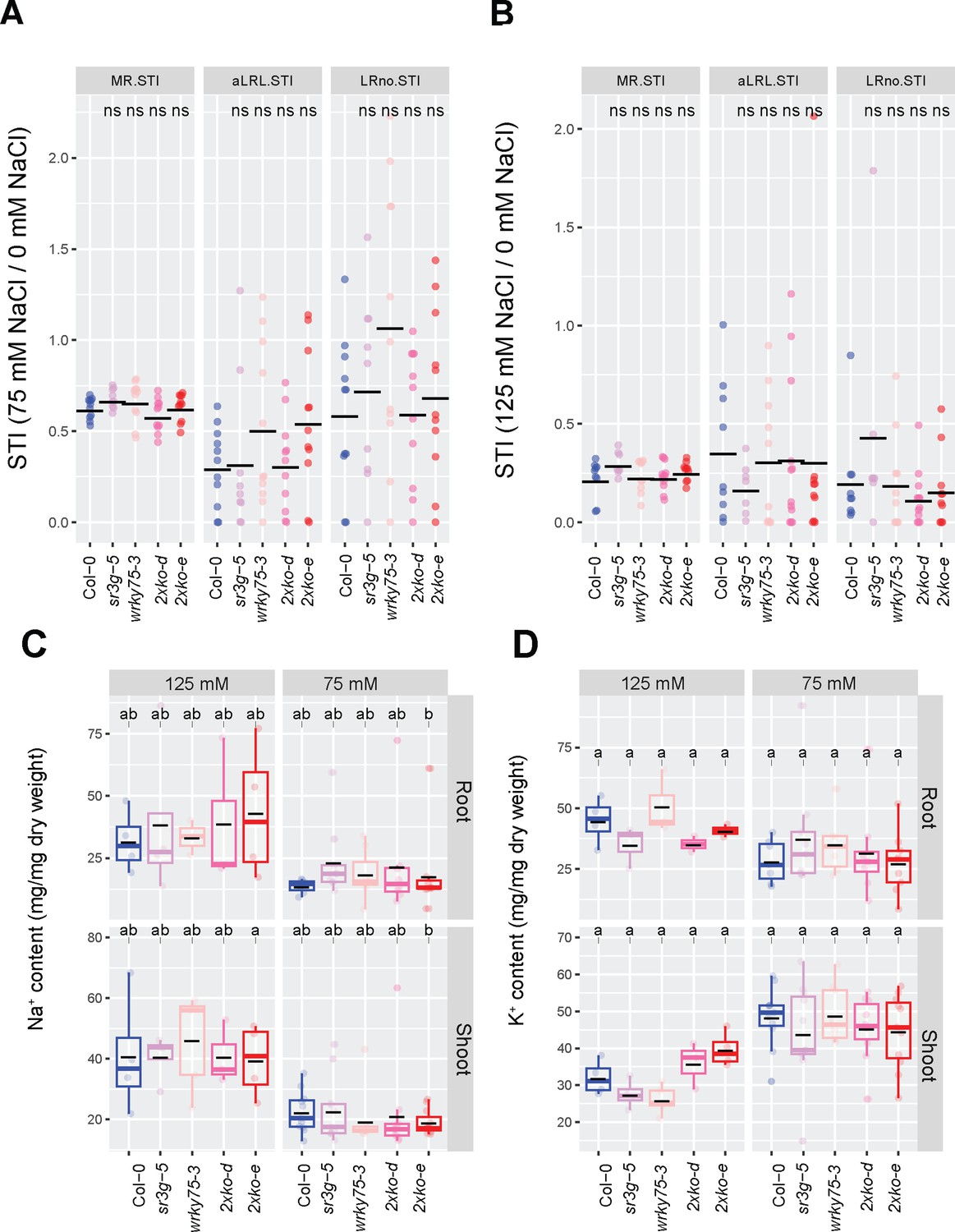
wrky75/sr3g double mutants exhibit higher shoot K+ contents compared to Col-0 and each individual single mutant in the presence of 125 mM salt.
(A) Salt Tolerance Index (STI) for the main root length (MR.STI), average lateral root length (aLRL.STI), and lateral root number (LRno.STI) at 75 mM and (B) 125 mM NaCl are shown here. The STI was calculated by dividing the growth rate measured under salt stress by the growth rate measured under control condition for each genotype. The STI reported here were correspond to root system architecture analysis in Figure 10. Each dot in (A) and (B) represents individual replicate per genotype. In (A) and (B), significant differences between Col-0 and the other genotypes was determined by the Student’s t-test: *p<0.05. ns stands for not significant. (C) Na+ and (D) K+ contents in root and shoot of Col-0, sr3g-5, wrky75-3, and two double mutants under various concentrations of NaCl are shown here. Statistical analysis in (A-B) was done by comparison of the means for all pairs using Tukey–Kramer HSD test. Levels not connected by the same letter are significantly different (p<0.05).
Figure 10—figure supplement 2
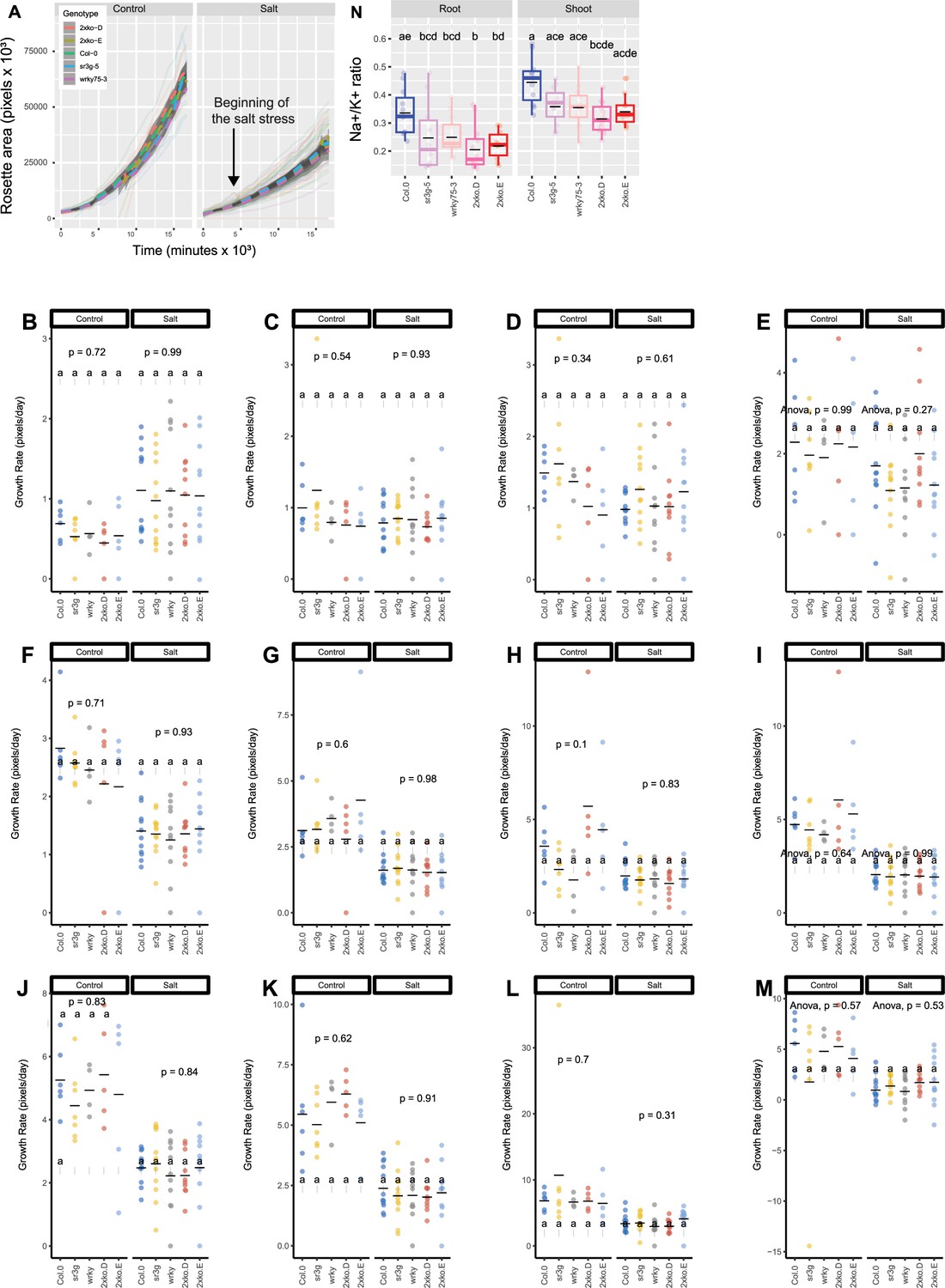
wrky75/sr3g double mutants exhibit no noticeable differences in rosette area compared to their individual single mutants when grown in saline soil.
The salt stress responses of 2-wk-old soil-grown plants that were exposed to a final concentration of 100 mM NaCl were examined here. Rosette area (A) and growth rate for each individual day were shown (B-M). Growth rate is shown for 1 (B), 2 (C), and 3 (D) d before the stress, respectively. Growth rate is shown for 1 (E), 2 (F), 3 (G), 4 (H), 5 (I), 6 (J), 7 (K), 8 (L), and 9 (M) d of stress application, respectively. Na+/K+ (N) ratio in shoot were measured in 4-wk-old plants. Each line in (A) represents the trajectory per genotype through time. The effect of the genotype within individual treatment in (B-M) was tested using ANOVA, and the significantly different genotypes were additionally determined using Tukey HSD test (p-value <0.05). Statistical analysis in (N) was done by comparison of the means for all pairs using Tukey–Kramer HSD test. Levels not connected by the same letter are significantly different (p<0.05).
Author response image 1

Additional files
-
Supplementary file 1
The individual traits recorded and calculated from the HapMap accessions exposed to the three different levels of salt stress.
The root and shoot projected area were quantified using a custom developed tool. The change in the root and shoot area over time was used to calculate Growth Factor (GF) by fitting an exponential function. The broad sense heritability (H2) was calculated for each trait over individual days and conditions using MVApp. This file supplements Figure 1.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp1-v1.xlsx
-
Supplementary file 2
Genotypic mean data used as an input for genome-wide association study (GWAS).
The genotypic mean data was calculated from at least four biological replicates per genotype per condition. The root and shoot area were extracted using a custom developed tool. The Growth Factors (GF) for root and shoot were calculated using exponential growth function. Total seedling size (Tot) was calculated by adding root and shoot area for an individual day. Shoot per Total (SpT) ratio was calculated for individual days by dividing genotype-specific shoot area by the total seedling area for that day and condition. Root per Shoot ratio (RpS) was calculated by dividing genotype-specific root area by the shoot area for that day and condition. This file supplements Figure 1.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp2-v1.xlsx
-
Supplementary file 3
Significant associations identified with genome-wide association study (GWAS) of all used traits.
The location of individual SNPs is listed according to its location on Chromosome (Chr) and position (Pos). The individual SNP Minor Allele Count (MAC) and Minor Allele Frequency (MAF) is calculated based on specific SNP set used (4 Million or 250 Thousand – listed in ‘Mapping’ column). The traits associated with individual SNPs are abbreviated as RpS for Root-per-Shoot, SpT for Shoot-per-Total seedling area, Tot for Total seedling area, GR for growth rate, and SHIIT for Shoot Ion Independent Tolerance index (Salt/Control). This file supplements Figure 2.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp3-v1.xlsx
-
Supplementary file 4
Most important genome-wide association study (GWAS) associations.
The most important associations identified through GWAS (Supplementary file 3) were inspected further for their functions using either T-DNA or RNAi mutant lines. List of oligos used for genotyping, RT-qPCR, cloning, and Y1H assay are provided. This file supplements Figure 2.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp4-v1.xlsx
-
Supplementary file 5
The gene ID, species, and gene names used for identification of SR3G orthologs and paralogs.
This file supplements Figure 3.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp5-v1.xlsx
-
Supplementary file 6
Background and foreground values of omega.
This file supplements Figure 3.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp6-v1.xlsx
-
Supplementary file 7
Links to cloning construct maps generated in this study.
This file supports Figure 5.
- https://cdn.elifesciences.org/articles/98896/elife-98896-supp7-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/98896/elife-98896-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Natural variation in salt-induced changes in root:shoot ratio reveals SR3G as a negative regulator of root suberization and salt resilience in Arabidopsis
eLife 13:RP98896.
https://doi.org/10.7554/eLife.98896.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}