Chromatin endogenous cleavage provides a global view of yeast RNA polymerase II transcription kinetics
- Department of Molecular Biosciences, Northwestern University, United States
- Department of Engineering Sciences and Applied Mathematics, Northwestern University, United States
Figures
Figure 1

Schematic of RNA polymerase II (RNAPII)-mediated transcription in Saccharomyces cerevisiae.
Two alternative mechanisms of RNAPII recruitment are shown: (1) direct recruitment to the promoter and (2) recruitment to the upstream activating sequences (UASs) facilitated by sequence-specific transcription factors (ssTFs) and coactivators such as Mediator, followed by transfer to the promoter. After RNAPII associates with the promoter, TFIIH is recruited, leading to phosphorylation of Serine 5 (inset) in the carboxyl terminal domain by the TFIIH-associated kinase Kin28 and initiation. RNAPII elongation through the transcribed region is associated with phosphorylation of Serine 2 in the carboxyl terminal domain. RNAPII pauses during cleavage and polyadenylation before dissociating. Created with BioRender.com.
Figure 2 with 2 supplements
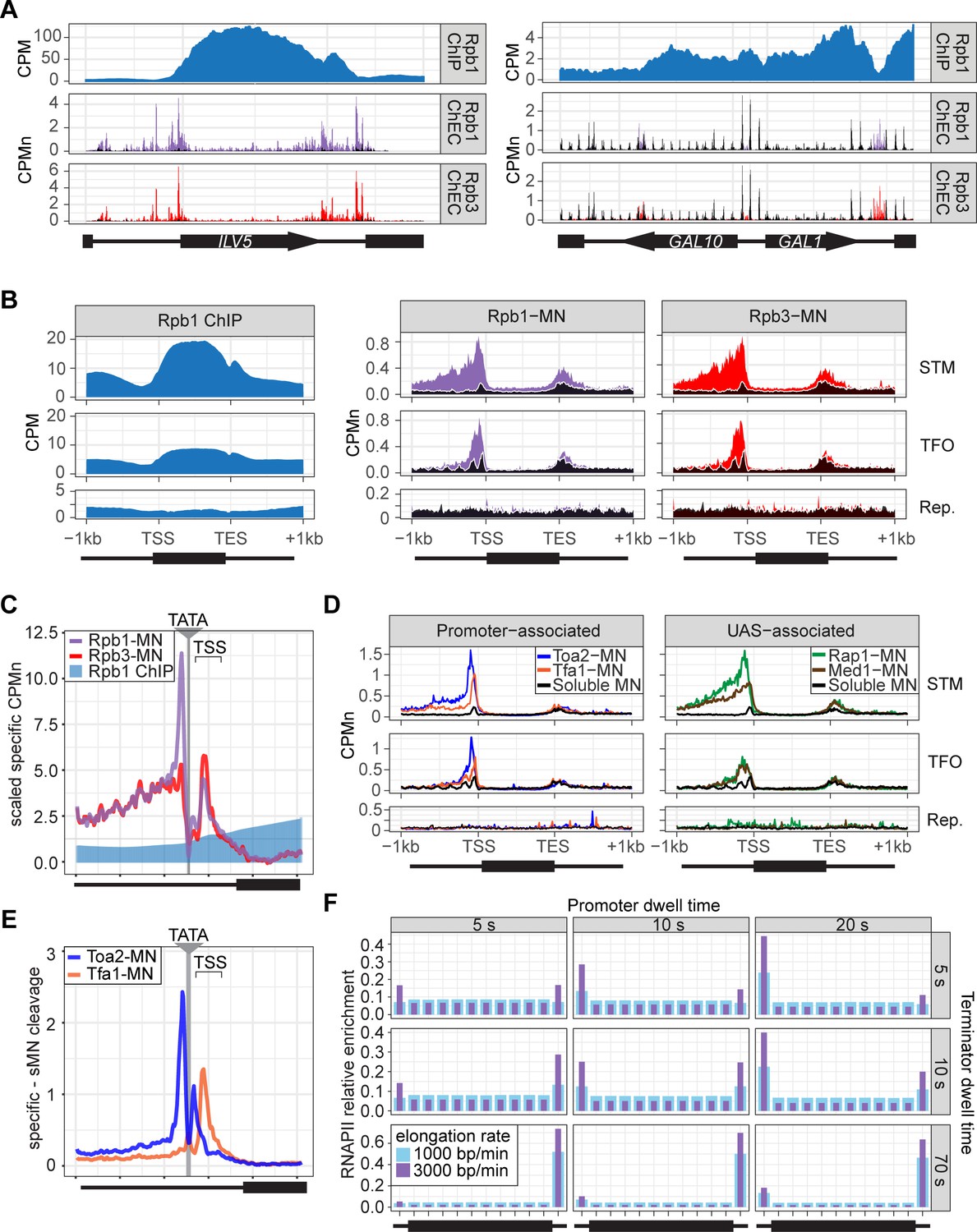
ChEC-seq2 and ChIP-seq reveal distinct RNA polymerase II (RNAPII) interactions with the genome.
(A) Gene plots displaying mean counts per million reads (CPM; Vijjamarri et al., 2023a) for ChIP-seq for Rpb1; or CPM-normalized cleavage frequency (CPMn) for ChEC-seq2 for Rpb1-MN or Rpb3-MN over the ILV5 and GAL1-10 loci±1 kb. Plots are smoothed using a window of 10 bp and a step size of 5 bp. Signal from soluble micrococcal nuclease (sMNase) is shown in black. (B) Metagene plots showing average signal over subsets of genes with distinct expression levels and mechanisms of regulation. The average signal from 150 genes with highest expression from STM and TFO classes (Rossi et al., 2021) and 84 repressed genes is plotted, along with sMNase (black; genes listed in Supplementary file 1). A length-normalized transcript (rectangle), 1 kb upstream of the transcription start site (TSS), and 1 kb downstream of the transcription end site (TES) is shown. Rpb1 ChIP-seq (left), ChEC-seq2 with Rpb1-MN (middle), or Rpb3-MN (right). (C) ChIP (Rpb1) and ChEC (Rpb1-MN and Rpb3-MN) signal over 597 TATA boxes from expressed genes ±250 bp (Supplementary file 1; Rhee and Pugh, 2012). The location of the TATA sequence is indicated with a gray bar and the TSS is 50 bp±39 bp to the right of the center of the TATA. For ChEC data, sMNase was subtracted from the respective specific CPMn. (D) Metagene plots as in (B) from ChEC-seq2 with Toa2-MN (TFIIA) and Tfa1-MN (TFIIE; left) or the sequence-specific transcription factor (ssTF) Rap1-MN and Med1-MN (Mediator; right) from 287 STM genes containing Rap1-peaks, top 150 expressed TFO genes, or 84 repressed genes. (E) Mean CPMn at TATA genes as in (C) from ChEC-seq2 with Toa2-MN (blue) or Tfa1-MN (orange). (A–E) The averages from three biological replicates. (F) Predicted occupancy of RNAPII based on a range of promoter dwell times (5–20 s), elongation rates (1000–3000 bp/min), and termination times (5–70 s). The transcribed region is 1200 bp divided into 10×120 bp bins, flanked by an upstream promoter bin and downstream terminator bin. RNAPII occupancy was simulated using a minimal stochastic model. RNAPII was assumed to be immediately present at the promoter and progressed to the transcript region with a rate inverse to the promoter dwell time. It then progressed along a 1200 bp coding region with the indicated elongation rate and terminated transcription with a rate inverse to the terminator dwell time.
Figure 2—figure supplement 1
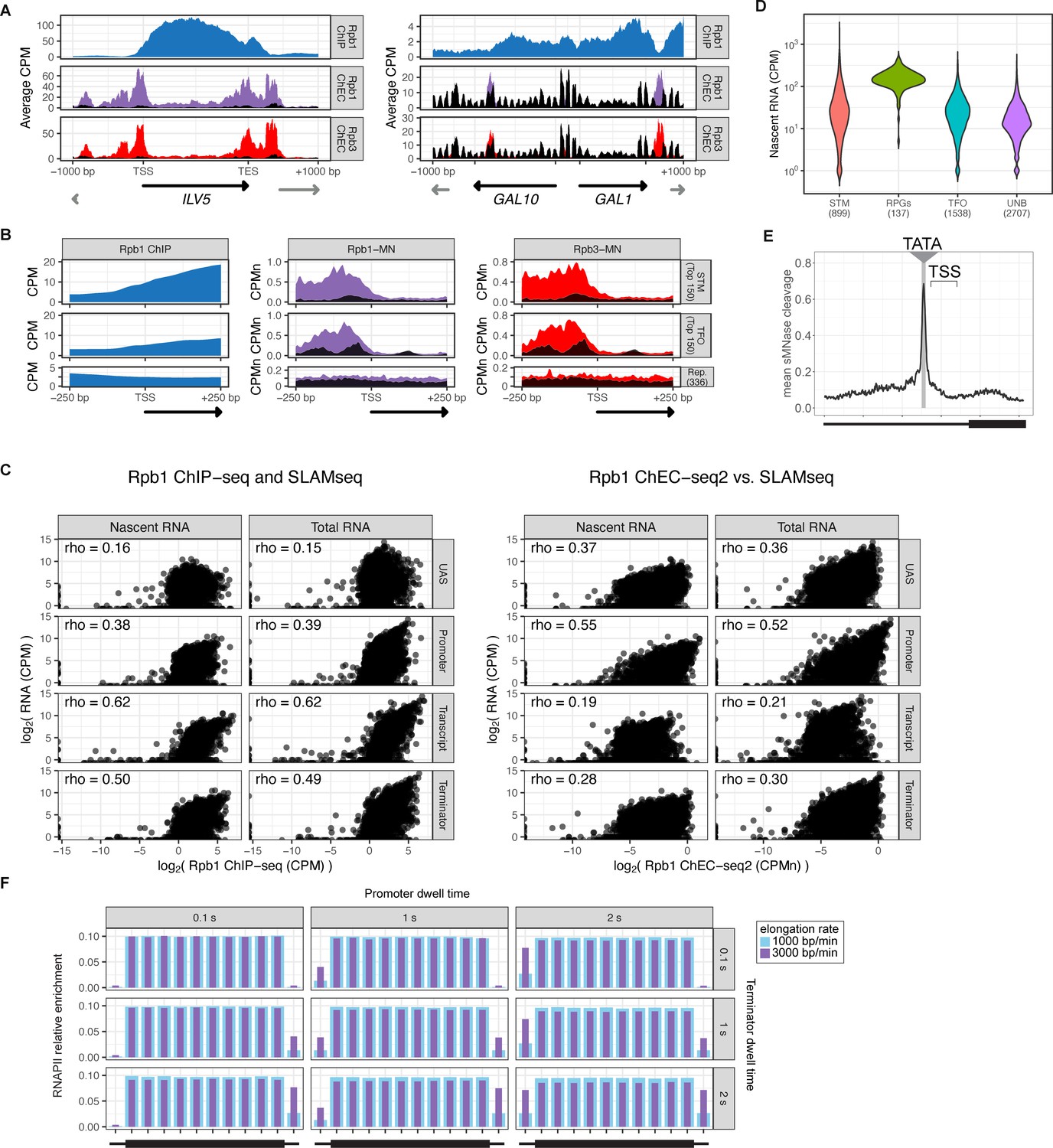
RNA polymerase II (RNAPII) ChEC vs. ChIP.
(A) Gene plots displaying mean counts per million reads (CPM) from Rpb1 ChIP-seq (Vijjamarri et al., 2023a) or ChEC-seq2 with Rpb1-MN or Rpb3-MN over the ILV1 and GAL1-10 loci. A region spanning 1 kb upstream and downstream of each locus is displayed and arrows mark the transcription start site (TSS) and transcription end site (TES). Truncated arrows represent neighboring genes that continue outside of the displayed range. Plots are smoothed with a step size of 5 and window of 10. Signal from the soluble micrococcal nuclease (sMNase) control is shown in black. (B) Metapromoter plots showing average signal flanking the TSS±250 bp from 150 genes with highest expression from STM and TFO classes (Rossi et al., 2021) and 84 repressed (Rep.) genes (Supplementary file 1). (C) Correlation of nascent or total mRNA levels (measured by SLAM-seq) and either ChIP-seq (left) or ChEC-seq2 (right) signal over the indicated regions of each gene. Spearman correlation coefficients for each are shown. (D) Nascent RNA levels from SLAM-seq for each class of genes from Rossi et al., 2021. (E) Metasite plot of 597 expressed, mRNA-encoding genes aligned by their TATA sequence (genes listed in Supplementary file 1; Rhee and Pugh, 2012). Average signal from sMNase grown in rich medium is plotted. A window spanning ±250 bp around the TATA sequence, with the TSS to the right, is shown. The location of the TATA sequence is indicated with a gray bar. The range encompassing TSSs is indicated and the rectangle below the plot designates the approximate location of the CDS. (F) Predicted occupancy of RNAPII based on a range of promoter dwell times (0.1–2 s), elongation rates (1000–3000 bp/min), and termination times (1–2 s). The transcribed region is 1200 bp divided into 10×120 bp bins, flanked by an upstream promoter bin and downstream terminator bin. RNAPII occupancy was simulated using a minimal stochastic model. RNAPII was assumed to be immediately present at the promoter and progressed to the transcript region with a rate inverse to the promoter dwell time. It then progressed along a 1200 bp coding region with the indicated elongation rate and terminated transcription with a rate inverse to the terminator dwell time.
Figure 2—figure supplement 2
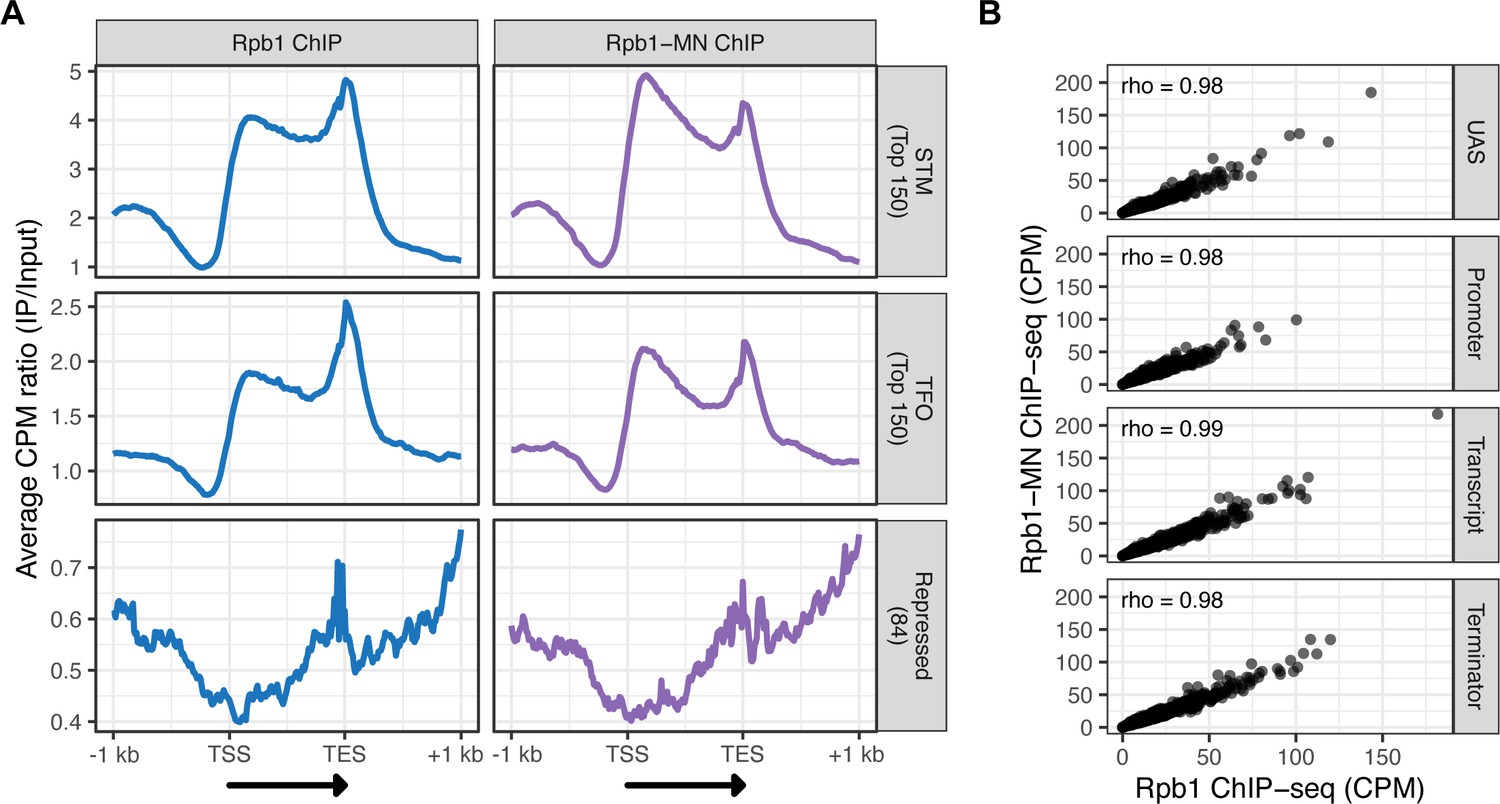
ChIP-seq against Rpb1 vs Rpb1-MN.
(A) Metagene plots showing the ratio of signal between IP and input fractions (IP/Input) over subsets of genes with distinct expression levels and mechanisms of regulation. The average signal from 150 genes with highest expression from STM and TFO classes (Rossi et al., 2021) and 84 repressed genes is plotted (genes listed in Supplementary file 1). A length-normalized transcript (arrow), 1 kb upstream of the transcription start site (TSS), and 1 kb downstream of the transcription end site (TES) is shown. Rpb1 ChIP-seq (left; blue), Rpb1-MN ChIP-seq (right; purple). (B) Correlation of ChIP-seq against Rpb1 vs. Rpb1-MN. Signal (counts per million reads [CPM]) over the indicated regions of each gene are compared. Spearman correlation coefficients for each are shown. The average of three biological replicates is shown in (A) and (B).
Figure 3 with 1 supplement
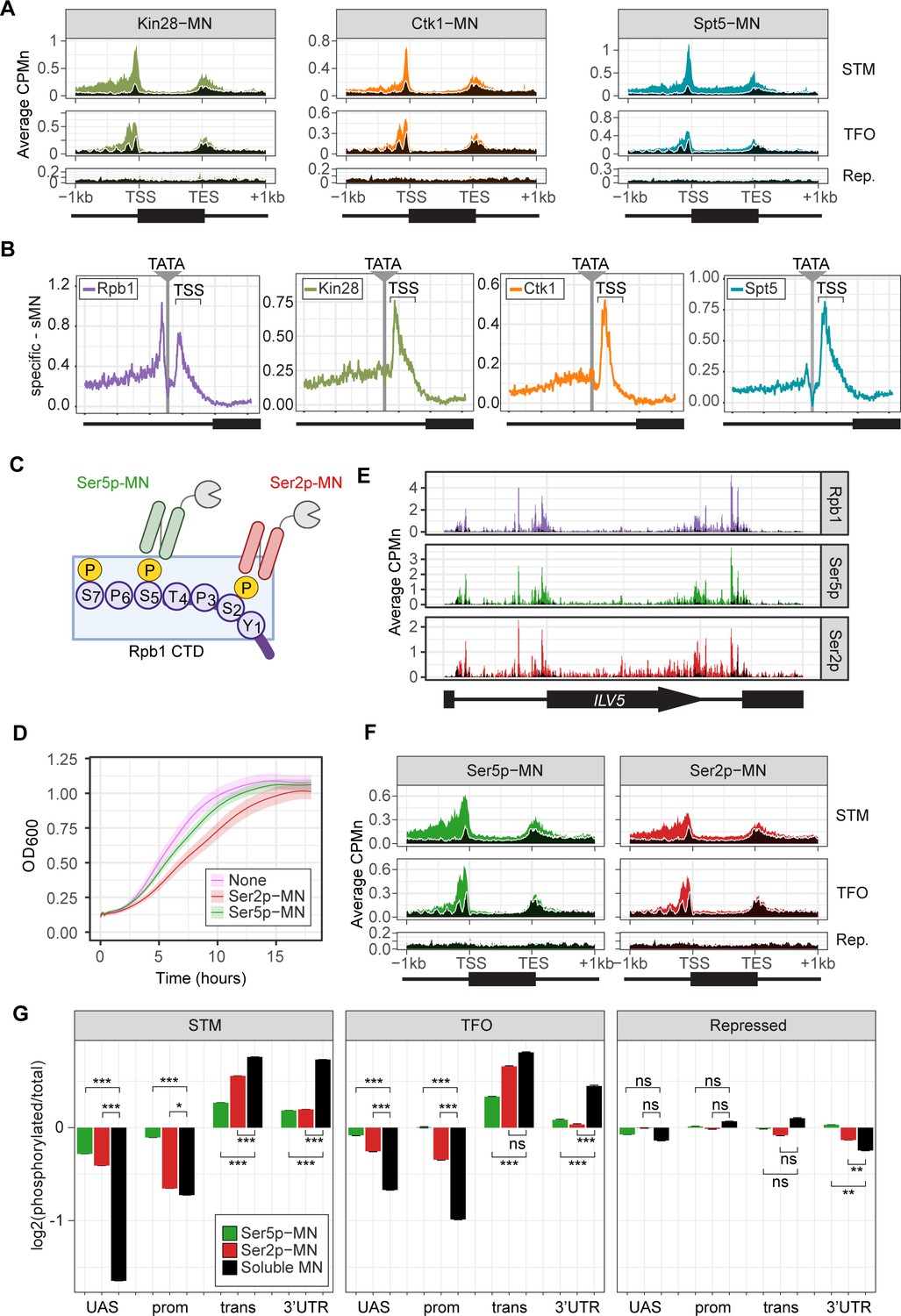
ChEC-seq2 to monitor initiation and elongation.
(A) Metagene plots showing average CPMn for ChEC with the indicated proteins over subsets of genes with distinct expression levels and mechanisms of regulation. The average signal from 150 genes with highest expression from STM and TFO classes (Rossi et al., 2021) and 84 repressed genes are plotted (Supplementary file 1). Signal from the soluble micrococcal nuclease (sMNase) control is shown in black with a white line for contrast. (B) Average CPMn for the indicated proteins over 597 TATA boxes ± 250 bp from expressed genes (Supplementary file 1; Rhee and Pugh, 2012). The location of the TATA sequence is indicated with a gray bar and the transcription start site (TSS) is 50 bp ± 39 bp to the right of the TATA. The signal for sMNase was subtracted from each. (C) Schematic for Mintbody-MNase constructs. Two single chain variable fragments of IgG specific to phosphorylation of Serine 5 (Ser5p) or Serine 2 (Ser2p) of the carboxy terminal domain (CTD) of RNA polymerase II (RNAPII) (Ohishi et al., 2022; Uchino et al., 2022) were tagged at their C-termini with MNase. (D) OD600 of the parent strain (pink), strain expressing Ser5p-MN (green), and strain expressing Ser2p-MN (red) over time in culture (average ± standard deviation). (E) Average CPMn from ChEC-seq2 with Rpb1-MN (purple), α-Ser5P-MN (green), and α-Ser2P-MN (red) at ILV5 ± 1 kb. Plots were smoothed with a step size of 5 and window of 10. (F) Metagene plots as in (A), but with signal from Ser5p-MN (green; left) and Ser2p-MN (red; right). (G) For each protein, the relative enrichment at upstream activating sequence (UAS), promoter, transcript, and 3’UTR regions was calculated and normalized by region length for each gene. The resulting values were normalized to those values for Rpb1-MN and the average from all genes in each group is plotted. Error bars represent the estimated variance between biological replicates from standard deviation (n=3). Differences between ratios and estimated variance were used to calculate a z score and p-value; *p<0.05, **p<0.01, ***p<0.001. All panels represent the average from three biological replicates. Panel C was created with BioRender.com.
Figure 3—figure supplement 1
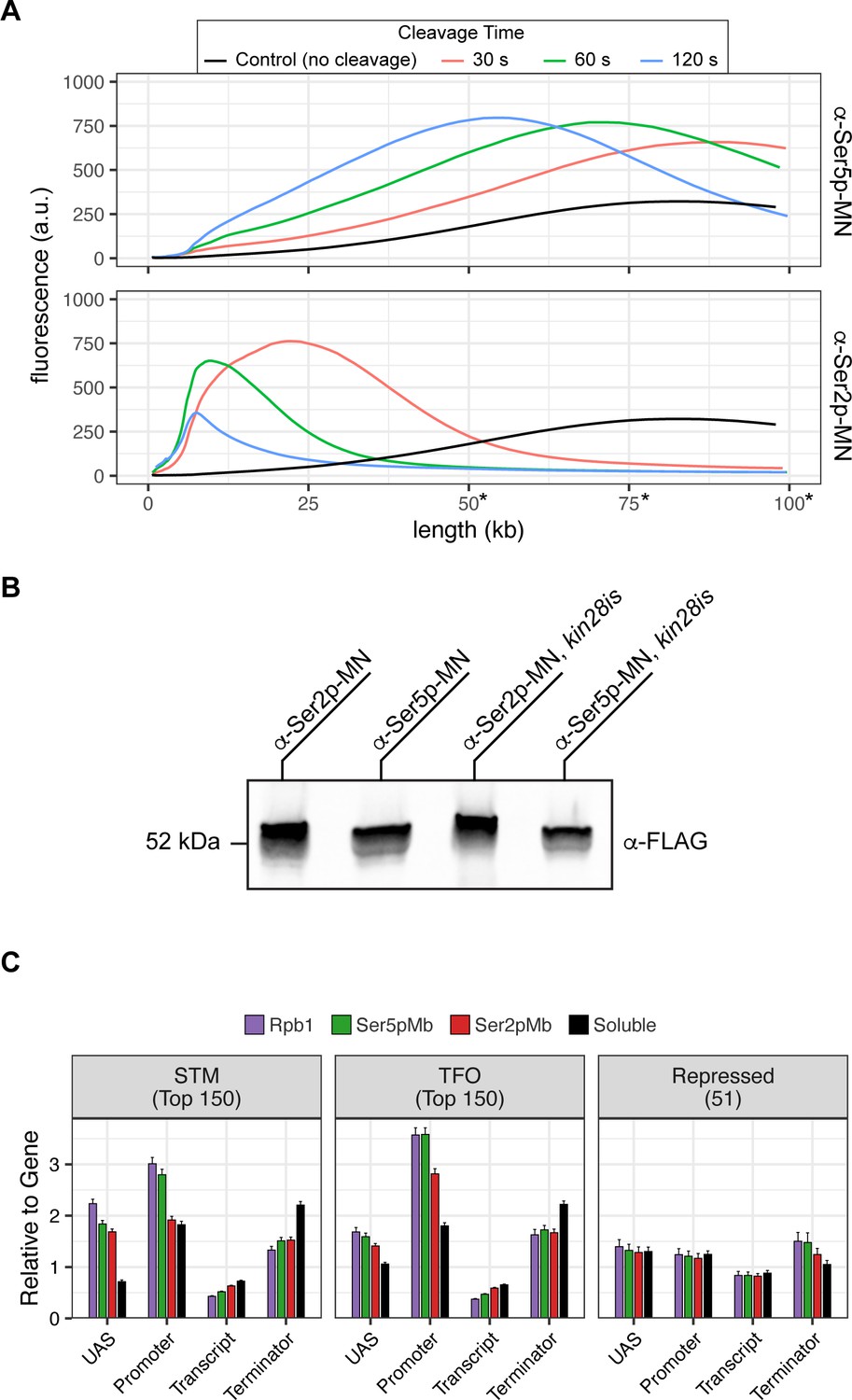
Mintbody-directed ChEC.
(A) Genomic DNA isolated from strains expressing Ser5p-MN (JVY305) and Ser2p-MN (JVY302) was analyzed on a TapeStation 4150. Micrococcal nuclease (MNase) was activated and cleavage proceeded for 30 s (red), 60 s (green), or 120 s (blue). Genomic DNA isolated from cells where no cleavage occurred is shown in black. Note: absolute determination of molecular weight above 50 kb is not possible with this assay and is shown here to highlight relative changes in molecular weight between samples. (B) Chemiluminescent western blot of strains expressing Mintbody-MNase constructs specific to Ser2 phosphorylation (a-Ser2p-MN, JVY302) or Ser5 phosphorylation (α-Ser5p-MN, JVY305) of the carboxy terminal domain (CTD) of RNA polymerase II (RNAPII). Strains expressing each construct on the kin28is background are also shown (α-Ser2p-MN, JVY314; α-Ser5p-MN, JVY317). (C) The relative enrichment at upstream activating sequence (UAS), promoter, transcript, and 3’UTR regions was calculated and normalized by region length for each gene. The average from all genes in each group is plotted. Error bars represent the standard error of the mean between three biological replicates.
-
Figure 3—figure supplement 1—source data 1
TIFF file containing original immunoblots from Figure 3—figure supplement 1B.
- https://cdn.elifesciences.org/articles/100764/elife-100764-fig3-figsupp1-data1-v1.zip
-
Figure 3—figure supplement 1—source data 2
Original files for immunoblot from Figure 3—figure supplement 1B.
- https://cdn.elifesciences.org/articles/100764/elife-100764-fig3-figsupp1-data2-v1.zip
Figure 4
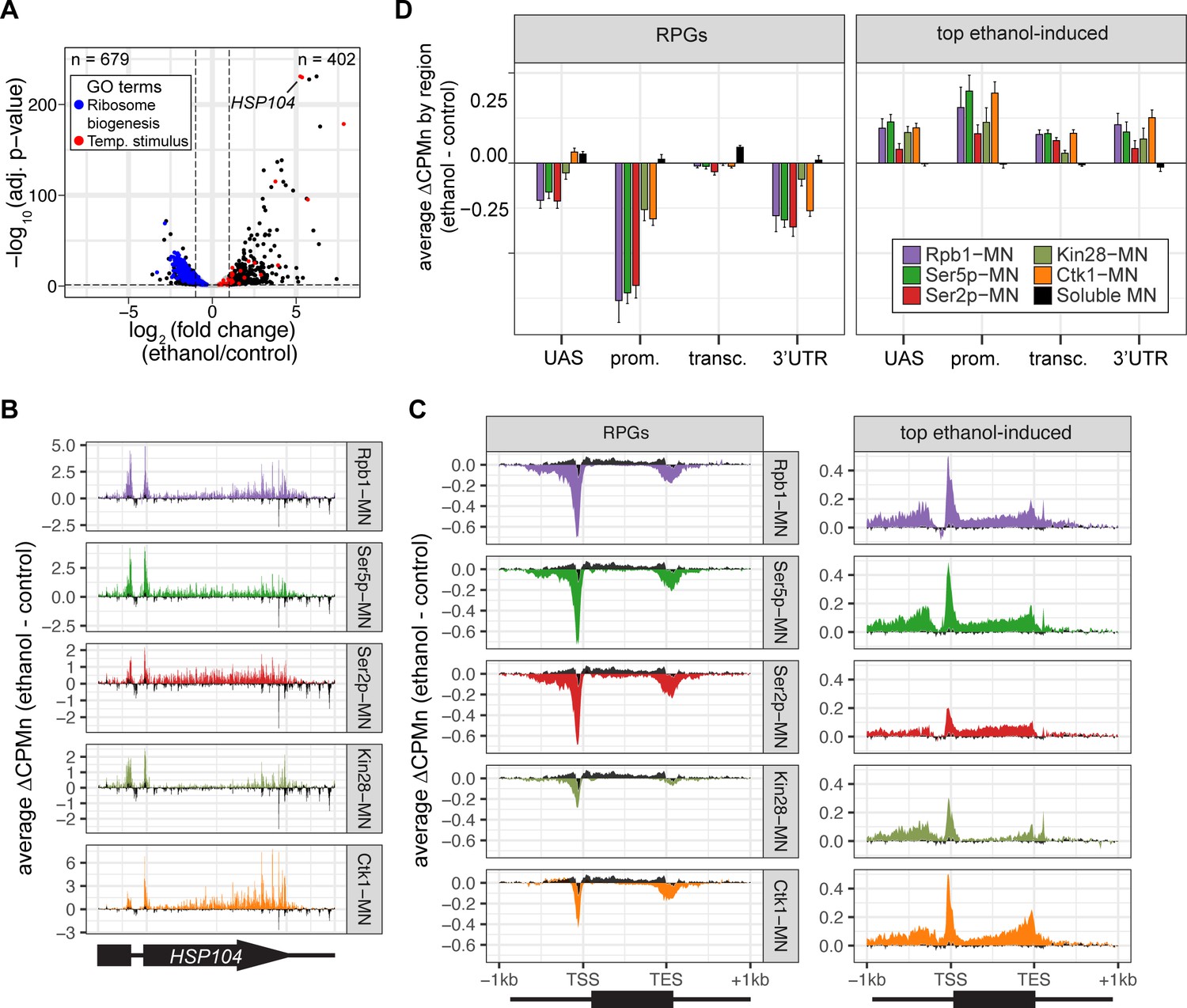
Transcriptional response to ethanol stress results in widespread changes in RNA polymerase II ChEC-seq2.
(A) Volcano plot of fold-change vs. -log10 of adjusted p-values of 5295 mRNAs comparing cells treated with 10% ethanol for 1 hr vs. untreated cells. The mRNAs belonging to the most statistically significant terms from Gene Ontology Enrichment analysis of the 402 upregulated mRNA (response to temperature stimulus, GO: 0009266; red) or 679 downregulated mRNA (ribosome biogenesis, GO: 0042254; blue) are highlighted. (B) Average change in CPMn over HSP104 ± 1 kb (ethanol - untreated) from ChEC-seq2 with Rpb1-MN, Mintbody-MNase constructs (α-Ser5p -MN, α-Ser2p-MN), Kin28-MN, and Ctk1-MN. Data were smoothed with a step size of 5 and window of 10. (C) Metagene plots showing the average change in CPMn (ethanol - untreated) from ChEC-seq2 of the top ethanol-induced genes (100 genes, right; Supplementary file 1) and the downregulated ribosomal protein genes (137 genes, left; Supplementary file 1). CPMn for soluble micrococcal nuclease (sMNase) is shown in black. (D) Gene-region enrichment of each protein relative to Rpb1-MN. For each protein and each gene, the average change in CPMn (ethanol - untreated) was calculated. Signal was binned into gene regions and normalized by region length. The length-normalized region signal relative to total signal (all gene regions) was calculated. The average for 137 RPGs and the top 100 ethanol-induced ± SEM is plotted. For all panels, the average from three biological replicates is shown.
Figure 5 with 1 supplement
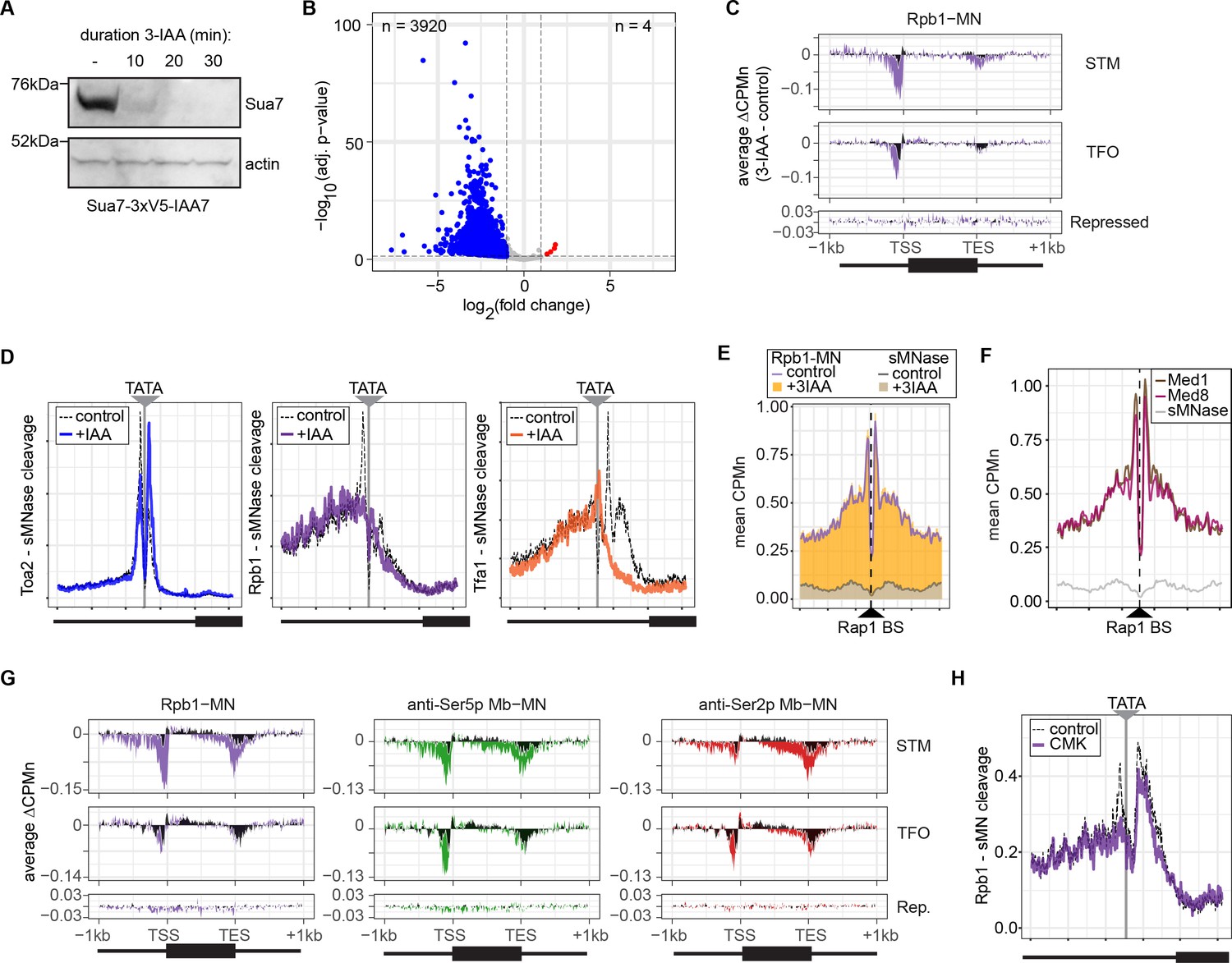
Conditional depletion of TFIIB and inhibition of TFIIH kinase cause distinct effects on promoter-associated RNA polymerase II (RNAPII).
(A) Chemiluminescent immunoblot of Sua7-3V5-IAA7 at the indicated time points following addition of 3-IAA. Signal from actin is shown as a loading control. (B) Volcano plot of log2 fold-change (LFC) in nascent RNA vs. the -log10 of the adjusted p-value following degradation of Sua7-3V5-IAA7 via treatment with 3-IAA for 60 min. Of 5295 mRNAs, 3920 mRNAs were significantly decreased (blue; LFC ≤ –1 and adj. p<0.05) and 4 mRNAs were significantly increased (red; LFC ≥1 and adj. p<0.05). Cells were grown in synthetic complete medium. (C) Metagene plots showing the average change in CPMn (3-IAA - control) from ChEC-seq2 with Rpb1-MN upon degradation of Sua7-3V5-IAA7 for 20 min over 150 genes with highest expression from STM and TFO classes (Rossi et al., 2021) and 84 repressed genes are plotted (Supplementary file 1). Cells were grown in SDC. (D) Metasite plot over the TATA boxes ±250 bp from 597 expressed, mRNA-encoding genes (Supplementary file 1; Rhee and Pugh, 2012). In each case, soluble micrococcal nuclease (sMNase) CPMn was subtracted from the specific CPMn and the untreated control is shown in gray for comparison. The location of the TATA sequence is indicated with a gray bar. Cells were grown in YPD and Sua7-3V5-IAA7 was depleted for 60 min. (E) Metasite plot of average CPMn from Rpb1-MN and sMNase cleavage over 896 high-confidence Rap1 sites (VanBelzen et al., 2024). Purple and dark gray lines represent mean Rpb1-MN and sMNase cleavage in untreated cells; orange and gray columns represent mean Rpb1-MN and sMNase cleavage upon Sua7 depletion for 60 min. (F) Metasite plots of average CPMn from Med1-MN (brown), Med8-MN (magenta), sMNase (gray) over Rap1 sites as in (E). (G) Metagene plots as in (C) of average change in signal (CMK - control) upon inhibition of kin28is for Rpb1-MN (purple), Ser5p-MN (green), Ser2p-MN (red). Cells were grown in SDC and treated with 5 µM CMK for 60 min. (H) Metasite plot over TATA boxes as in (D) for the sMNase-corrected signal from Rpb1-MN before (gray) and after (purple) inhibition of kin28-is with 5 µM CMK for 60 min. For all panels, the average of three biological replicates is plotted.
-
Figure 5—source data 1
PDF file containing labeled western blot membrane corresponding to Figure 5A.
Three biological replicates are shown. Samples were collected 0, 10, 20, and 30 min following the addition of 3-IAA. Rainbow molecular weight marker is loaded in the lane immediately to the left of each 0 min time point. Biological replicate #1 is shown in Figure 5A.
- https://cdn.elifesciences.org/articles/100764/elife-100764-fig5-data1-v1.zip
-
Figure 5—source data 2
Original western blot membrane corresponding to Figure 5A.
- https://cdn.elifesciences.org/articles/100764/elife-100764-fig5-data2-v1.zip
Figure 5—figure supplement 1
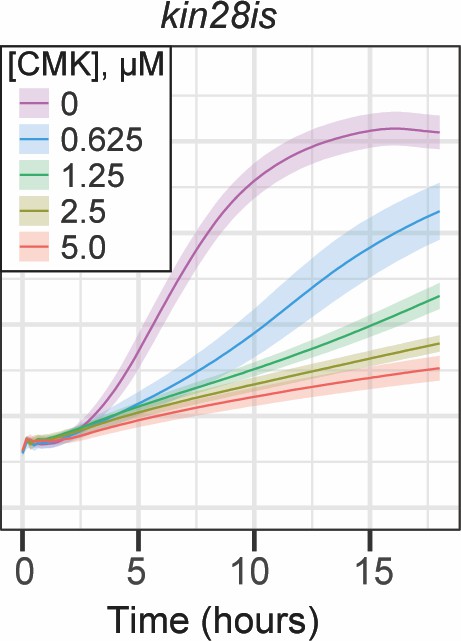
Growth effect of CMK treatment in wild-type and kin28is cells.
OD600 of kin28is strain grown at 30°C in synthetic complete medium with the indicated concentrations of CMK. The average ± standard deviation is plotted.
Figure 6 with 1 supplement
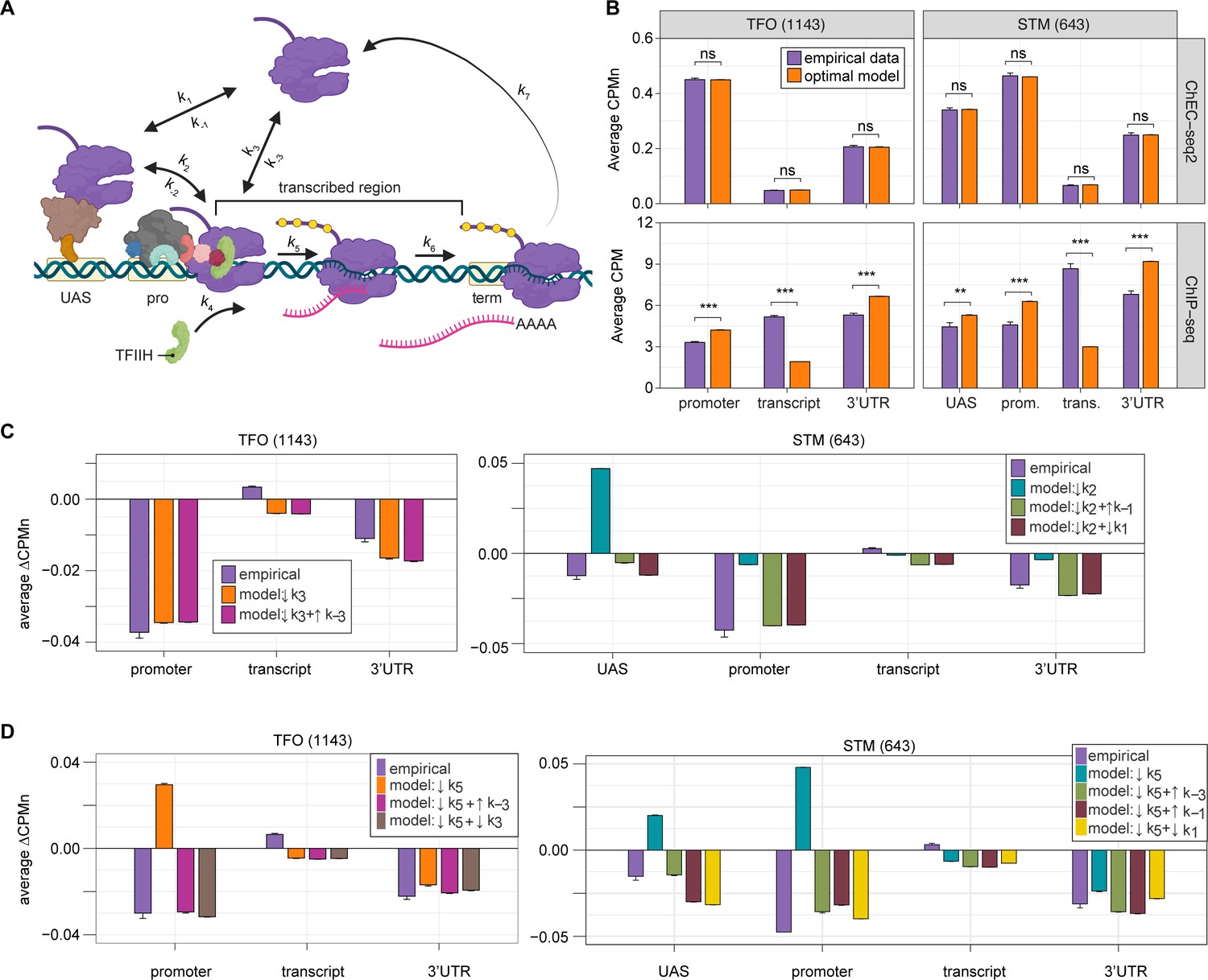
Global kinetic model for RNA polymerase II (RNAPII) transcription.
(A) Schematic for a model of the global kinetics of transcription by RNAPII in S. cerevisiae. Two alternative mechanisms of RNAPII recruitment are shown: (1) direct recruitment to TFO promoters governed by rates k3 and k-3 and (2) recruitment to the STM upstream activating sequences (UASs) facilitated by a sequence-specific transcription factors (ssTFs) and coactivators such as Mediator (k1 and k-1), followed by transfer to the promoters (k2 and k-2). After RNAPII arrives at the promoter it can dissociate at rate k-3 until TFIIH is recruited (k4), followed by initiation (k5). RNAPII elongation (k6) across the transcribed region produces mRNA. Pausing during termination is determined by the dissociation rate k7. Transcription is modeled as a stochastic, processive process with successful recruitment of TFIIH representing the committed step. Processes involving rates k4, k5, k6, and k7 are irreversible. (B) The average Rpb1 signal (purple) from ChEC-seq2 (top) and ChIP-seq (bottom) over the indicated regions from 1143 TFO-class genes and 643 STM-class genes that are expressed in SDC (Supplementary file 1). Rates k2, k-2, k-3, and k4 were explored to fit the empirical data for each dataset. The remaining rates were drawn from published values (see Table 1). RNAPII occupancy was simulated across gene regions. UAS, promoter, and 3’UTR were represented by a single 120 bp bin and the transcript region was composed of 10 sequential bins to represent a 1200 bp transcript. The average predicted occupancy for RNAPII over each region from the models (i.e. sets of rates) that best matched the empirical data are shown (see Methods). For Rpb1 ChEC-seq2, 789 STM models and 371 TFO models fit the empirical data. Using the same fit thresholds for ChIP-seq data produced no models. Instead, the average predicted occupancy from an equal number of the top-performing ChIP-seq models as used in ChEC-seq2 simulations (i.e. 789 for STM and 371 for TFO) was used to generate the predictions shown (Table 1). (C, D) The average change in Rpb1-MN by ChEC-seq2 at each gene region following conditional depletion or inactivation of preinitiation complex (PIC) components (purple) for 1143 TFO-class genes and 643 STM-class genes that are expressed in SDC is shown. The rates from the ensemble of best models in (B) were adjusted to model the observed changes in Rpb1-MN over each gene region. (C) The average change in Rpb1-MN (3-IAA - control) following conditional depletion of TFIIB (purple, from Figure 5C). For TFO-class genes, a decrease in promoter recruitment (↓k3) with or without an increase in promoter dissociation (↓k3 + ↑k-3) fit the empirical findings. For STM genes, a decrease in transfer from UAS (↓k2) combined with an increase in dissociation from UAS (↓k2 + ↑k-1) or decrease in UAS recruitment (↓k2 + ↓k1) fit the observed changes (Table 2). (D) The average change in Rpb1-MN (CMK - control) following inhibition of TFIIH kinase (purple, from Figure 5G). For TFO-class genes, a decrease in initiation (↓k5) combined with an increase in dissociation from promoter (↓k5 + ↑k-3) or decrease in promoter recruitment (↓k5 + ↓k3) fit the observed changes. For STM, a decrease in initiation (↓k5) combined with either an increase in dissociation from promoter (↓k5 + ↑k-3), an increase in dissociation from UAS (↓k5 + ↑k-1), or a decrease in UAS recruitment (↓k5 + ↓k1) fit the observed changes (Table 2). Panel A was created with BioRender.com.
Figure 6—figure supplement 1
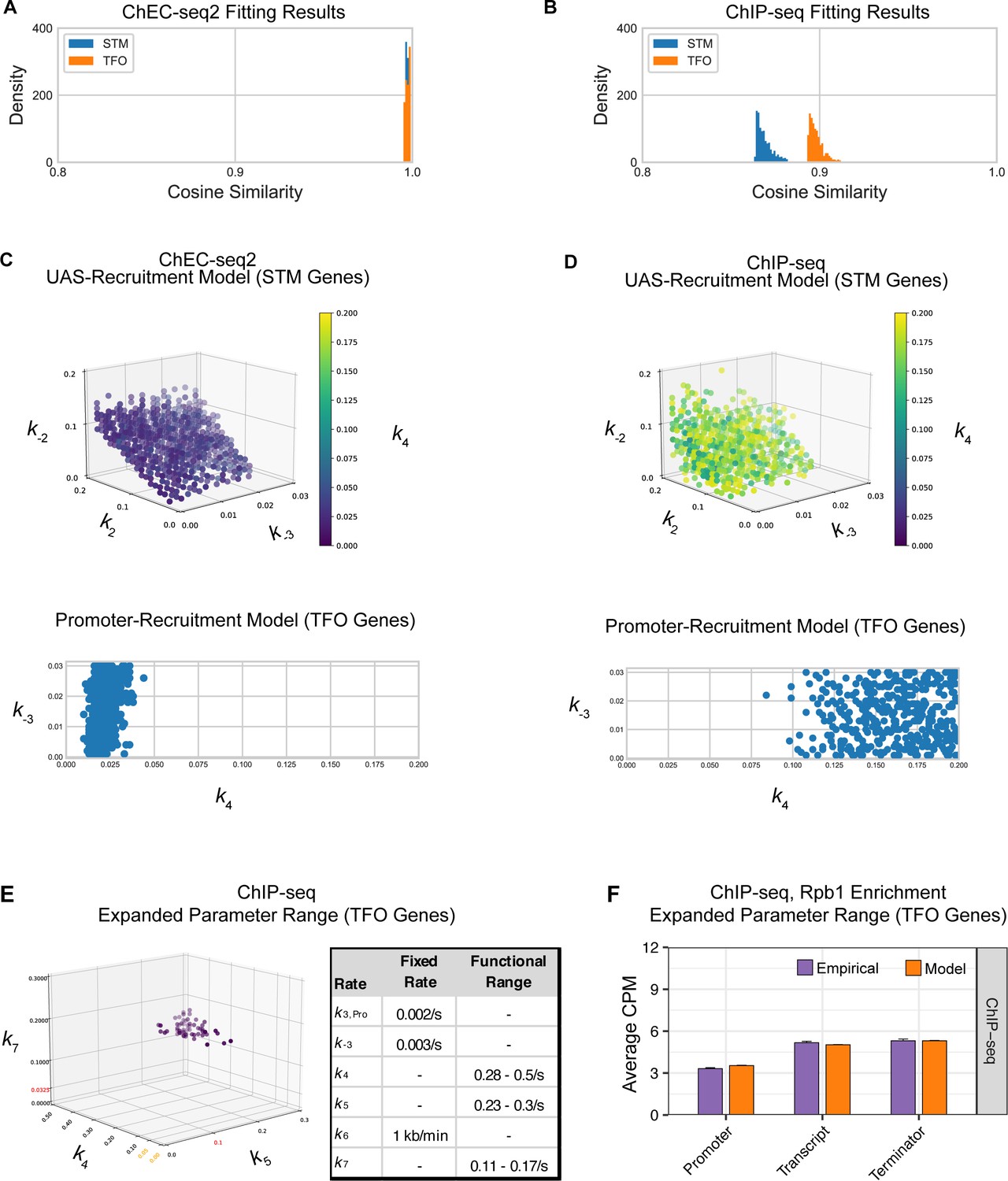
Parameter fitting of unknown transcription rates.
Rates with no known value were fit to RNA polymerase II (RNAPII) occupancies from either ChEC-seq2 or ChIP-seq data using a grid search (see Methods). (A–C) For STM model, rates k2, k-2, and k4 were explored in the range [0, 0.2] and k-3 in the range [0, 0.03]. For TFO model, only rates k-3, and k4 were fit, in the same range. (A) Distribution of cosine similarity for the model ensemble when fit to ChEC-seq2 data. Cosine similarity of 1 indicates perfect alignment, 0 indicates no correlation, and –1 indicates perfect inverse alignment. (B) Distribution of cosine similarity for the model ensemble when fit to ChIP-seq data. (C) Rate combinations that fit the empirical data ChEC-seq2 data (Rpb1-MN). This resulted in 789 rate combinations for the STM model and 371 rate combinations for the TFO model. (D) No rate combinations resulted in a satisfactory fit to the empirical ChIP-seq data (Rpb1). Instead, an equal number of rate combinations (best-fit) as shown in (C) is displayed. (E) In an attempt to identify rates that fit the RNAPII enrichment seen by ChIP-seq, we used the promotor-recruitment model (TFO genes) and loosened previously fixed rates k5 and k7 and expanded the search range for k4 while fixing k-3. Published rates for k5 and k7 are displayed in red on the axes. Range from parameter fit k4 from ChEC-seq2 data (C) is shown in orange on the k4 axis. The range of values for k4, k5, and k7 that fit the ChIP-seq data are shown in the table (functional range). In the idealized case k-3=0 and k4 is instantaneous then k7 should be equal to the product of k6 and the ratio between the average occupancy given by ChIP in the coding region and terminator of the gene (approximately 0.14 s–1), and k5 should be equal to the product of k6 and the ratio between the average occupancy given by ChIP in the coding region and the terminator of the gene (approximately 0.2 s–1). The functional ranges shown agree with this, as rate k5 is bounded below by the idealized approximation, and rate k7 is centered around its idealized approximation. (F) The average Rpb1 signal (purple) from ChIP-seq over the indicated regions from TFO-class genes that are expressed in SDC. RNAPII enrichment resulting from rate combinations shown in (E) were modeled in combination with fixed rates from the literature shown in Table 1. Upstream activating sequence (UAS), promoter, and 3’UTR were represented by a single 120 bp bin and the transcript region was composed of 10 sequential bins to represent a 1200 bp transcript. The average predicted occupancy for RNAPII over each region from the models (i.e. sets of rates) that best matched the empirical data are shown (see Methods). For Rpb1 ChIP-seq, 55 rate combinations from the promoter model fit the empirical data from TFO-class genes. Empirical and model outcomes were compared for each gene region with a Student’s t-test, which reported no significant differences (p>0.05).
Figure 7 with 1 supplement
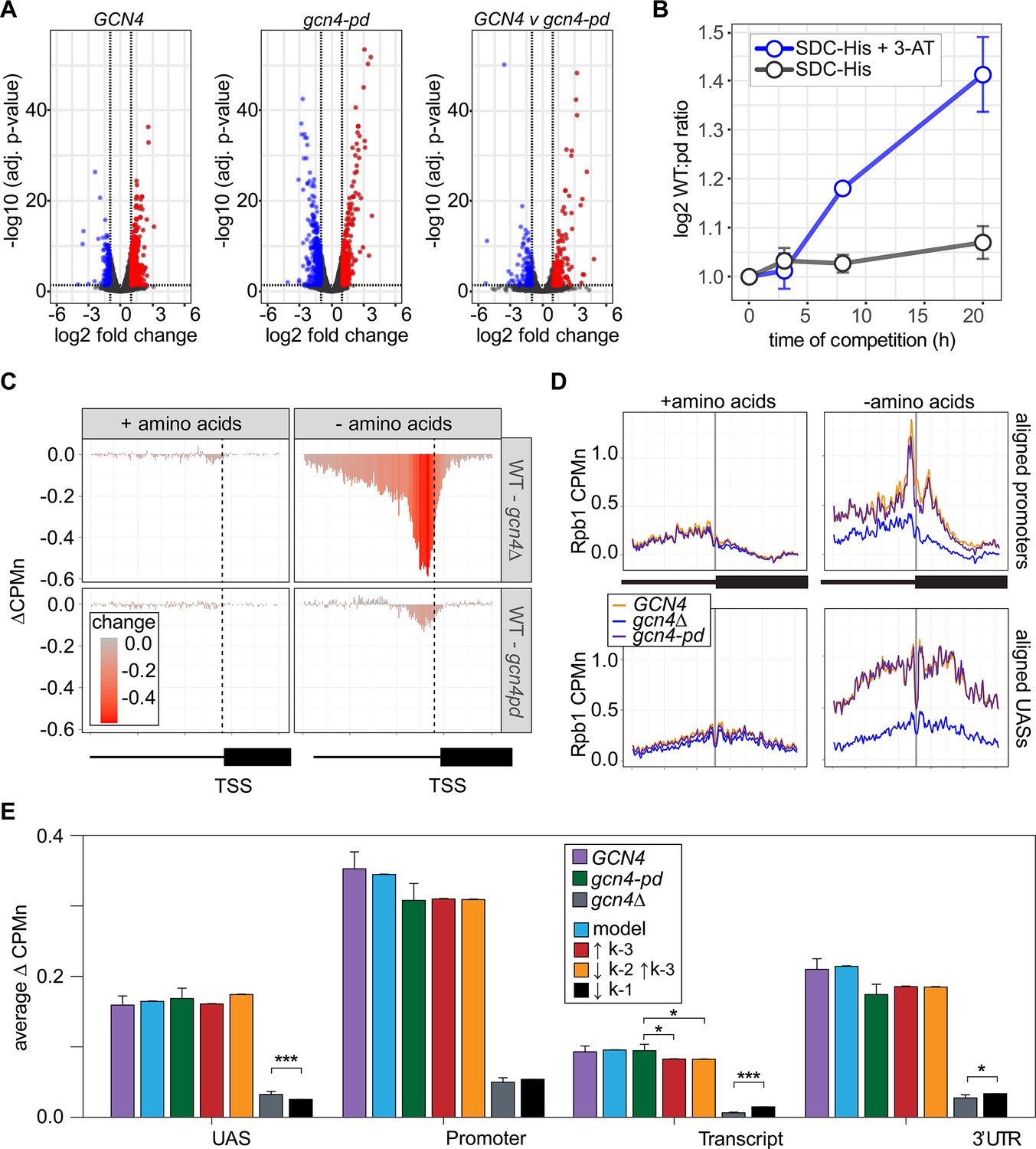
The Gcn4 positioning domain stabilizes RNAPII association with the promoter without affecting recruitment to the upstream activating sequence (UAS).
(A) Volcano plot of log2 fold-change in nascent RNA vs. -log10 adjusted p-values from cells starved for histidine for 1 hr vs. cells in complete medium. Nascent RNA counts for a total of 5295 mRNAs were measured in GCN4 (left) and gcn4-pd (middle). The log2 fold-change in nascent RNA vs. -log10 adjusted p-values in GCN4 vs. gcn4-pd from cells grown in the absence of histidine (right). Significantly downregulated (blue; LFC ≤ –1 and adj. p<0.05), upregulated (red; LFC ≥1 and adj. p<0.05) genes are highlighted. (B) Relative abundance of GCN4 and gcn4-pd strains in a mixed culture, determined by quantifying the relative abundance of the two alleles in the population (Sump et al., 2022) in either SDC-His or SDC-His+10 mM 3-amino triazole (3-AT). (C) Average difference in Rpb1-MN between gcn4∆ (top) and gcn4-pd (bottom; mutant - GCN4, ∆CPMn) upstream of 246 Gcn4-target genes for cells grown + amino acids (left) or - amino acids for 1 hr (Supplementary file 1). A region spanning 700 bp upstream and 400 bp downstream of the transcription start site (TSS) (hashed vertical line) is displayed and the color scale reflects the difference between wild-type and each mutant. (D) Metasite plot showing average Rpb1-MN CPMn from cells grown + amino acids (left) or - amino acids (right) for gcn4∆ (blue), gcn4-pd (purple), and GCN4 (yellow) strains. Top: Rpb1-MN-sMNase CPMn over TATA boxes±250 bp upstream of 173 Gcn4-dependent genes (Supplementary file 1; Rhee and Pugh, 2012). Bottom: Rpb1-MN CPMn over 284 Gcn4 binding sites (Gcn4BS)±250 bp upstream of 130 Gcn4 target genes (Supplementary file 1). (E) The average change in Rpb1-MN by ChEC-seq2 at each gene region in cells shifted into media lacking amino acids (SD+uracil) vs. cells shifted into media with amino acids (SDC) is plotted (SD+uracil - SDC, ∆CPMn) for each strain (GCN4, purple; gcn4-pd, green; gcn4∆, gray). Rates k2, k-2, k-3, and k4 were re-fit to the observed Rpb1-MN occupancy at 287 Gcn4-dependent genes in wild-type cells grown in the absence of amino acids and yielded 1057 STM models (blue). The rates from these best-fit models were adjusted to fit the observed changes in Rpb1-MN over each gene region in gcn4-pd and gcn4∆ strains. For gcn4-pd, an increase in dissociation from the promoter fit the empirical findings (↑k-3, red; Table 2) or a combined increase in promoter dissociation and decrease in transfer from UAS to promoter (↓k-2↑k-3, orange; Table 2). For gcn4∆, a decrease in recruitment to UAS fit in the model and the empirical findings (↓k1, black; Table 2).
Figure 7—figure supplement 1

Parameter fitting of unknown transcription rates in upstream activating sequence (UAS)-recruitment model for Gcn4 target genes.
Rates with no known value were parameter fit using a grid search (see Methods). We used the UAS-recruitment model and explored rates k2, k-2, and k4 in the range [0, 0.2] and k-3 in the range [0, 0.03]. Rate combinations that fit the Rpb1-MN ChEC-seq2 data from 287 Gcn4-target genes under amino acid starvation conditions. The fitting procedure resulted in 1057 rate combinations that fit the empirical data.
Author response image 1
Author response image 2
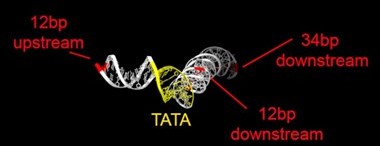
Cleavage at -12, +12 and +34.
Author response image 3
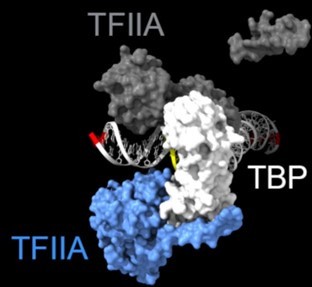
Highlighted sites corresponding to the peaks in TFIIA assembled with TBP.
Author response image 4

The complete PIC, protecting the +12 site, but leaving the +34 site exposed.
Videos
Video 1
Animation of the accessibility of DNA to cleavage during assembly of the closed preinitiation complex, based on PDB 7nvs (Aibara et al., 2021).
Tables
Table 1
Kinetic parameters from stochastic model.
| Rate | Published Values | Standard Growth - ChEC-seq | Standard Growth - ChIP-seq | GCN4 | Rate Description | |||
|---|---|---|---|---|---|---|---|---|
| Selected Rate | Functional Range | Selected Rate | Functional Range | Selected Rate | Functional Range | |||
| k1, UAS | 0.0019–0.0027 /s (Rosen et al., 2020) | 0.002 /s | - | 0.002 /s | - | 0.002 /s | - | Recruitment of RNAPII to UAS |
| k-1, UAS | 0.001–0.005 /s (Rosen et al., 2020) | 0.003 /s | - | 0.003 /s | - | 0.003 /s | - | Dissociation of RNAPII from UAS |
| k2, UAS | None Identified | - | 0.03–0.1 /s | - | 0.03–0.1 /s | - | 0.03–0.1 /s | Transfer of RNAPII from UAS to Promoter. Requirement for assembly early PIC components at Promoter is incorporated |
| k-2, UAS | None Identified | - | 0.0–0.07 /s | - | 0.0–0.1 /s | - | 0.0–0.04 /s | Transfer of RNAPII from Promoter to UAS |
| k3, Promoter | None identified. Used k1 | 0.002 /s | - | 0.002 /s | - | NA | - | Recruitment of RNAPII to Promoter |
| k-3 | None Identified | - | 0.0–0.03 /s | - | 0.0–0.03 /s | - | 0.0–0.03 /s | Dissociation of RNAPII from Promoter |
| k4 | None Identified | - | 0.0075–0.0275 /s | - | 0.025–0.1 /s | - | 0.01–0.03/s | Recruitment of TFIIH |
| k5 | 0.1 /s (TFIIH residency, Nguyen et al., 2021) | 0.1 /s | - | 0.1 /s | - | 0.1 /s | - | Initiation and phosphorylation of RNAPII CTD by TFIIH Kinase (Kin28) |
| k6 | 1–3 kb/min | 1 kb/min | - | 1 kb/min | - | 1 kb/min | - | Transcription elongation |
| k7 | 0.009–0.034/s (Larson et al., 2011 on MDN1) | 0.0325 /s | - | 0.0325 /s | - | 0.0325 /s | - | Terminantion pause |
Table 2
Kinetic parameters from modeling genetic perturbations of transcription.
| Experiment | Gene Class | Model # | k1, UAS | k-1, UAS | k2, UAS | k-2, UAS | k3, Promoter | k-3 | k4 | k5 | k6 | k7 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.002 /s | 0.003 /s | 0.03–0.1 /s | 0.0–0.07 /s | 0.002 /s | 0.0–0.03 /s | 0.0075–0.0275 /s | 0.1 /s | 1 kb/min | 0.0325 /s | |||
| TFIIB Degradation, 20 min | STM | 1 | 0.004–0.04 | - | ||||||||
| TFIIB Degradation, 20 min | STM | 2 | 0.04 | 0.004–0.04 | - | |||||||
| TFIIB Degradation, 20 min | STM | 3 | 0.0006 | 0.004–0.04 | - | |||||||
| TFIIB Degradation, 20 min | TFO | 1 | - | - | - | - | 0.0008 | |||||
| TFIIB Degradation, 20 min | TFO | 2 | - | - | - | - | 0.0008 | 0.0–0.12 | ||||
| TFIIB Degradation, 60 min | STM | 1 | 0.002–0.02 | - | ||||||||
| TFIIB Degradation, 60 min | STM | 2 | 0.06 | 0.002–0.02 | - | |||||||
| TFIIB Degradation, 60 min | STM | 3 | 0.0005 | 0.002–0.02 | - | |||||||
| TFIIB Degradation, 60 min | TFO | 1 | - | - | - | - | 0.0004 | |||||
| TFIIB Degradation, 60 min | TFO | 2 | - | - | - | - | 0.0004 | 0.0–0.24 | ||||
| TFIIH Inhibition | STM | 1 | - | 0.02 | ||||||||
| TFIIH Inhibition | STM | 2 | - | 0.0–0.24 | 0.02 | |||||||
| TFIIH Inhibition | STM | 3 | 0.06 | - | 0.02 | |||||||
| TFIIH Inhibition | STM | 4 | 0.0005 | - | 0.02 | |||||||
| TFIIH Inhibition | TFO | 1 | - | - | - | - | 0.02 | |||||
| TFIIH Inhibition | TFO | 2 | - | - | - | - | 0.0–0.24 | 0.02 | ||||
| TFIIH Inhibition | TFO | 3 | - | - | - | - | 0.0005 | 0.02 | ||||
| Experiment | Gene Class | Model # | k1, UAS | k-1, UAS | k2, UAS | k-2, UAS | k3, Promoter | k-3 | k4 | k5 | k6 | k7 |
| 0.002 /s | 0.003 /s | 0.03–0.1 /s | 0.0–0.03 /s | 0.002 /s | 0.0–0.03 /s | 0.0075–0.0275 /s | 0.1 /s | 1 kb/min | 0.0325 /s | |||
| gcn4pd | STM | 1 | 0.0–0.048 | |||||||||
| gcn4pd | STM | 2 | 0.009–0.09 | 0.0–0.039 | ||||||||
| gcn4null | STM | 1 | 0.0006 |
Additional files
-
Supplementary file 1
Lists of gene subsets used in this study.
- https://cdn.elifesciences.org/articles/100764/elife-100764-supp1-v1.xlsx
-
Supplementary file 2
Yeast strains used in this study.
- https://cdn.elifesciences.org/articles/100764/elife-100764-supp2-v1.xlsx
-
Supplementary file 3
Plasmids and oligonucleotides used in this study.
- https://cdn.elifesciences.org/articles/100764/elife-100764-supp3-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/100764/elife-100764-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Chromatin endogenous cleavage provides a global view of yeast RNA polymerase II transcription kinetics
eLife 13:RP100764.
https://doi.org/10.7554/eLife.100764.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}