Regulatory roles of Escherichia coli 5' UTR and ORF-internal RNAs detected by 3' end mapping
- Division of Molecular and Cellular Biology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, United States
- Postdoctoral Research Associate Program, National Institute of General Medical Sciences, National Institutes of Health, United States
- Wadsworth Center, New York State Department of Health, United States
- Bioinformatics and Scientific Programming Core, Eunice Kennedy Shriver National Institute of Child Health and Human Development, United States
- Department of Biomedical Sciences, School of Public Health, University at Albany, United States
Abstract
Many bacterial genes are regulated by RNA elements in their 5´ untranslated regions (UTRs). However, the full complement of these elements is not known even in the model bacterium Escherichia coli. Using complementary RNA-sequencing approaches, we detected large numbers of 3´ ends in 5´ UTRs and open reading frames (ORFs), suggesting extensive regulation by premature transcription termination. We documented regulation for multiple transcripts, including spermidine induction involving Rho and translation of an upstream ORF for an mRNA encoding a spermidine efflux pump. In addition to discovering novel sites of regulation, we detected short, stable RNA fragments derived from 5´ UTRs and sequences internal to ORFs. Characterization of three of these transcripts, including an RNA internal to an essential cell division gene, revealed that they have independent functions as sRNA sponges. Thus, these data uncover an abundance of cis- and trans-acting RNA regulators in bacterial 5´ UTRs and internal to ORFs.
eLife digest
In most organisms, specific segments of a cell’s genetic information are copied to form single-stranded molecules of various sizes and purposes. Each of these RNA molecules, as they are known, is constructed as a chain that starts at the 5´ end and terminates at the 3´ end.
Certain RNAs carry the information present in a gene, which provides the instructions that a cell needs to build proteins. Some, however, are ‘non-coding’ and instead act to fine-tune the activity of other RNAs. These regulatory RNAs can be separate from the RNAs they control, or they can be embedded in the very sequences they regulate; new evidence also shows that certain regulatory RNAs can act in both ways.
Many regulatory RNAs are yet to be catalogued, even in simple, well-studied species such as the bacterium Escherichia coli. Here, Adams et al. aimed to better characterize the regulatory RNAs present in E. coli by mapping out the 3´ ends of every RNA molecule in the bacterium.
This revealed many new regulatory RNAs and offered insights into where these sequences are located. For instance, the results show that several of these RNAs were embedded within RNA produced from larger genes. Some were nested in coding RNAs, and were parts of a longer RNA sequence that is adjacent to the protein coding segment. Others, however, were present within the instructions that code for a protein.
The work by Adams et al. reveals that regulatory RNAs can be located in unexpected places, and provides a method for identifying them. This can be applied to other types of bacteria, in particular in species with few known RNA regulators.
Introduction
The expression of many bacterial genes is controlled by elements in the 5´ untranslated regions (UTRs) of mRNAs. Changes in the secondary structures of these cis-acting RNA elements lead to altered expression of the associated gene(s) by modulating accessibility of ribosomes to sites of translation initiation, accessibility of RNases, or premature transcription termination. The RNA secondary structure changes can occur in response to temperature (RNA thermometers), translation of small upstream open reading frames (uORFs), or the binding of trans-acting factors such as metabolites (riboswitches), tRNAs, RNA-binding proteins such as CsrA, or small base-pairing RNAs (sRNAs) (reviewed in Breaker, 2018; Kreuzer and Henkin, 2018; Loh et al., 2018; Orr et al., 2020; Romeo and Babitzke, 2019; Storz et al., 2011).
Some of the regulatory events in 5´ UTRs are associated with premature transcription termination, which occurs by one of two mechanisms: intrinsic (Rho-independent) or Rho-dependent (reviewed in Roberts, 2019). Intrinsic termination requires only RNA polymerase and an RNA hairpin followed by a U-rich tract in the nascent RNA. Rho-dependent termination requires the loading of the hexameric Rho protein complex onto nascent, untranslated RNA at Rho utilization (Rut) sites that are typically C-rich, G-poor, and unstructured sequences (reviewed in Mitra et al., 2017). Rho translocates along the RNA until the protein catches RNA polymerase and promotes transcription termination, typically between 100 and 200 nt downstream of the Rut site, leading to 3´ ends that are processed by 3´ to 5´exonucleases (Dar and Sorek, 2018b; Wang et al., 2019).
Several studies have sought to identify sites of transcription termination across the E. coli genome by sequencing RNA 3´ ends or by mapping the distribution of transcribing RNA polymerase (Dar and Sorek, 2018b; Ju et al., 2019; Peters et al., 2012; Peters et al., 2009; Yan et al., 2018). The vast majority of identified termination sites are in 3´ UTRs. In some studies, termination was compared in cells grown with/without the Rho inhibitor, bicyclomycin (BCM), facilitating the identification of Rho termination sites (Dar and Sorek, 2018b; Ju et al., 2019; Peters et al., 2012). These data provided evidence for Rho termination of mRNAs in 3´ UTRs and spurious transcripts initiated within genes. While termination within 5´ UTRs was noted in some of these studies, extensive global characterization of premature termination has not been performed. Many of the uncharacterized 3´ ends in 5´ UTRs or open reading frame (ORF)-internal regions are likely to be the result of regulatory events, given that riboswitches (Bastet et al., 2017; Hollands et al., 2012), attenuators (Ben-Zvi et al., 2019; Gall et al., 2016; Herrero Del Valle et al., 2020; Konan and Yanofsky, 1997; Kriner and Groisman, 2015), RNA-binding proteins (Baniulyte et al., 2017; Figueroa-Bossi et al., 2014), and sRNAs (Bossi et al., 2012; Sedlyarova et al., 2016) have all been implicated in affecting premature Rho termination events.
In addition to being the product of a regulatory event, RNA fragments generated by premature termination or RNase cleavage themselves can have functions as regulatory sRNAs. sRNAs commonly base pair with trans-encoded mRNAs, frequently with the assistance of the RNA chaperone protein Hfq, resulting in changes in the stability or translation of the target mRNA (reviewed in Hör et al., 2020). Most sRNAs characterized to date are transcribed independent of other genes or are processed from mRNA 3´ UTRs, though a few 5´ UTR-derived sRNAs have been reported (reviewed in Adams and Storz, 2020). RNA fragments entirely internal to coding sequences (Dar and Sorek, 2018a) also have been suggested to function as regulators, though this has not been tested. While sRNAs generally base pair with mRNA targets, a few small transcripts have been shown to have roles as competing endogenous RNAs (ceRNAs) also known as ‘sponges’, which base pair primarily with sRNAs, targeting the sRNAs for degradation or blocking their interactions with mRNA targets (reviewed in Denham, 2020; Figueroa-Bossi and Bossi, 2018; Grüll and Massé, 2019).
To systematically identify new regulatory elements in E. coli, we globally mapped RNA 3´ ends, and specifically characterized those ends in 5´ UTRs and ORF-internal regions. We compared this 3´ end dataset with another dataset where BCM treatment was used to identify sites of Rho termination. Using these approaches, we detected hundreds of RNA 3´ ends within 5´ UTRs and internal to ORFs, likely generated by premature transcription termination or RNase processing. We propose the majority of these 3´ ends are the consequence of regulatory events, and we document regulation for multiple examples. For instance, we show 3´ ends are associated with the translation of uORFs, or result from the binding of some sRNAs to mRNA 5´ UTRs. Furthermore, we demonstrate that RNA fragments generated by premature transcription termination and from within coding sequences function as independent sRNA regulators; one as part of an autoregulatory loop and another that connects cell division to the cell envelope stress response. These findings reveal extensive and diverse regulation through premature transcription termination and RNase processing of mRNAs, which can lead to the generation of RNA by-products with independent functions.
Results
Global mapping of 3´ ends in E. coli
Two independent cultures of wild-type E. coli MG1655 (WT) were grown to OD600 ~0.4 in rich (LB) medium, OD600 ~2.0 in LB, and OD600 ~0.4 in minimal (M63) glucose medium. Total RNA was isolated and analyzed using modified RNAtag-seq (Shishkin et al., 2015) (total RNA-seq) and 3´ end (Term-seq) protocols (Dar et al., 2016; Figure 1—figure supplement 1). The replicate total RNA-seq and Term-seq datasets were highly correlated (Figure 1—figure supplement 2A). Using the Term-seq data, we curated a list of dominant RNA 3´ ends (Supplementary file 1). The total numbers of identified 3´ ends were 1175 and 882 for cells grown in LB to OD600 ~0.4 or 2.0, respectively, and 1053 for cells grown in M63 glucose to OD600 ~0.4 (Figure 1—figure supplement 2B). The detected 3´ ends were further subclassified (see Materials and methods for details) according to their locations relative to annotated genes (Figure 1A). 3´ ends that could not be assigned to one unique category were counted in multiple categories. For cells grown to OD600 ~0.4 in LB, this analysis revealed that, while 23% of 3´ ends mapped <50 bp downstream of an annotated gene (primary 3´ ends), hundreds (58%) were classified as orphan and internal 3´ ends, many mapping upstream of, and within ORFs (Figure 1B).
Figure 1 with 2 supplements see all
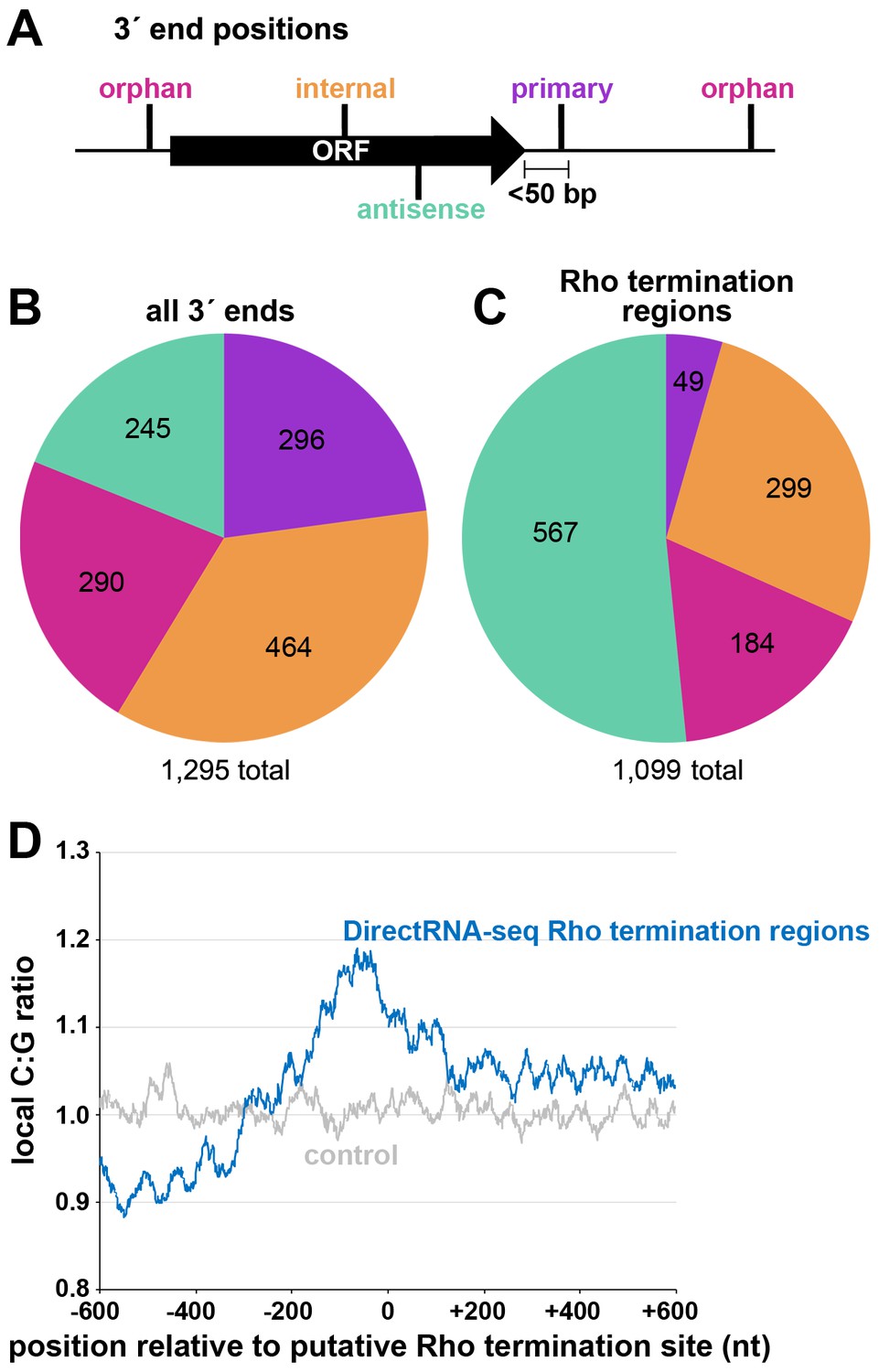
Distribution of 3´ ends and putative sites of Rho termination.
(A) Schematic of classification of Term-seq 3´ ends and Rho termination sites relative to an annotated ORF. 3´ ends and termination sites were defined as: primary (purple colored, located on the same strand within 50 bp downstream of the 3´ end of an annotated gene (mRNA ORF, tRNA, rRNA, or sRNA)), antisense (aquamarine colored, located on the opposite strand within 50 bp of a gene start and end coordinates), internal (orange colored, located on the same strand within a gene) and orphan (fuchsia colored, located in a 5´ UTR, long 3´ UTR or not falling in any of the previous classes). The black arrow represents an ORF. (B) Distribution of Term-seq 3´ ends relative to annotated genes. Some 3´ ends fit the criteria for two different categories; 99 are primary and antisense, eight are primary and internal and 13 are internal and antisense. (C) Distribution of Rho termination sites relative to annotated genes based on DirectRNA-seq after BCM treatment. Some fit the criteria for two different categories; 12 are primary and antisense and six are primary and internal, three are antisense and internal. (D) C:G ratio of sequences surrounding predicted Rho termination sites. Nucleotide proportions were calculated by scanning 600 nt upstream and downstream of Rho 3´ ends (Supplementary file 2) using 25 nt windows. Plotted values represent the average ratios for all 1078 regions (blue). Control plot (gray) represents average C:G ratios calculated in the same manner for 1078 random E. coli MG1655 genomic positions.
We compared the detected 3´ ends to those identified by three other RNA-seq based studies (Dar and Sorek, 2018b; Ju et al., 2019; Yan et al., 2018). Note that the number of overlapping 3´ ends differs depending on the direction of the comparison (is non-commutative) because multiple 3´ ends from one dataset can be close to a single 3´ end from the other dataset. While there was significant overlap between the 3´ ends identified in our work and those identified in each of the other studies (Figure 1—figure supplement 2C,E,F; hypergeometric test p<2.2e−16 in all three cases), the majority were unique to our study. Given that a previous E. coli Term-seq study only reported 3´ ends downstream of annotated genes (Dar and Sorek, 2018b), we used our 3´ end calling algorithm to re-analyze the sequencing data from this study (Figure 1—figure supplement 2D). Again, there was significant overlap (hypergeometric test p<2.2e−16), but 51% were unique to our study. Here, we focused on, and characterized 3´ ends mapping upstream of, and internal to, coding sequences, most of which were not detected in the other RNA-seq studies.
Global mapping of Rho termination regions in E. coli
Concurrently, we mapped instances of Rho-dependent transcription termination. One E. coli MG1655 culture grown to OD600 ~0.5 in LB was split with half left untreated and the other half treated with 100 µg/ml BCM for 15 min. Total RNA was isolated and analyzed using a DirectRNA-seq protocol (Ozsolak and Milos, 2011; Figure 1—figure supplement 1), a method optimized for sequencing very short reads (20–30 nt) directly from RNA, an advantage for examining the effect of Rho on the generation of small RNA fragments. Using these data, we calculated a ‘Rho score’ for each genomic position by comparing DirectRNA-seq coverage in 800 nt windows upstream and downstream in the treated (+BCM) and untreated (-BCM) samples (see Materials and methods). This ratio reflects the degree of transcriptional Rho readthrough in the +BCM cells, where a score of >2.0 is indicative of at least a two-fold increase in readthrough in the +BCM compared to the -BCM sample. 1078 genomic regions were putatively associated with a Rho termination event (Supplementary file 2). We note that these genomic positions are likely closer to processed RNA 3´ ends than the termination sites, since Rho-terminated transcripts typically are processed by 3´ to 5´ exonucleases (Dar and Sorek, 2018b; Wang et al., 2019). Hence, we refer to the identified genomic locations as ‘Rho termination regions’. As for 3´ ends mapped by Term-seq, we classified Rho termination regions by their position relative to annotated genes (Figure 1C). 3´ ends antisense to annotated genes represented the largest category (51.5%), consistent with significant Rho termination of antisense RNAs (Peters et al., 2012). Only a small percentage (4.5%) of the Rho termination regions are <50 nt downstream of an annotated gene (primary), likely because Rho loading and termination typically requires >50 nt untranslated RNA (reviewed in Mitra et al., 2017). As for the 3´end-mapping by Term-seq, many Rho termination regions (44%) were classified as orphan and internal, frequently mapping upstream of, or within ORF sequences.
The C:G nucleotide usage was calculated for the putative Rho termination regions (Supplementary file 2), as well as for a control group of randomly selected genomic coordinates. Relative to the control group, there was a higher local C:G ratio within 200 nt of the 3´ ends associated with Rho termination regions (Figure 1D), consistent with enrichment for C-rich, G-poor sequences attributed to Rut sites (reviewed in Mitra et al., 2017).
We also compared the Rho termination regions (Supplementary file 2) to the sites of Rho termination reported in three previous genome-wide studies (Dar and Sorek, 2018b; Ju et al., 2019; Peters et al., 2012; Figure 1—figure supplement 2G). Again, there was significant overlap with each of the three previous studies (hypergeometric test p<2.2e−16 in all cases), though many (43%) of the putative Rho termination regions we identified are >500 bp away from any of the previously identified Rho termination regions (Dar and Sorek, 2018b; Ju et al., 2019; Peters et al., 2012). The large sets of Rho termination regions that differ between the studies suggest that much remains to be learned about Rho-dependent termination. The comparison of our Term-seq LB 0.4 dataset (Supplementary file 1) with our DirectRNA-seq LB 0.5 dataset (Supplementary file 2) revealed that there was significant overlap (hypergeometric test p<2.2e−16), with 34.6% of the Term-seq 3´ ends within 500 nt of a Rho termination region (Figure 1—figure supplement 2H). This suggests that these 3´ ends are associated with Rho-terminated transcripts.
Known regulatory events are associated with 3´ ends and Rho termination regions in 5´ UTRs
Many 3´ ends identified by Term-seq mapped upstream or internal to annotated mRNAs (Supplementary file 1). We specifically focused on these 3´ ends (see Materials and methods), since we hypothesized they could be reflective of regulatory events. Our analysis identified 665 and 507 3´ ends for the LB 0.4 and LB 2.0 samples, respectively, and 580 3´ ends for the M63 0.4 sample (Supplementary file 3). Among the 3´ ends in 5´ UTRs, several correspond to sites of known cis-acting RNA regulation (annotated in Supplementary file 3). These include mRNAs that previously have been shown to be regulated by premature Rho-dependent termination, such as the riboswitch-regulated genes thiM (Bastet et al., 2017; Figure 2—figure supplement 1A), mgtA (Hollands et al., 2012), ribB (Hollands et al., 2012) and lysC (Bastet et al., 2017), and the translationally-repressed genes ilvX (Lawther and Hatfield, 1980) and topAI (Baniulyte and Wade, 2019). We also noticed that even some 5´ UTRs where regulation has only been reported to be at the level of translation, such as the RNA thermometer upstream of rpoH (Morita et al., 1999a; Morita et al., 1999b) and at ribosomal protein operons (reviewed in Zengel and Lindahl, 1994), harbored defined 3´ ends in 5´ UTRs.
For the LB 0.4 Term-seq dataset, for which the growth conditions were most similar to those of the DirectRNA-seq dataset, we determined whether each 3´ end was associated with a detected Rho termination event. Significant Rho scores are listed in Supplementary file 3 (see Materials and methods for details). It should be noted that some genes containing 3´ ends with significant Rho scores in Supplementary file 3 are absent from Supplementary file 2, because the genomic position with the highest Rho score in that region is located in a neighboring gene/region, and thus given a different gene designation.
We also used a previously described quantitative model to score putative intrinsic terminators, where a score >3.0 is predictive of intrinsic termination (Chen et al., 2013). Based on these analyses for the 3´ ends for the LB 0.4 dataset in Supplementary file 3, we predict that 20% have secondary structures and sequences consistent with intrinsic termination (intrinsic terminator score ≥3.0), 11% have detectable Rho termination, and the remaining 3´ ends are likely the result of RNA processing. However, the number of Rho termination sites could be an underestimate, because inhibition of Rho leads to extensive readthrough transcription from very strongly transcribed genes that can mask overlapping transcripts, and 3´ ends generated by Rho termination are typically unstable (Dar and Sorek, 2018b; Wang et al., 2019). Some 3´ends also may be generated by a combination of mechanisms. Nonetheless, overall, our data suggest that modulation of premature transcription termination in 5´ UTRs or ORFs is a widespread regulatory mechanism.
Novel sites of regulation are predicted by 3´ ends and Rho termination regions in 5´ UTRs
Several genes harboring Rho-dependent 3´ ends within the 5´ UTR or ORF have not been previously described as being regulated by Rho but are associated with characterized cis-acting RNA regulators. Examples are the sugE (gdx) and moaA genes, which are preceded by the guanidine II riboswitch (Sherlock et al., 2017) and molybdenum cofactor riboswitch (Regulski et al., 2008), respectively. A browser image of the RNA-seq data for the sugE locus in the LB 0.4 condition documents a predominant 3´ end 76 nt downstream the sugE transcription start site (TSS) (Figure 2A). This region was associated with significant readthrough in the +BCM DirectRNA-seq sample and a 3´ end Rho score of 3.7 (Supplementary file 3), strongly suggesting that the riboswitch impacts Rho-dependent premature termination, as is the case for other riboswitches.
Figure 2 with 1 supplement see all

Experimental validation of premature Rho termination.
(A) RNA-seq screenshot of the sugE (gdx) locus displaying sequencing reads from LB 0.4 total RNA-seq, LB 0.4 Term-seq and DirectRNA-seq ±BCM treatment. Total and Term-seq tracks represent an overlay of two biological replicates. Read count ranges are shown in the top of each frame. The chromosome nucleotide coordinates, relative orientation of the sugE and blc ORFs (wide black arrows), dominant 3´ end in the sugE 5´ UTR from Supplementary file 3 (small black arrow labeled 3´ end), and sugE TSS (Thomason et al., 2015) (bent green arrow) are indicated. (B) RNA-seq screenshot of the mdtJI locus, labeled as in (A). (C) RNA-seq screenshot of the ispU (uppS) locus, labeled as in (A). (D) β-galactosidase activity for sugE, cfa, cyaA, mdtJ, eptB and ispU 5´ UTR + ORF transcriptional fusions to lacZ in WT (AMD054) and rhoR66S mutant (GB4). All gene-reporter fusions initiate from the same high expression promoter and were assayed at OD600 ~0.4–0.6 (see Materials and methods for details). Values represent the mean of at least three independent replicates (indicated by black dots). Error bars represent one standard deviation from the mean. The rhoR66S vs WT fold change is reported above the values for each 5´ UTR. A speA 5´ UTR + ORF-lacZ could not be assayed because cells did not grow, likely because of toxicity associated with overexpression of the full-length gene product. (E) β-galactosidase activity for sugE, cfa, cyaA, speA, mdtJ, eptB, and ispU 5´ UTR transcriptional fusions to lacZ in WT (AMD054) and rhoR66S mutant (GB4). Experiments were performed and data analyzed as in (D). DirectRNA-seq Rho scores for the dominant 3´ end in the 5´ UTR (from Supplementary file 3) of these loci are: 3.7 for sugE, 2.6 for cfa, 2.0 and 2.6 for cyaA (there are two 3´ ends), 3.1 for speA, 2.3 for mdtJ, 2.3 for eptB and 0.7 for ispU. Rho termination regions were also identified in these genes, or neighboring genes, for all loci except ispU (Supplementary file 2). See Supplementary file 4 for oligonucleotides used in cloning to delineate regions in each lacZ fusion. (F) Northern analysis for sugE, cfa, cyaA, speA, mdtJ, eptB and ispU 5´ UTRs in WT (GSO989) and rhoR66S mutant (GSO990) cells. Cells were grown to OD600 ~0.4 or 2.0 after a dilution of the overnight culture and lysed. Total RNA was extracted, separated on an acrylamide gel, transferred to a membrane and probed for the indicated RNAs (RNAs were probed sequentially on the same membrane). Blot was also probed for 5S (Figure 2—figure supplement 1C). Size markers are indicated for all RNAs. Asterisks signify the transcript predicted to correlate to the 3´ end in Supplementary file 3. Arrow points to expected full-length sugE transcript.
While the involvement of 5´ UTRs in sugE and moaA regulation was known, most of the genes for which we found 3´ ends in 5´ UTRs or ORFs have not been reported to have RNA-mediated regulation (Supplementary file 3). In some cases, such as the mdtJI mRNA, encoding a spermidine efflux pump, we observed a novel 3´ end that is clearly Rho-dependent (Figure 2B) with a Rho score of 2.3. In other cases, such as ispU (uppS), encoding the undecaprenyl pyrophosphate synthase, a novel 3´ end was observed with no readthrough upon Rho inhibition (Figure 2C; Rho score of 0.7). Rather, this 3´ end had an intrinsic terminator score (Chen et al., 2013) of 14.1 (Supplementary file 3), and we predicted a 6 bp stem-loop followed by eight U residues, consistent with intrinsic termination.
To further test whether genes associated with 3´ ends in 5´ UTRs or ORFs are indeed regulated by premature Rho-dependent transcription termination, we generated lacZ transcriptional reporter fusions using the entire 5´ UTR and ORF for 27 genes (Supplementary files 2 and 3) arbitrarily chosen for a range of calculated Rho scores and possible regulation. All constructs had the same constitutive promoter. β-galactosidase activity was assayed in the context of a WT E. coli background or a mutant strain with an R66S substitution in Rho (rhoR66S), which disrupts the primary RNA-binding site (Baniulyte et al., 2017; Bastet et al., 2017; Martinez et al., 1996). A fusion to thiM, which harbors a 5´ UTR Rho-dependent terminator (Bastet et al., 2017), exhibited significantly higher levels of β-galactosidase activity in the Rho mutant strain, indicative of a disruption in Rho-dependent termination (Figure 2—figure supplement 1B). Northern analysis identified an RNA consistent with the 3´ end identified by Term-seq (Figure 2—figure supplement 1C), though we noted the abundance of this 5´ RNA fragment and extent of readthrough in the Rho R66S mutant varied with growth phase.
Among the other constructs assayed, the expression of 14 fusions was >2-fold higher in rhoR66S compared to WT cells (Figure 2D and Figure 2—figure supplement 1D), consistent with Rho termination in the 5´ UTR or ORF for sugE, cfa, cyaA, mdtJ, add, cspB, cspG, moaA, pyrG, yhaM, ydjL, yhiI, ytfL, and yajO. The effect of the Rho mutation on the eptB, chiP, and crp-yhfK fusions was intermediate (1.6- to 1.8-fold) (Figure 2D), while the fusions to ispU (Figure 2D), as well as mnmG, rpsJ, argT, srkA, and trmL (Figure 2—figure supplement 1D), displayed similar levels of β-galactosidase activity (1.0- to 1.4-fold) for rhoR66S compared to WT cells. These assays support the notion that the 3´ end observed in the ispU 5´ UTR (Figure 2C) is generated by intrinsic termination. It is unclear why we did not detect evidence for Rho termination of mnmG, rpsJ, argT, srkA or trmL, despite these genes having significant Rho scores. Interestingly, fusions to the ompA, yebO, glpF, and rimP 5´ UTR and ORF had decreased expression in the Rho mutant background (Figure 2—figure supplement 1D). This may be a consequence of additional levels of regulation or indirect effects of Rho inhibition. The ompA and glpF genes were not associated with Rho termination by DirectRNA-seq.
Transcriptional lacZ reporter gene fusions to only the 5´ UTR (i.e. without the ORF) were also generated for seven genes, to distinguish between Rho termination in the ORF and in the 5´ UTR. The effect of rhoR66S was eliminated for the shorter cyaA and eptB fusions (Figure 2E), suggesting that Rho-dependent termination occurs within the coding sequence of these genes. Regulation of Rho termination in 5´ UTRs is probably associated with the accessibility of Rut sequences, whereas Rho termination within coding sequences is probably associated with regulated translation initiation (reviewed in Kriner et al., 2016), with translational repression indirectly leading to Rho termination.
Finally, using RNA extracted from WT and rhoR66S strains grown to OD600 ~0.4 and 2.0 in LB, northern analysis was performed with probes for the 5´ UTRs of sugE, cfa, cyaA, speA, mdtJ, eptB and ispU (Figure 2F) as well as with probes for the coding sequences of cfa, cyaA, speA and mdtJ (Figure 2—figure supplement 1E). For all of the mRNAs, we detected 5´ fragments that likely correspond to the 3´ ends detected by Term-seq (indicated with an asterisk) (Supplementary file 3). For sugE and cfa, however, the dominant band on the northern blot was not necessarily the most dominant 3´ end sequenced using Term-seq. The enrichment for the 5´ fragments as well as longer transcripts likely corresponding to the full-length sugE, cfa, cyaA, speA and mdtJ mRNAs (indicated with an arrow) in the rhoR66S samples reflect transcriptional readthrough in the mutant background. This is consistent with Rho-dependent termination as seen for the lacZ fusions. The effect of rhoR66S on eptB was intermediate, while no effect was observed for ispU. For all of the Rho-terminated genes, we were surprised to observe the significant increase in the levels of short transcripts, as seen most strikingly for speA. This suggests that the detected 3´ ends can be generated by RNA processing from both Rho-terminated and full-length transcripts, with the increased abundance of longer transcripts in the rhoR66S samples leading to higher levels of the processed product. We also noted that the effects of the rho mutation varied under the different growth conditions tested, as is most apparent for cfa and cyaA. Collectively, these data validate premature termination in 5´ UTRs and, in several cases, suggest complex regulation.
Premature Rho termination of mdtJI is dependent on spermidine and translation of a uORF mdtU
The mdtJI mRNA, encoding a spermidine exporter, has a long 5´ UTR (TSS located 278 nt from the start codon) (Figure 3A) and a 3´ end that mapped six nt into the ORF (Figure 2B). Additionally, the transcript is subject to premature Rho termination (Figure 2D–F, Figure 2—figure supplement 1E). The levels of the mdtJI transcript were previously reported to increase in response to high levels of spermidine, a polyamine (Higashi et al., 2008a), though a mechanism for this regulation was not described. Polyamines play important roles in RNA-mediated regulation, reported to cause structural changes to the 5´ UTR of the oppA mRNA (Higashi et al., 2008b; Yoshida et al., 1999) and induce ribosome stalling at a uORF in the 5´ UTR of the speFL mRNA (Ben-Zvi et al., 2019; Herrero Del Valle et al., 2020). In light of these studies, we examined the effect of spermidine on mdtJI mRNA levels. Total RNA was extracted from cells grown in LB medium with or without 10 mM spermidine at either neutral or high pH (spermidine and other polyamines have a stronger negative effect on cell growth at high pH Yohannes et al., 2005). Northern analysis of these samples probed for the mdtJI mRNA revealed a ~280 nt transcript, consistent with the 3´ end detected by Term-seq (Supplementary file 3), that was susceptible to readthrough upon the addition of spermidine for cells grown at high pH (Figure 3B). We therefore hypothesized that spermidine inhibits premature Rho termination of the mdtJI mRNA.
Figure 3 with 1 supplement see all
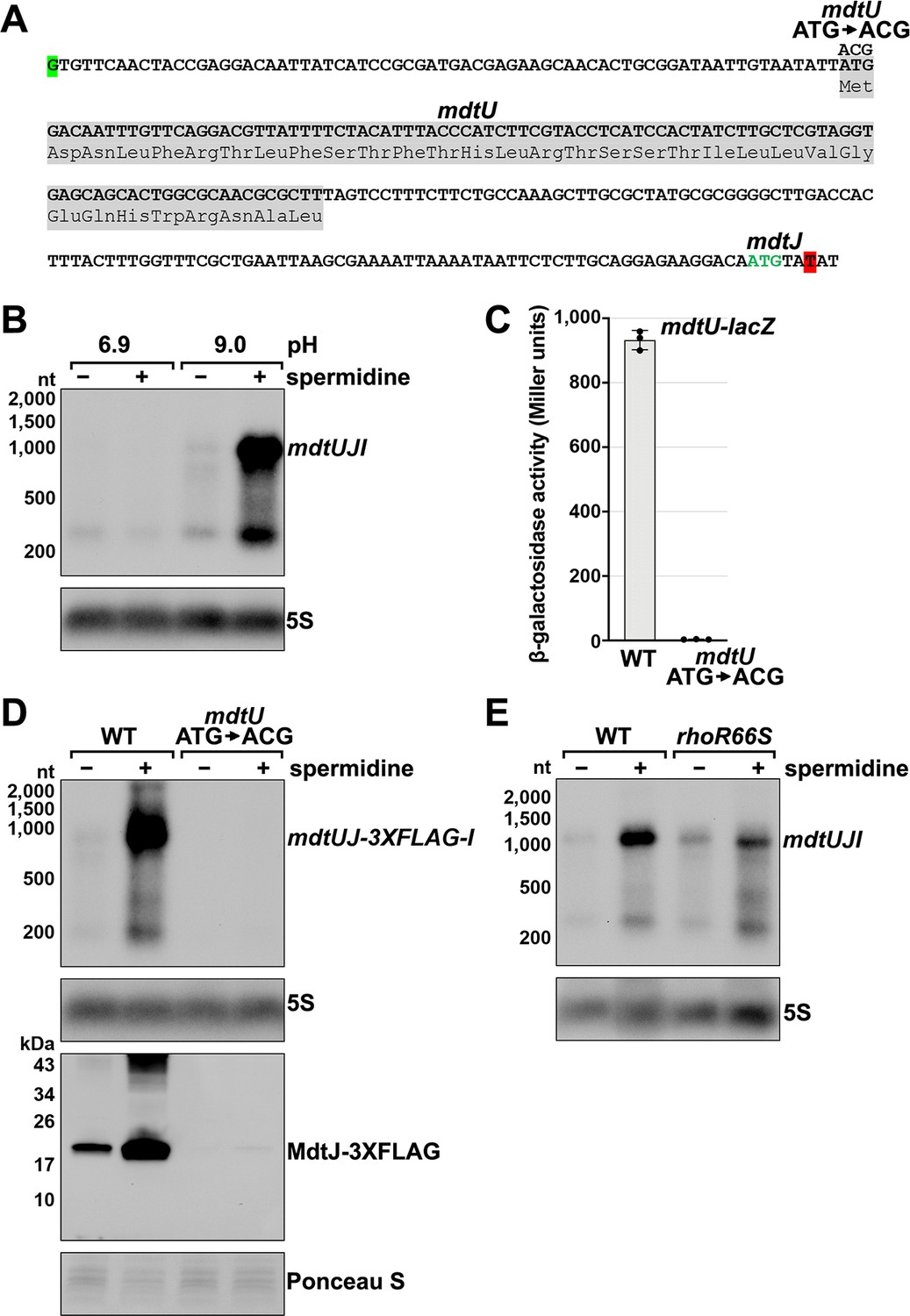
Effect of spermidine on mdtUJI expression.
(A) Sequence of the mdtJI 5´ UTR. The transcription start site (green shaded nucleotide) determined by dRNA-seq (Thomason et al., 2015) and 3´ end (red shaded nucleotide) determined by Term-seq (current study) are indicated. Sequence encoding the mdtU uORF is highlighted in gray. Start codon of the mdtJ ORF is indicated with green text. (B) Northern analysis of effects of spermidine on mdtUJI mRNA levels. WT (GSO989) cells were grown for 150 min after a dilution of the overnight culture, ±10 mM spermidine in either LB pH 6.9 or LB pH 9.0. Total RNA was extracted, separated on an agarose gel, transferred to a membrane and sequentially probed for the mdtJI 5´ UTR and 5S. (C) β-galactosidase activity of a mdtU translational lacZ fusion. WT mdtU (pASW1) and start codon (ATG→ACG) mutant (pGB337) were assayed and analyzed as in Figure 2D. Constructs included the native mdtUJI TSS and full-length mdtU ORF. (D) Northern and western analyses of the effect of the mdtU uORF mutant on mdtJ-3XFLAG-mdtI mRNA and MdtJ-3XFLAG levels. WT mdtU (GSO991) and start codon (ATG→ACG) mutant (GSO992) cells harboring an mdtUJ-3XFLAG-I were grown for 150 min after a dilution of the overnight culture, ±10 mM spermidine in LB pH 9.0. Total RNA was analyzed as in (B). Protein extracts were separated on a Tris-Glycine gel, transferred to a membrane, stained using Ponceau S stain, and probed using ɑ-FLAG antibodies. We do not know the identity of the higher molecular weight bands observed for the WT sample in the western analysis. They could be due to multimeric MdtJ or MdtJ association with the membrane. (E) Northern analysis of Rho effect on mdtUJI mRNA levels in the presence of spermidine. WT (GSO989) and rhoR66S mutant (GSO990) cells were grown for 150 min after a dilution of the overnight culture, ±10 mM spermidine in LB pH 9.0. Total RNA was analyzed as in (B).
Closer inspection of the mdtJI 5´ UTR revealed a putative upstream small ORF (uORF) of 34 codons with the stop codon 106 nt upstream of the mdtJ AUG (Figure 3A). Translation of this uORF was previously detected by ribosome profiling, with expression of the corresponding small protein (YdgV) documented by western blot analysis (Weaver et al., 2019). To independently verify translation of the uORF, the coding sequence was translationally fused to lacZ together with the upstream sequence and native mdtUJI promoter. Robust β-galactosidase activity was detected for cells carrying the fusion (Figure 3C). By contrast, no β-galactosidase activity was observed for cells carrying an equivalent fusion with the uORF start codon mutated (ATG→ACG), supporting the conclusion that the uORF (ydgV), here renamed mdtU, is indeed translated.
To investigate the role of mdtU in spermidine-mediated regulation of mdtUJI, the mdtU start codon mutation (ATG→ACG) was introduced on the chromosome of a strain where a 3XFLAG tag was translationally fused to C-terminus of MdtJ. Northern and western blot analysis of strains encoding mdtUJ-3XFLAG-I showed mRNA and protein levels were strongly induced by spermidine, which was abolished in the strain with the mdtU start codon mutation (Figure 3D). We suggest that translation of MdtU is critical for spermidine-mediated expression of MdtJ. Northern analysis was also carried out to determine if Rho termination in the mdtJI 5´ UTR impacts the induction by spermidine. In the rhoR66S strain, the spermidine-dependent increase in full-length mdtUJI mRNA levels was substantially reduced relative to WT cells (Figure 3E). However, inhibition of Rho did not completely abolish the stimulatory effect of spermidine on mdtUJI mRNA levels, perhaps because growth in spermidine and high pH may increase transcription initiation of mdtUJI. Together, these data support the model that spermidine, Rho, and translation of the mdtU uORF affect the levels of MdtJI and hence spermidine transport, though the mechanisms deserve further study. A screen for uORFs upstream of mdtJ orthologs in other gammaproteobacterial species showed that MdtU, particularly the C-terminal region, is conserved in at least 17 genera (Figure 3—figure supplement 1). This suggests that mdtU-dependent regulation of mdtUJI expression is a conserved process that may depend on the sequence of the MdtU C-terminus.
The presence of 3´ ends downstream of putative uORFs could be a way to identify new uORF-dependent regulatory sequences. A search for 3´ ends located <200 nt downstream of experimentally validated, but uncharacterized potential uORFs (Hemm et al., 2008; VanOrsdel et al., 2018; Weaver et al., 2019) revealed nine examples: ybgV-gltA, yhiY-yhiI, ykiE-insA-7, yliM-ompX, ymdG-putP, ymiC-acnA, yqgB-speA, and ytgA-iptF (Supplementary file 3), consistent with this suggestion.
5´ mRNA fragments can be generated by sRNA-mediated regulation
The vast majority of characterized regulatory binding sites for sRNAs are in 5´ UTRs, and we observed several RNA 3´ ends in 5´ UTRs near positions of documented sRNA base pairing (Supplementary file 3). For instance, the eptB, ompA and chiP mRNAs, which are targets of MgrR (Moon and Gottesman, 2009), MicA (Udekwu et al., 2005) and ChiX (Figueroa-Bossi et al., 2009), respectively, all had 3´ ends directly downstream of the sequences involved in sRNA base pairing (Figure 4A and Figure 4—figure supplement 1). The presence of RNA 3´ ends at these positions suggested that stable 5´ mRNA fragments could be generated or perhaps protected by sRNA-mediated regulation. Indeed, 5´ transcripts for eptB and chiP were previously detected by total RNA-seq (Dar and Sorek, 2018a), and the chiP 5´ transcript was reported to accumulate in the absence of the 3´-to-5´ phosphorolytic exoribonuclease PNPase (Cameron et al., 2019).
Figure 4 with 1 supplement see all
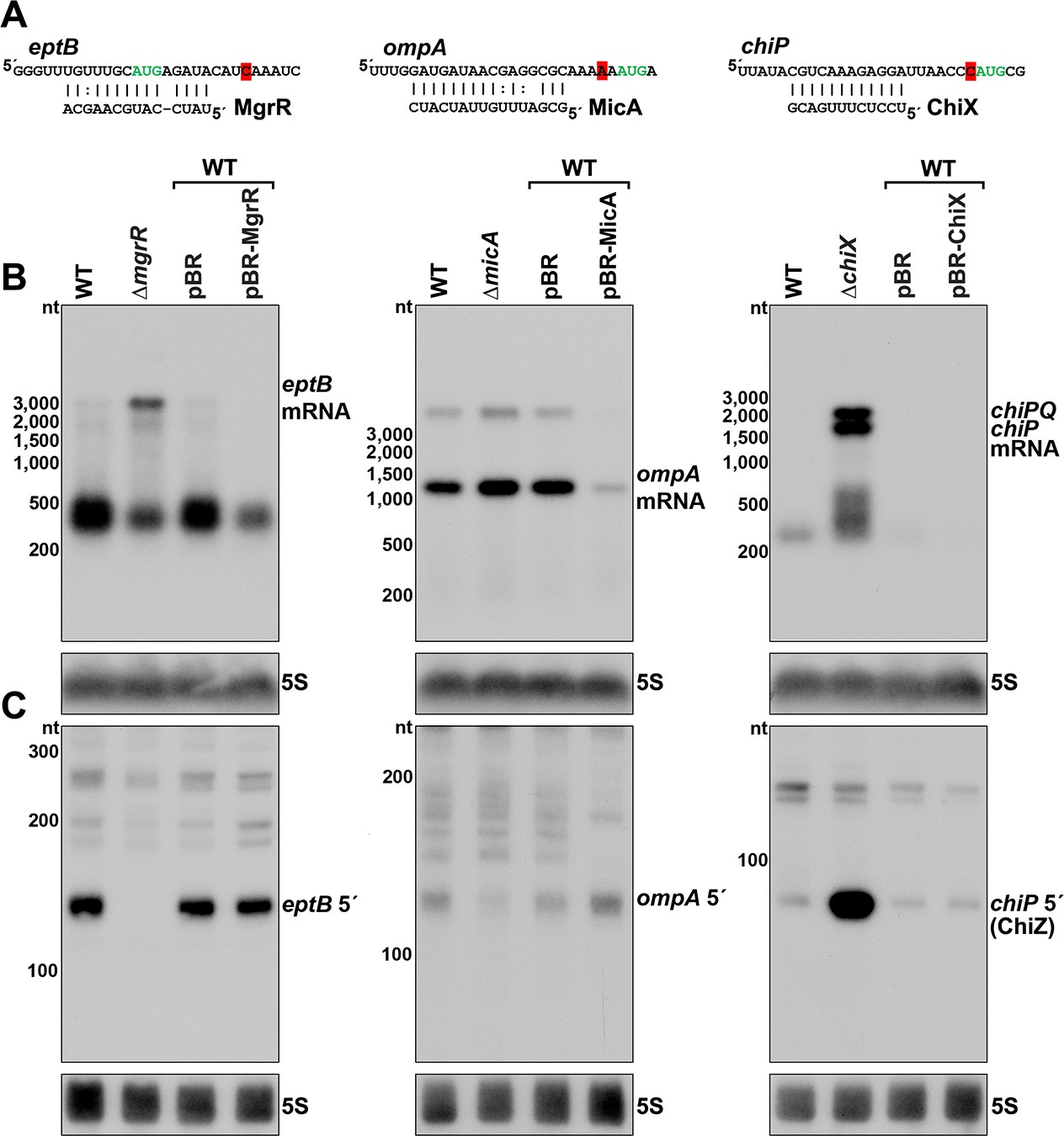
Effect of sRNA deletions on eptB, ompA, and chiP fragments.
(A) Sequence of documented region of sRNA-mRNA pairing. 3´ end determined by Term-seq is highlighted in red. Start codon of the corresponding ORF is indicated with green text. (B) Northern analysis of eptB, ompA, and chiPQ mRNAs. WT (GSO982) without and with indicated plasmids and ΔmgrR (GSO993), ΔmicA (GSO994), and ΔchiX (GSO995) cells were grown for 150 min after a dilution of the overnight culture. Total RNA was extracted, separated on an agarose gel, transferred to a membrane and sequentially probed for specific mRNAs and 5S. Size markers are indicated for all RNAs. (C) Northern analysis of eptB, ompA, and chiP 5´ UTR fragments. The same RNA as in (B) was separated on an acrylamide gel, transferred to a membrane and probed for specific 5´ UTR fragments and 5S. Size markers are indicated for all RNAs.
To examine how sRNAs impacted the 5´ derived mRNA fragments of eptB, ompA, and chiP, we used northern analysis to examine the consequences of deleting or overexpressing the cognate sRNA gene. Two oligonucleotide probes, one within the coding sequence (downstream of the 3´ end identified using Term-seq) and one within the 5´ fragment, were used to determine the relative levels of the full-length mRNAs (Figure 4B) and 5´ fragments (Figure 4C). As expected, given the known sRNA-mediated downregulation of eptB, ompA and chiP, the levels of the target mRNAs were elevated in the sRNA deletion background compared to the WT strain and decreased with sRNA overexpression (Figure 4B). Some other RNA species were detected for eptB (~400 nt), ompA (<3000 nt), and chiP (~200–500 nt), but these did not match the expected sizes for the mRNAs, and may be degradation and/or readthrough products. For the 5´ UTR region of eptB, there was a reciprocal effect of the ∆mgrR background, with a strong decrease in the abundance of a ~140 nt band (Figure 4C). Given that we observed a moderate effect of Rho mutation on eptB expression (Figure 2D,F), we speculate that MgrR base pairing both promotes Rho termination and protects the resultant RNA from exonucleases. For the 5´ UTR region of ompA, which showed only modest de-repression in the ∆micA strain, there was a decrease in the abundance of an ~120 nt fragment in the deletion strain (Figure 4C), and the levels of this fragment increased upon MicA overexpression. These effects are consistent with MicA sRNA-directed cleavage of the ompA mRNA generating the fragment, or the sRNA base pairing protecting the 3´ end from exonucleolytic processing. The effect of the ∆chiX background on the chiP 5´ fragment was strikingly different. Instead of decreasing, there was a large increase in the levels of an ~90 nt RNA (Figure 4C). Given the strong signal detected for this transcript, we hypothesized that this RNA might have an independent role as a regulatory RNA.
ChiZ and IspZ sRNA sponges derive from 5´ UTRs
To test the hypothesis that 5´ UTR transcripts with defined bands have independent functions as sRNAs, we carried out further studies on the ~90 nt chiP 5´ UTR transcript (Figure 4C), which we renamed ChiZ, and the 81 and 60 nt ispU 5´ UTR transcripts (likely expressed from two TSSs with a shared 3´ end, Figure 2C and F), denoted IspZ (Figure 5A).
Figure 5 with 1 supplement see all
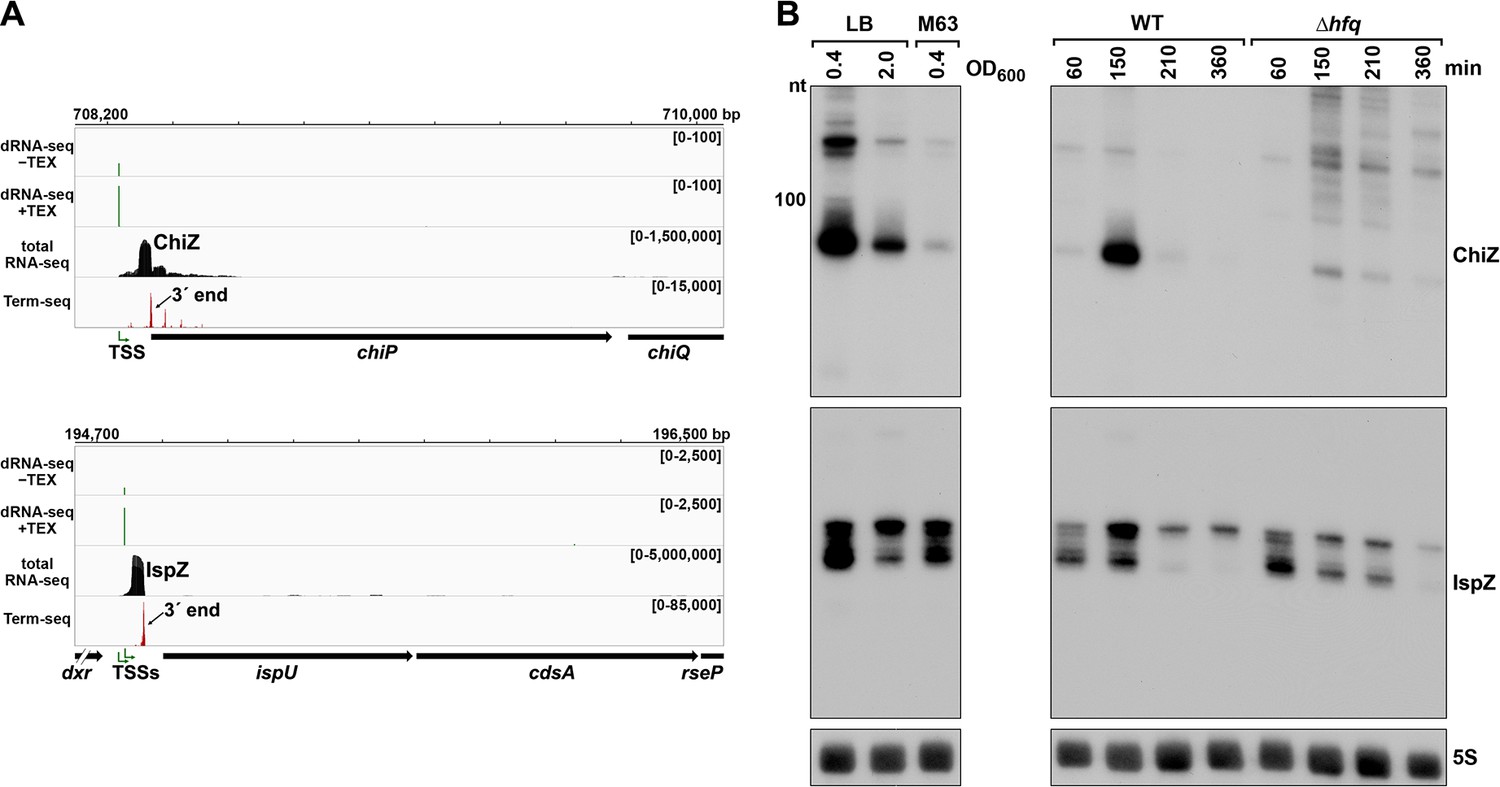
5´ UTR-derived sRNAs ChiZ and IspZ.
(A) RNA-seq screenshot of the ChiZ and IspZ loci displaying sequencing reads from the LB 0.4 growth condition from dRNA-seq (Thomason et al., 2015, HS2 samples), total RNA-seq and Term-seq. Total and Term-seq tracks represent an overlay of two biological replicates. Read count ranges are shown in the top of each frame. The chromosome nucleotide coordinates, relative orientation of the ORF (wide black arrow), dominant 3´ end from Supplementary file 3 (small black arrow) and TSS (green bent arrow) as determined by the ratio of reads between ±TEX tracks, are indicated. (B) Northern analysis of ChiZ and IspZ. Left: the same WT (GSO988) RNA samples used for total RNA-seq and Term-seq in (A). Right: RNA was extracted from WT (GSO982) and Δhfq (GSO954) cells at specific times after dilution of the overnight culture, (60, 150, 210, and 360 min) corresponding to early, middle, and late exponential and stationary phase. The Δhfq strain reaches a lower final OD600, yet exhibits a similar pattern of growth (Melamed et al., 2020). Total RNA was separated on an acrylamide gel, transferred to a membrane and probed for the indicated RNAs (RNAs were probed sequentially on the same membrane). The position of the 100 nt size marker is indicated for ChiZ (the region of the northern below 100 nt is shown for IspZ).
To obtain more information about the expression of these putative sRNAs, we performed northern analysis using the same RNA analyzed in the Term-seq experiment (Figure 5B). Distinct bands were detected for the two 5´ UTRs, consistent with the generation of stable RNAs with predicted stems protecting the ends (Figure 5—figure supplement 1A–B). While ChiZ was most abundant in cells grown to exponential phase in LB, IspZ was abundant in LB and M63 glucose medium in both exponential and stationary phase. Since many sRNA levels are negatively affected by the lack of the RNA chaperone Hfq (reviewed in Hör et al., 2020), we also conducted northern analysis using RNA extracted from WT or Δhfq cells across growth in LB (Figure 5B). Similar to other base-pairing sRNAs, and consistent with Hfq binding, ChiZ abundance was low in the ∆hfq background. IspZ levels were only slightly affected by the absence of Hfq, even though this RNA is bound by Hfq (Melamed et al., 2020). Given that the binding of the sRNA ChiX to the mRNA chiP (containing ChiZ) increases Rho-mediated regulation of chiP (Bossi et al., 2012), we tested the role of Rho on ChiZ levels (Figure 6A). The effects of the rhoR66S mutant were dependent on growth phase, with a decrease in ChiZ for cells grown in LB to OD600 ~0.4, when ChiZ levels are highest. In contrast, IspZ levels were not affected by the rhoR66S mutant (Figure 2F), and IspZ is likely subject to intrinsic termination as stated previously.
Figure 6

5´ UTR-derived sRNAs ChiZ and IspZ act as sRNA sponges.
(A) Northern analysis for ChiZ in WT (GSO989) and rhoR66S mutant (GSO990) cells. Cells were grown to OD600 ~0.4 or 2.0 after a dilution of the overnight culture. Total RNA was extracted, separated on an acrylamide gel, transferred to a membrane and probed for ChiZ and 5S. This is the same blot depicted in Figure 2F and Figure 2—figure supplement 1C. (B) Northern analysis of ChiZ effect on chiP mRNA. RNA was extracted from WT (GSO982) and Δhfq (GSO955) cells at 150 min after dilution of the overnight culture. Total RNA was separated on an acrylamide or agarose gel, transferred to a membrane and probed for the indicated RNAs (RNAs were probed sequentially on the same membrane). (C) β-galactosidase activity for chiP translational fusions to lacZ in WT (DJS2979) and ΔchiX (DJS2991) strains. Cells were grown and assayed 150 min after dilution of the overnight culture (see Materials and methods for details). Values represent the mean of three independent replicates (indicated by black dots). Error bars represent one standard deviation from the mean. (D) Model of ChiZ effects on ChiX, with indirect effects on the chiP mRNA. ChiZ (derived from the 5´ end of chiP) is blue and ChiX is red. (E) Northern analysis of IspZ effect on OxyS upon oxidative stress. WT (GSO982) and Δhfq (GSO955) cells were grown for 150 min after dilution of the overnight culture, and WT (-H2O2) samples were collected. To induce OxyS, 0.2 mM H2O2 was spiked into the cultures for 20 min, and WT and Δhfq samples were collected. Total RNA was extracted and separated on an acrylamide gel, transferred to a membrane and probed for the indicated RNAs (RNAs were probed sequentially on the same membrane). (F) Predicted base pairing between IspZ and OxyS with mutations assayed. (G) Test of direct interaction between IspZ and OxyS. RNA was extracted from WT (GSO982) and oxyS-M1 (GSO996) cells transformed with the pBR plasmids at 150 min after dilution of the overnight culture and 20 min incubation with 0.2 mM H2O2. Northern analysis was performed on total RNA as in (E). A smaller processed product was detected for the chromosomal OxyS-M1mutant, possibly due to secondary structural changes brought about by the M1 mutation.
Given their association with Hfq, we tested the independent functions of ChiZ and IspZ as base-pairing sRNAs. Since the region of chiP encoding ChiZ has been documented to be a target for base pairing with the sRNA ChiX through compensatory mutations (Figueroa-Bossi et al., 2009; Overgaard et al., 2009), we postulated that ChiZ reciprocally regulates ChiX, sponging its base-pairing activity. To test this possibility, we assayed the effects of ChiZ overexpression. As for chromosomally-encoded ChiZ (Figure 5B), longer transcripts were observed for plasmid-encoded ChiZ, likely due to readthrough, but only the levels of the 90 nt ChiZ band were strongly reduced in the ∆hfq mutant (Figure 5—figure supplement 1C). Upon ChiZ overexpression in the WT background, we observed increased levels of the chiP mRNA, without a change in ChiX levels (Figure 6B). The levels of the chiP mRNA overall were higher in the Δhfq mutant background, but we no longer observed an increase upon ChiZ overexpression, likely due to the instabilities of ChiX and ChiZ. We also observed upregulation of a PBAD-chiP-lacZ chromosomal translational fusion (Schu et al., 2015) upon ChiZ overexpression in a WT but not a ∆chiX background (Figure 6C). These observations support a novel sRNA regulatory network in which an mRNA (chiP) that is the target of an sRNA (ChiX) produces an RNA fragment (ChiZ) that reciprocally sponges the sRNA (ChiX) (Figure 6D).
We expected IspZ also might function as a base-pairing sRNA, and thus searched for potential targets identified by RIL-seq (Melamed et al., 2020; Melamed et al., 2016), an approach where RNAs in proximity on an RNA-binding protein are identified by co-immunoprecipitation, ligation, and sequencing of the chimeras. The predominant target for IspZ in these datasets is the oxidative stress-induced sRNA OxyS (Altuvia et al., 1997). This observation led us to test whether IspZ might act as a sponge of OxyS. As for chromosomally-encoded IspZ (Figure 5), and in contrast to ChiZ, little readthrough and no effect of ∆hfq was observed for plasmid-expressed IspZ (Figure 5—figure supplement 1D). We examined the effect of IspZ overexpression on OxyS levels in cells treated with 0.2 mM hydrogen peroxide, a condition known to induce OxyS expression (Altuvia et al., 1997). High levels of IspZ were associated with slightly lower OxyS levels, in line with sponging activity (Figure 6E). To obtain evidence for direct base pairing between IspZ and OxyS, IspZ was mutated on the overexpression plasmid (IspZ-M1), and compensatory mutations were introduced into the chromosomal copy of OxyS (OxyS-M1). Consistent with the predicted pairing (Figure 6F), IspZ-mediated down-regulation was eliminated with IspZ-M1 or OxyS-M1 alone but was restored when both mutations were present (Figure 6G).
Putative ORF-internal sRNAs
We also noted examples of abundant 3´ ends internal to ORFs (Supplementary file 3), downstream of nearby 5´ ends previously identified by dRNA-seq (Thomason et al., 2015), and associated with a strong signal in total RNA-seq (Figure 7). A previous study inferred from total RNA-seq data that some sRNAs might be derived from sequences internal to ORFs (Dar and Sorek, 2018a). To test whether we could detect defined transcripts for these internal (int) signals, we selected candidate RNAs derived from the ftsI (renamed FtsO), aceK, rlmD, mglC, and ampG ORFs for further investigation (Figure 7A). Analysis of dRNA-seq data (Thomason et al., 2015) suggested that the FtsO, aceK int and ampG int 5´ ends likely are generated by RNase processing of the overlapping mRNA, whereas rlmD int and mglC int likely are transcribed from promoters internal to the overlapping ORFs. In nearly all cases, the RNA 3´ ends are not predicted to be due to Rho-dependent transcription termination events (Supplementary file 3), strongly suggesting they are generated by RNase processing or, for aceK int, intrinsic termination (intrinsic terminator score = 5.9, Supplementary file 3).
Figure 7

Detection of ORF-internal sRNAs.
(A) RNA-seq screenshots of the ftsI, aceK, rlmD, mglC, and ampG mRNAs containing putative internal (int) sRNAs. Sequencing reads from the LB 0.4 dRNA-seq (Thomason et al., 2015, HS2 samples), total RNA-seq and Term-seq are displayed. Total RNA-seq and Term-seq tracks represent an overlay of two biological replicates. Read count ranges are shown in the top of each frame. The chromosome nucleotide coordinates, relative orientation of ORFs (wide black arrows), dominant 3´ end from Supplementary file 3 (small black arrows labeled 3´ ends), and TSS (green bent arrows) or 5´ processed end (small black arrow labeled 5´ ends) as determined by the ratio of reads between ±TEX tracks, are indicated. (B) Northern analysis of ORF-internal sRNAs. Left: the same WT (GSO988) RNA samples used for total RNA-seq and Term-seq in (A); this is the same blot as depicted in Figure 5B. Right: the same WT (GSO982) and Δhfq (GSO954) RNA samples collected from cells at specific times after dilution of the overnight culture, (60, 150, 210, and 360 min) corresponding to early, middle, and late exponential and stationary phase as in Figure 5B (same blot with RNAs probed sequentially on the same membrane). Size markers are indicated for all RNAs (the region of the northern below 100 nt is shown for ampG int).
Northern analysis was performed for these RNAs using the same RNA analyzed in the Term-seq experiment (Figure 7B); distinct bands were detected for all the RNAs tested. While FtsO and ampG int were relatively abundant under all growth conditions tested, mglC int and rlmD int were only expressed in cells grown in LB, and aceK int was most abundant in LB at OD600 ~ 2.0. We also conducted northern analysis using RNA extracted from WT or Δhfq cells across growth (Figure 7B). The aceK int transcript was strongly dependent upon hfq, whereas the other RNAs were unaffected by hfq deletion, though all five transcripts have been reported to co-immunoprecipitate with Hfq (Melamed et al., 2020; Melamed et al., 2016). Additionally, the ORF-internal sRNAs are found in chimeras with other putative mRNA and sRNA targets in the Hfq RIL-seq datasets (Melamed et al., 2020; Melamed et al., 2016): FtsO and RybB, aceK int and ompF, rlmD int and MicA, mglC int and ArcZ, ampG int and CyaR. Significant chimeras also were detected between aceK int and gatY in the RIL-seq data set for the ProQ RNA chaperone (Melamed et al., 2020). Collectively, these data suggest these ORF-internal transcripts have independent regulatory functions.
ORF-internal FtsO is an sRNA sponge
To test for the suggested regulatory function, we focused on FtsO, which is encoded internal to the coding sequence of the essential cell division protein FtsI, and exhibited high levels across growth (Figure 7B). The predominant target for FtsO in the RIL-seq datasets was the sRNA RybB (Figure 8A) followed by the sRNA CpxQ (Melamed et al., 2020). The Hfq-mediated FtsO-RybB interaction was also detected in an independent CLASH dataset (Iosub et al., 2020). We hypothesized that FtsO functions as a sponge for RybB and CpxQ, which are induced by misfolded outer membrane proteins and inner membrane proteins, respectively, and down-regulate the corresponding classes of proteins (reviewed in Hör et al., 2020). This model was tested by overexpressing FtsO in WT or Δhfq cells grown to exponential and/or stationary phase (150 and 360 min after subculturing). For the 360 min time point when the levels of both RybB and CpxQ are highest, a reduction was observed for both sRNAs in cells overexpressing FtsO (Figure 8B). As reported previously, the levels of RybB and CpxQ are significantly lower in the ∆hfq strain, though some FtsO-dependent downregulation of RybB is still detected. Interestingly, we also observed that RybB overexpression had the reciprocal effect of decreasing FtsO levels, though only at the 360 min timepoint (Figure 8—figure supplement 1A).
Figure 8 with 2 supplements see all
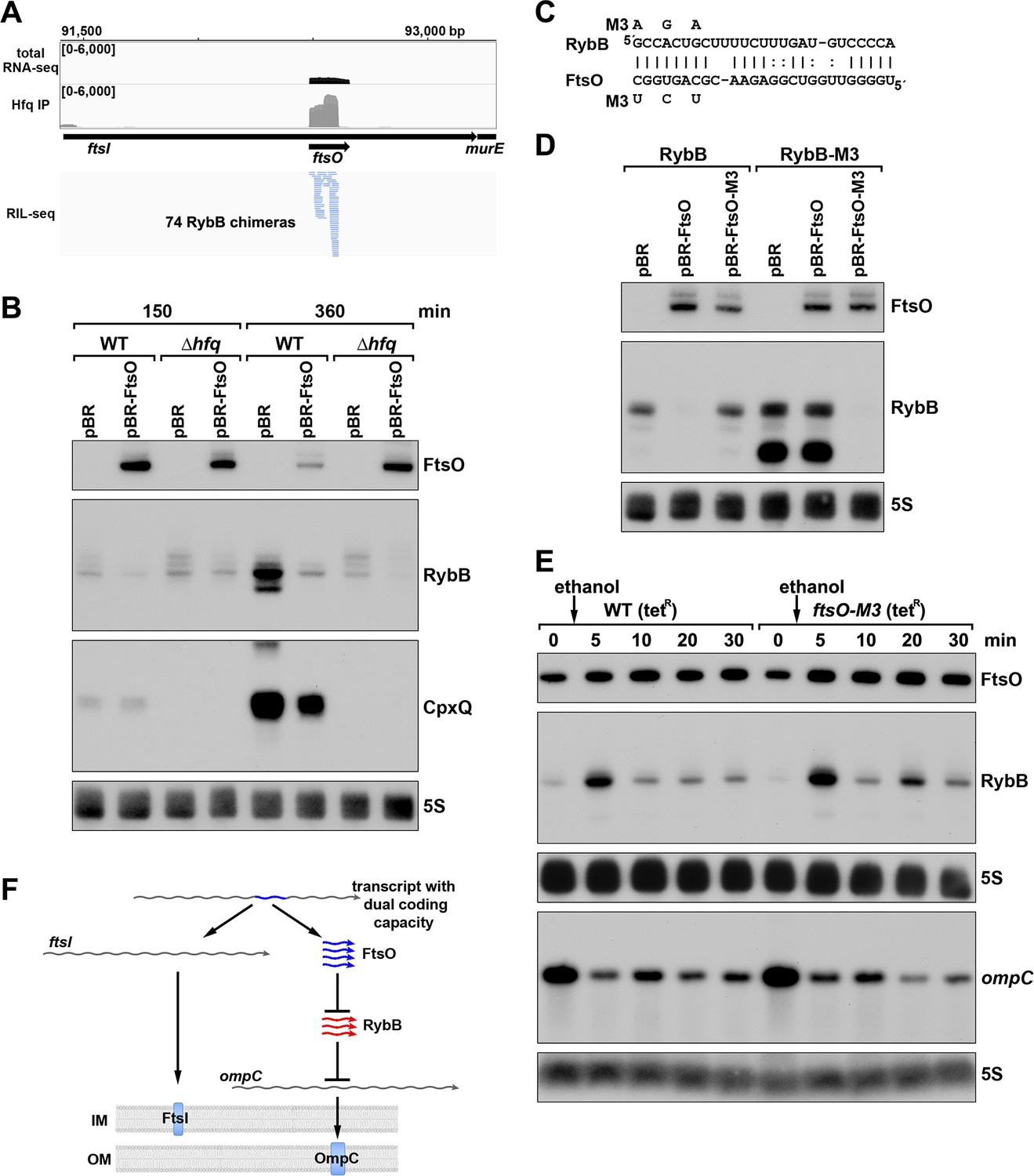
ORF-internal sRNA FtsO acts as a sponge of the RybB sRNA.
(A) RIL-seq screenshot showing RybB chimeras at the ftsO locus. Data are from Hfq-FLAG LB RIL-seq performed 150 min after a dilution of the overnight culture, (Melamed et al., 2020, RIL-seq experiment 2). Top: signals for total RNA (dark gray) and Hfq RIL-seq single fragments with two biological repeats overlaid (light gray). Read count ranges are shown in the upper left of each frame. Bottom: chimeras with FtsO, blue lines indicate FtsO is the second RNA in the chimera. (B) RybB and CpxQ levels decrease in the presence of FtsO. RNA was extracted from WT (GSO982) and Δhfq (GSO954) cells transformed with the indicated pBR plasmids at 150 and 360 min after dilution of the overnight culture. Total RNA was separated on an acrylamide gel, transferred to a membrane and probed for the indicated RNAs (RNAs were probed sequentially on the same membrane). (C) Predicted base pairing between FtsO and RybB with mutations assayed. (D) Test of direct interaction between FtsO and RybB. RNA was extracted from WT (GSO982) and rybB-M3 (GSO997) cells transformed with the indicated pBR plasmids at 360 min after dilution of the overnight culture. Northern analysis was performed on total RNA as in (B). (E) Chromosomally-encoded FtsO mutant dysregulates RybB levels under membrane stress. WT tetR (GSO998) and ftsO-M3 tetR (GSO999) cells were grown for 120 min after dilution of the overnight culture prior to the addition of EtOH to a final concentration of 5%. Cells were collected and extracted for RNA at the indicated time points after addition of EtOH. Northern analysis was performed on total RNA separated on either acrylamide or agarose gels as in Figure 4B and C. (F) Model showing how same DNA sequence can encode two different gene products. The ftsI mRNA encodes the essential FtsI protein, found in the inner membrane (IM). This transcript also encodes the FtsO sRNA (blue), which blocks the activity of the RybB sRNA (red), induced by cell envelope stress, to down-regulate the synthesis of outer membrane (OM) porins such as OmpC.
To confirm the direct interaction between FtsO and RybB, we mutated three nucleotides in the site of predicted base pairing (Figure 8C) on the plasmid copy of ftsO. This mutation (FtsO-M3) abolished the effect of FtsO overexpression on RybB levels (Figure 8D). The repressive effect was similarly abolished by mutating the predicted site of base pairing in the chromosomal copy of rybB but was restored by combining the complementary mutations in ftsO and rybB (Figure 8D).
To test the downstream effect of sponging RybB, we examined the levels of the known RybB mRNA-target ompC (Johansen et al., 2006) compared the effects of RybB to the effects of the previously characterized RybB sRNA sponge, RbsZ (Melamed et al., 2020; Figure 8—figure supplement 1B). Both FtsO and RbsZ overexpression led to a decrease in RybB levels and a concomitant increase in ompC mRNA levels, though the effect of RbsZ was stronger. With both sRNA sponges, the level of OmpC protein was not obviously changed, likely because of the high levels of this protein.
Finally, we mutated the chromosomal copy of ftsO to introduce the same nucleotide substitutions (Figure 8C) that are silent with respect to the FtsI amino acid sequence. WT cells and cells carrying these mutations were treated with ethanol, causing outer membrane stress, which is known to induce RybB (Peschek et al., 2019). In both strains, a transient increase in RybB levels was observed 5 min after ethanol addition (Figure 8E). In WT cells, RybB levels then were decreased for 30 min after ethanol treatment. By contrast, in cells with mutant ftsO, RybB levels again increased at 20 min following ethanol treatment. This effect was also observed in a second experiment (Figure 8—figure supplement 1C) that documented higher RybB levels up to 60 min following ethanol treatment in ftsO-M3 cells. We also assessed the consequences of FtsO sponging RybB on the levels of ompC for the same RNA samples. In both experiments, the ompC mRNA levels decreased after cells were treated with ethanol but for the ftsO-M3 mutant strain there was a further decrease at the 20 min time point (Figure 8E). Along with RbsZ and the 3´ETSleuZ tRNA fragment (Lalaouna et al., 2015), FtsO is the third documented sponge of RybB. It is possible the other sponges mask some of the effects of FtsO.
The conservation of ftsO was examined by aligning ftsI orthologs across 18 species of gammaproteobacteria, revealing a striking degree of ftsO conservation (91% average identity) at ftsI wobble positions compared to the entire ftsI mRNA wobble positions (67% average identity) (Figure 8—figure supplement 2A). The region of base pairing parallels the conservation of the RybB seed sequence and is predicted to be within an unstructured region of FtsO (Figure 8—figure supplement 2B and C). Collectively, our data suggest that FtsO base pairing with RybB is conserved, and lowers RybB levels and activity following outer membrane stress (Figure 8F). The work demonstrates stable regulatory sRNAs can be derived from sequences internal to ORFs such that the same DNA sequence encodes two different functional molecules.
Discussion
Widespread premature transcription termination
Through our transcriptome-wide mapping of 3´ ends and Rho-dependent termination, we uncovered extensive RNA-mediated regulation and sRNA regulators encoded by 5´ UTRs and internal to ORFs. Other studies have previously identified RNA 3´ ends and regions of Rho-dependent termination in E. coli (Dar and Sorek, 2018b; Ju et al., 2019; Peters et al., 2012; Yan et al., 2018). While there was considerable overlap between our work and these prior studies, there were also substantial differences. For example, reporter gene fusion data and northern analysis supported Rho termination of sugE, cfa, cyaA, speA, and mdtJ, of which, only sugE was detected in the previous genome-wide surveys (Dar and Sorek, 2018b; Ju et al., 2019; Yan et al., 2018). Some of the differences between studies can be attributed to differences in the E. coli strains, growth conditions, and methods used. Indeed, we previously found that small methodological differences have a large impact on the identification of mapped TSSs (Thomason et al., 2015). Like any RNA-seq method, a few 3´ ends also could be due to RNA degradation during library preparation. Nevertheless, our follow-up experiments confirmed the biological relevance of several 3´ ends in 5´ UTRs and internal to ORFs. Given that strain and growth conditions used for our Term-seq and total RNA-seq match those of the previous dRNA-seq analysis (Thomason et al., 2015) in which we identified TSSs and 5´ processed ends, the combined sets represent a valuable resource for examining the E. coli transcriptome (see Materials and methods for links to interactive browsers).
The majority of the 3´ ends that we identified were classified as ‘internal’ or ‘orphan’, most of which map within 5´ UTRs or internal to ORFs, and a significant number of which are predicted to be generated by premature transcription termination. This notion of widespread premature transcription termination has been underappreciated in other studies that detected RNA 3´ ends and Rho-mediated termination. It is generally not possible to identify the exact position of Rho termination due to post-transcriptional RNA processing. Nevertheless, our reporter assays showed that in most cases tested, Rho termination could be localized to the 5´ UTR, suggesting that modulation of Rut accessibility in 5´ UTRs could be a common mechanism of regulation.
Multiple levels of regulation at 5´ UTRs
Presuming that many premature termination events are regulatory, we documented and characterized examples of novel, diverse regulatory events for several of the 3´ ends. Undoubtedly, additional unique regulatory mechanisms exist for many of the other 3´ ends. We propose that the identification of 3´ ends in 5´ UTRs and ORFs is an effective approach to discover novel regulatory elements. Classically, these regulators, such as riboswitches and attenuators, have been identified by serendipity, studies of individual genes, or searches for conserved RNA structures (reviewed in Breaker, 2018), but these approaches may miss regulatory RNA elements if the function of the downstream gene is unknown or the region is not broadly conserved. Given that Term-seq is a sensitive, relatively unbiased, and genome-wide approach, it is another means of obtaining evidence for regulation in 5´ UTRs. As an example, the Term-seq data showed that transcripts from the 5´ UTR of the E. coli glycerol facilitator glpF have different 3´ ends under LB and M63 growth conditions, which could be due to uncharacterized regulation. 3´ end-mapping applied to E. coli grown under other conditions or other bacterial species should lead to the characterization of many more regulators, particularly in organisms such as the Lyme disease pathogen Borrelia burgdorferi (Adams et al., 2017) that lack any known cis-acting RNA elements. Consistent with broad applicability, Term-seq previously led to the identification of known riboregulators in Bacillus subtilis and Enterococcus faecalis, and a novel attenuator in Listeria monocytogenes (Dar et al., 2016).
A number of 3´ ends in 5´ UTRs and ORFs were found to be associated with uORFs. Our characterization of the mdtU uORF suggests that regulation of premature mdtJI transcription termination occurs in response to ribosome stalling induced by polyamines. Two recent studies showed that the polyamine ornithine can stall ribosomes immediately upstream of the stop codon of the speFL uORF, affecting Rho binding and the secondary structure of the speFL mRNA (Ben-Zvi et al., 2019; Herrero Del Valle et al., 2020). Strikingly, conservation of MdtU is strongest at the C-terminus, and overlaps a region of the mdtU RNA that is predicted to base pair with the mdtJI ribosome binding site. Thus, a mechanism similar to the one found for speFL may regulate mdtJI induction by spermidine, a hypothesis that deserves further study, together with other examples where 3´ ends are located downstream of uORFs.
We also documented three instances of 3´ ends that localized a short distance downstream of known trans-acting sRNA base-pairing sites. These 3´ ends could be generated by endonuclease processing as a result of sRNA base pairing, or could be due to protection against exonucleases as a result of sRNA pairing. An examination of sRNA base-pairing sites predicted by RIL-seq points toward other instances of this type of regulation. For example, Term-seq identified 3´ ends immediately downstream of the predicted sRNA base-pairing regions for the uncharacterized mRNA-sRNA interactions rbsD-ArcZ, dctA-MgrR, and yebO-CyaR detected by RIL-seq chimeras (Melamed et al., 2020). In all these instances, the 3´ ends could be a result of the sRNA regulatory effect, and in some cases, may result in the formation of a new sRNA, as we observed for ChiZ. In general, our data further illustrate the complex regulation that occurs once transcription has initiated.
Generation of sRNAs from 5´ UTRs and ORF-internal sequences
Previous studies have shown that intergenic regions and mRNA 3´ UTRs are major sources of regulatory sRNAs, with a few characterized examples of sRNAs derived from 5´ UTRs, and no characterized ORF-internal sRNAs (reviewed in Adams and Storz, 2020). Our data document that 5´ UTRs and ORFs can indeed encode functional base-pairing sRNAs. However, our work also raises important questions, including the mechanisms by which 5´ UTR-derived and ORF-internal sRNAs are generated.
Given that sRNAs derived from 5´ UTRs only require the generation of a new 3´ RNA end (likely sharing a TSS with their cognate mRNA), and are not usually constrained by codon sequences, they could evolve rapidly. We documented the formation of several 5´ UTR fragments by cis-regulatory events. These by-products of regulation could obtain independent regulatory functions, as has been reported for a few riboswitches and attenuators (DebRoy et al., 2014; Melior et al., 2019; Mellin et al., 2014). The sRNA 3´ end can be formed by intrinsic termination or Rho-dependent termination and/or processing. Importantly, for the downstream mRNA to be expressed, there needs to be some transcriptional readthrough. RNA structure predictions strongly suggest the IspZ 3´ end is generated by intrinsic termination (Supplementary file 3) for which we observed very little readthrough. In contrast, the ChiZ 3´ end is generated by Rho-dependent termination with significant readthrough that would allow chiP expression. The mechanisms that drive formation of either 5´-derived sRNAs or the corresponding full-length mRNAs could be regulated and are an interesting topic for future work.
Less is known about both the 5´ and 3´ ends of the ORF-internal sRNAs. The ends might be generated by ORF-internal promoters, termination, or RNase processing. In cases where one or both sRNA ends are generated by processing, this is presumably coupled with down-regulation of the overlapping mRNA. Strikingly, the number of sequencing reads for FtsO is orders of magnitude higher than that for the ftsI mRNA. How and when FtsO is produced are interesting questions for future studies. It is possible that the ftsI mRNA is protected from cleavage by ribosomes during cell division such that FtsO is only generated in the absence of mRNA translation. Other coding sequence-derived sRNAs, such as the putative regulator internal to mglC, likely have their own TSS. In some cases, such as rlmD int and ampG int, a transcript originating from a TSS internal to the cognate mRNA could be processed to form the 5´ end of the sRNA. These observations underscore how the interplay of transcription initiation, transcription termination, and RNase processing leads to many short transcripts that have the potential to evolve independent regulatory functions.
Roles of 5´ UTR-derived and ORF-internal sRNAs
We identified and characterized three sRNA sponges that have 3´ ends either in 5´ UTRs or internal to the coding sequence. The first example, ChiZ-ChiX-chiP, represents a novel reciprocal autoregulatory loop. ChiZ is generated from the chiP 5´ UTR encompassing the site of pairing with the ChiX sRNA. We found ChiZ is formed by Rho-dependent termination, and in the absence of ChiX base pairing with chiP. When cells utilize chitobiose as a carbon source and ChiX levels are naturally low, there are higher levels of chiP (Overgaard et al., 2009) and likely also higher levels of ChiZ. In this model, when chitooligosaccharides need to be imported, ChiZ prevents ChiX from base pairing, but does not promote degradation of this sRNA. When metabolic needs shift, ChiX could be released from ChiZ allowing ChiX to regulate chiP and other targets. This may work in competition, conjunction, or at separate times from the chbBC intergenic mRNA sequence, which also sponges ChiX, but results in ChiX decay (Overgaard et al., 2009). It will be interesting to see if other 5´ UTRs and sRNAs form similar autoregulatory loops, since 5´ UTRs are enriched for sRNA pairing sites, and we have shown that sRNA pairing is associated with distinct small transcripts from 5´ UTRs.
In a second example, we characterized IspZ, which is generated from the 5´ UTR of ispU (uppS), encoding the synthase for undecaprenyl pyrophosphate (UPP), a lipid carrier for bacterial cell wall carbohydrates (Apfel et al., 1999). We suggest IspZ may connect cell wall remodeling with the oxidative stress response. Cellular levels of toxic reactive oxygen species are increased when cell wall synthesis is blocked, and oxidative damage impedes ispU-related cell wall growth (Kawai et al., 2015). Thus, IspZ downregulation of the hydrogen peroxide-induced sRNA OxyS may dampen the oxidative response at a time when the response might be detrimental.
While small transcripts from within coding sequences have been noted previously (Dar and Sorek, 2018a; Reppas et al., 2006), and a homolog of FtsO (STnc475) has been detected for Salmonella enterica (Smirnov et al., 2016), we are the first to document a regulatory role for a bacterial ORF-internal sRNA. FtsO was found to base pair with, and negatively regulate the membrane stress response sRNA, RybB. The ftsI mRNA encodes a low-abundance but essential penicillin-binding protein that is localized to the inner membrane at the division site and cell pole (Weiss et al., 1997). The cell may need to alter its response to membrane stress during the division cycle when many membrane components are needed, and we suggest FtsO could facilitate crosstalk between cell division and membrane stress by regulating RybB activity. Intriguingly, we observed the greatest effect of ethanol addition on RybB levels in the ΔftsO background at 20 min, which is also the doubling time for E. coli MG1655. While it is reasonable to assume that regulatory sRNAs encoded by intragenic sequences are rare, due to the challenge of encoding two functions in one region of DNA, we think it is likely that other ORF-internal sRNAs have function.
Our work has significantly increased the number of sRNAs documented to modulate the activities of other sRNAs by sponging their activities, as found for ChiZ, or affecting their levels, as shown for IspZ and FtsO. The findings raise other questions including how many more short transcripts generated by termination or processing have regulatory functions. It also is intriguing that some abundant sRNAs are subject to regulation by multiple sponges, including ChiX, which is regulated by ChiZ and the chbBC intergenic region (Overgaard et al., 2009), and RybB, which is regulated by FtsO, RbsZ (Melamed et al., 2020) and the 3´ETSleuZ tRNA fragment (Lalaouna et al., 2015). Finally, little is known about how the activities and levels of the sponges themselves are regulated. The levels of some, but not others, are influenced by Hfq binding. A number appear to be constitutively expressed, such that target sRNA levels must increase to overcome the effects of the sponges.
Further identification and characterization of RNA fragments generated by premature termination or processing, detected by mapping the 5´ and 3´ ends of bacterial transcriptomes, will help elucidate the effects of regulatory RNAs. Our datasets point to a plethora of potential cis- and trans-acting regulatory elements in 5´ UTRs and ORF-internal regions, providing a valuable resource for further studies of gene regulation.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Escherichia coli) | MG1655 (WT) | this study | N/A | see Supplementary file 4 for derivatives |
| Antibody | mouse monoclonal anti-FLAG-M2-HRP | Sigma-Aldrich | Cat#A8592 | WB (1:2000) |
| Antibody | rabbit polyclonal anti-OmpC | Biorbyt | Cat#orb6940 | WB (1:500) |
| Antibody | donkey polyclonal peroxidase labeled anti-rabbit | GE Healthcare | Cat#NIF824 | WB (1:1000) |
| Recombinant DNA reagent | pBRplac | Guillier and Gottesman, 2006 | N/A | see Supplementary file 4 for derivatives |
| Recombinant DNA reagent | pNM46 (pBRplac-lacI) | this study | N/A | see Supplementary file 4 for derivatives |
| Recombinant DNA reagent | pKD46 | Datsenko and Wanner, 2000 | N/A | see Supplementary file 4 for derivatives |
| Recombinant DNA reagent | pMM1 | Stringer et al., 2014 | N/A | see Supplementary file 4 for derivatives |
| Sequence-based reagent | northern probes and primers | this study | N/A | see Supplementary file 4 |
| Chemical compound, drug | spermidine | Sigma-Aldrich | Cat#S2626-1G | |
| Other | TRIzol reagent | Thermo Fisher Scientific | Cat#15596018 | RNA extractions |
| Other | RNA-sequencing reagents | Melamed et al., 2018 | N/A | |
| Other | ureagel-8 | National Diagnostics | Cat#EC-838 | acrylamide northern solution |
| Other | ureagel complete | National Diagnostics | Cat#EC-841 | acrylamide northern solution |
| Other | NuSieve 3:1 agarose | Lonza | Cat#50090 | agarose for northern blotting |
| Other | 37% formaldehyde | Fisher Scientific | Cat#BP531-500 | |
| Other | RiboRuler high range RNA ladder | Thermo Fisher Scientific | Cat#SM1821 | |
| Other | RiboRuler low range RNA ladder | Thermo Fisher Scientific | Cat#SM1831 | |
| Other | Zeta-Probe blotting membrane | Bio-Rad | Cat#1620159 | northern membrane |
| Other | ULTRAhyb-oligo hybridization buffer | Thermo Fisher Scientific | Cat#AM8663 | |
| Other | γ-32P ATP | PerkinElmer | Cat#NEG035C010MC | |
| Other | T4 polynucleotide kinase | New England Biolabs | Cat#M0201L | |
| Other | Illustra MicroSpin G-50 columns | GE Healthcare | Cat#27533001 | |
| Other | mini-PROTEAN TGX gels | Bio-Rad | Cat#456–1086 | |
| Other | EZ-RUN pre-stained Rec protein ladder | Fisher Scientific | Cat#BP3603-500 | |
| Other | nitrocellulose membrane | Thermo Fisher Scientific | Cat#LC2000 | western membrane |
| Other | SuperSignal West Pico PLUS chemiluminescent substrate | Thermo Fisher Scientific | Cat#34580 | |
| Software, algorithm | lcdb-wf | ‘lcdb-wf’. Dale et al., GitHub Repository | v1.5.3 | https://github.com/lcdb/lcdb-wf |
| Software, algorithm | sra-tools | SRA Toolkit Development Team | v2.9.1_1 | http://ncbi.github.io/sra-tools/ |
| Software, algorithm | cutadapt | Martin, 2011 | v2.3 | https://github.com/marcelm/cutadapt |
| Software, algorithm | fastqc | Wingett and Andrews, 2018 | v0.11.8 | https://qubeshub.org/resources/fastqc |
| Software, algorithm | bwa | Li and Durbin, 2010 | v0.7.17 | http://bio-bwa.sourceforge.net/ |
| Software, algorithm | bowtie2 | Langmead and Salzberg, 2012 | v2.3.5 | http://bowtie-bio.sourceforge.net/bowtie2 |
| Software, algorithm | samtools | Li et al., 2009 | v1.9 | http://www.htslib.org/ |
| Software, algorithm | subread | Liao et al., 2013 | v1.6.4 | http://subread.sourceforge.net/ |
| Software, algorithm | multiqc | Ewels et al., 2016 | v1.7 | https://github.com/ewels/MultiQC |
| Software, algorithm | picard | ‘Picard Toolkit.’ 2019. Broad Institute, GitHub Repository | v2.20.0 | https://github.com/broadinstitute/picard |
| Software, algorithm | deeptools | Ramírez et al., 2016 | v3.2.1 | https://deeptools.readthedocs.io/ |
| Software, algorithm | termseq-peaks | ‘termseq-peaks.’ 2020. NICHD-BSPC, GitHub Repository | N/A | https://github.com/NICHD-BSPC/termseq-peaks |
| Software, algorithm | bedtools | Quinlan and Hall, 2010 | v2.27.1 | https://bedtools.readthedocs.io/ |
| Software, algorithm | pybedtools | Dale et al., 2011 | v0.8.0 | https://github.com/daler/pybedtools |
| Software, algorithm | ucsc-toolkit | Kent et al., 2010 | v357 | https://doi.org/10.1093/bioinformatics/btq351 |
| Software, algorithm | biopython SeqIO | Cock et al., 2009 | v1.73 | biopython.org |
| Software, algorithm | rhoterm-peaks | ‘rhoterm-peaks’ 2020. GitHub repository | N/A | https://github.com/gbaniulyte/rhoterm-peaks |
| Software, algorithm | CLG Genomics Workbench | Qiagen | v8.5.1 | alignment of DirectRNA-seq reads |
| Software, algorithm | CLUSTAL | Madeira et al., 2019 | v2.1 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Software, algorithm | BOXSHADE | v3.21 | https://embnet.vital-it.ch/software/BOX_form.html | |
| Software, algorithm | Sfold | Ding et al., 2004 | v2.2 | http://sfold.wadsworth.org/cgi-bin/srna.pl |
Bacterial strains and plasmids
Request a detailed protocolDerivatives of E. coli K12 MG1655 (WT) were used for all experimental studies. All strains, plasmids and oligonucleotides used are listed in Supplementary file 4. Engineered mutations and plasmid inserts were verified by sequencing.
The E. coli strains carrying mdtJ-3XFLAG were engineered using the FRUIT method (Stringer et al., 2012). Briefly, a 3XFLAG-thyA-3XFLAG tag/selection marker was PCR-amplified with primers JW9000 + JW9001 and recombineered into strain AMD061 and then counter-selected for thyA loss due to recombination of the FLAG tags, resulting in the intermediate strain YY18. This intermediate was used to create the mdtU start codon mutant (ATG→ACG) by recombineering the PCR-amplified thyA marker (using primers JW10309 + JW10310) into the mdtU gene and then replacing the thyA marker with the mdtU mutation (recombineering a PCR-amplified product made using primers JW10311 - JW10314). The native thyA locus was restored as described previously (Stringer et al., 2012). This resulted in the wild-type mdtU mdtJ-3XFLAG (YY20) or the mdtU start codon (ATG→ACG) mutant mdtJ-3XFLAG (AMD742) strain. The ΔmgrR::kan (GSO769), ΔmicA::kan (GSO157), and ΔchiX::kan (GSO169) deletions (Hobbs et al., 2010) were transduced into MG1655 (GSO982) by P1 transduction, resulting in GSO993, GSO994 and GSO995 respectively. The oxyS-M1::kan (GSO996) and rybB-M3::kan (GSO997) strains were constructed by PCR-amplifying the kanR sequence in pKD4 (Datsenko and Wanner, 2000) using primers PA313 + PA314 (oxyS-M1) or PA218 + PA219 (rybB-M3) and recombineering the product (Datsenko and Wanner, 2000; Yu et al., 2000) into the chromosome of E. coli NM400 (kind gift of Nadim Majdalani). The ftsO-M3::kan (GSO999) strain was constructed by first transforming a temperature sensitive ftsI Y380D mutant (PA215) (Dai et al., 1993), with pKD46 (Datsenko and Wanner, 2000). This strain was electroporated with an ftsO-M3 PCR product (amplified from the ftsO-M3 geneblock (Supplementary file 4) with primers PA216 + PA217) in the presence of 20 mM L-arabinose to induce the λ recombinase on pKD46. Colonies were selected by plating at 45 °C. Colony PCRs (using primers PA216 + PA217) and sequencing was performed to check for repair of the ftsI Y380D mutation and simultaneous ftsO-M3 incorporation (GSO999). A colony without the ftsO-M3 change was kept as a wild-type control (GSO998). All sRNA mutant alleles were subsequently transferred into E. coli MG1655 (GSO982) by P1 transduction.
The pMM1 β-galactosidase transcriptional reporter fusion plasmid was constructed by PCR-amplifying the high expression promoter KAB-TTTG (Burr et al., 2000) from pJTW064 (Stringer et al., 2014) using primers JW10252 + JW10253, and the DNA was ligated into pAMD-BA-lacZ plasmid (Stringer et al., 2014) digested with the NsiI and HindIII restriction enzymes. Either the entire 5´ UTR and annotated ORF region, or 5´ UTR region alone for selected genes was PCR-amplified using the oligonucleotides listed in Supplementary file 4, and cloned into the pMM1 vector, cut with NsiI and NheI restriction enzymes, using the NEBuilder HiFi kit (NEB). The mdtU-lacZ translational fusions were constructed by PCR-amplifying the mdtU gene from either a WT (E. coli MG1655) genomic template or mdtU start codon mutant (ATG→ACG) template (AMD742) using primers JW7269 + JW8934. These PCR products were subsequently ligated into the pAMD-BA-lacZ plasmid (Stringer et al., 2014), cut with SphI and HindIII restriction enzymes, using the NEBuilder HiFi kit (NEB). sRNAs were over expressed using the pBRplac plasmid (Guillier and Gottesman, 2006). sRNA sequences were PCR-amplified using the oligonucleotides listed in Supplementary file 4, digested with AatII and EcoRI, and cloned into pBRplac digested with the same restriction enzymes. The ChiZ overexpression construct was engineered using the NEBuilder kit (NEB), according to the manufacturer’s instructions with primers PA311 + PA312 and LW043 + LW044 and the pBRplac-lacI derivative, pNM46 (kind gift of Nadim Majdalani).
Growth conditions
Request a detailed protocolBacterial strains standardly were grown with shaking at 250 rpm at 37 °C in either LB rich medium or M63 minimal medium supplemented with 0.2% glucose and 0.001% vitamin B1. Ampicillin (100 µg/ml), kanamycin (30 µg/ml), chloramphenicol (30 µg/ml) and/or IPTG (1 mM) were added where appropriate. Unless indicated otherwise, overnight cultures were diluted to an OD600 of 0.05 and grown to the indicated OD600 or time point.
RNA isolation
Request a detailed protocolE. coli cells corresponding to the equivalent of 10 OD600 were collected by centrifugation, washed once with 1X PBS (1.54 M NaCl, 10.6 mM KH2PO4, 56.0 mM Na2HPO4, pH 7.4) and pellets snap frozen in liquid N2. RNA was isolated using TRIzol (Thermo Fisher Scientific) exactly as described previously (Melamed et al., 2020). RNA was resuspended in 20–50 µl DEPC H2O and quantified using a NanoDrop (Thermo Fisher Scientific).
Term-seq
Request a detailed protocolTwo biological replicates of E. coli MG1655 (GSO988) were diluted 1:500 from an LB overnight culture in either LB or M63 glucose media. Cells were collected at an OD600 ~0.4 and 2.0 for LB and an OD600 ~0.4 for M63 grown cultures. RNA was extracted as described above and analyzed using an Agilent 4200 TapeStation System to check the quality. Any contaminating DNA in the samples was removed by treating 15 µg of RNA with 10 U of DNase I (Roche) for 15 min at 37 °C in the presence of 80 U of recombinant RNase inhibitor (Takara Bio). Next, RNA was purified by mixing the sample with an equal volume of phenol stabilized:chloroform:isoamyl alcohol (25:24:1) and centrifugation at maximum speed in Heavy Phase Lock Gel tubes (5 PRIME). A volume of chloroform, equal to the original sample volume, was added to the same Heavy Phase Lock Gel tubes and spun again. The aqueous layer was removed and ethanol precipitated in the presence of 15 µg GlycoBlue (Ambion). RNA pellets were reconstituted in 10 µl DEPC H2O and analyzed using an Agilent 4200 TapeStation System to ensure DNase-treated RNA was at high quality. Term-seq libraries were prepared using a modified version of the RNAtag-seq methodology (Shishkin et al., 2015), based on the previously published Term-seq methodology (Dar et al., 2016). 1.5 µg of DNA-free RNA was first ligated at the 3´ end with 150 µM barcoded oligonucleotide adapters which were 5´phosphorylated and dideoxycytidine 3´ terminated (Supplementary file 4). RNA and 3´ adapters were incubated at 22 °C for 2.5 hr with 51 U of T4 RNA Ligase I (NEB) and 12 U of recombinant RNase inhibitor (Takara Bio) in 1X T4 RNA Ligase Buffer (NEB), 9% DMSO, 20% PEG 8000, and 1 mM ATP. 3´ ligated RNA was cleaned by incubating with 2.5X volume of RNAClean XP beads (Beckman Coulter) and 1.5X volume of isopropanol for 15 min, before separation on a magnetic rack. Bead-bound RNA was washed with 80% ethanol, air dried, and resuspended in DEPC H2O. RNA-containing-supernatants were removed and the same RNAClean XP bead cleanup protocol was repeated, with a final DEPC H2O elution of 9.5 µl. RNA was fragmented by incubating 9 µl of cleaned-up RNA with 1X Fragmentation Reagent (Invitrogen) for 2 min at 72 °C, followed by an addition of 1X Stop Solution (Invitrogen). Samples were stored on ice following individual fragmentation of each sample. Fragmented-RNA was pooled together and cleaned using the RNA Clean and Concentrator-5 kit (Zymo) according to the manufacturer’s instructions. Library construction continued following the bacterial-sRNA adapted, RNAtag-seq methodology starting at the rRNA removal step (Melamed et al., 2018). Term-seq RNA libraries were analyzed on a Qubit 3 Fluorometer (Thermo Fisher Scientific) and an Agilent 4200 TapeStation System prior to paired-end sequencing using the HiSeq 2500 system (Illumina).
Identification of 3´ ends from Term-seq
Request a detailed protocolRaw sequence reads were processed using lcdb-wf (lcdb.github.io/lcdb-wf/) according to the following steps. Raw sequence reads were trimmed with cutadapt 1.18 (Martin, 2011) to remove any adapters while performing light quality trimming with parameters ‘-a AGATCGGAAGAGC -q 20 –minimum-length = 25.’ Sequencing library quality was assessed with fastqc v0.11.8 with default parameters. The presence of common sequencing contaminants was evaluated with fastq_screen v0.11.3 with parameters ‘–subset 100000 –aligner bowtie2.’ Trimmed reads were mapped to the E. coli reference genome (MG1655 NC_000913.3) using BWA-MEM. Multimapping reads were filtered using samtools (Li et al., 2009). Uniquely aligned reads were then mapped to gene features using subread featureCounts v1.6.2 with default parameters. BedGraph files were generated using deepTools (Ramírez et al., 2016) on reads from each strand separately.
An initial set of termination peaks was called per sample on the bedGraph files from uniquely aligned reads using a novel signal processing approach combined with a statistically-informed method of combining multiple replicates. Briefly, the scipy.signal Python package was used to call peaks on each replicate in a manner which handled high, sharp peaks as found in Term-seq data, using the scipy.signal.find_peaks function with a width of (1, None), a prominence of (None, None), and a relative height of 0.75. Peaks for each replicate were then combined using the IDR framework (Landt et al., 2012) into a set of peaks that were reproducible (both strong and consistent) across replicates. The code for this can be found at https://github.com/NICHD-BSPC/termseq-peaks Adams, 2020 (copy archived at swh:1:rev:8bb48d2a034b22a312a7e848e0f8694a284a8324) and can be used in general for Term-seq peak-calling in other bacteria. Termination peaks were subsequently curated according to the following criteria. The single-bp peak coordinate was set to the strongest signal nucleotide within the boundary of the initial broader peak using multiBigWigSummary from deepTools 3.1.3. The most downstream position, relative to the peak orientation, was chosen when several positions were equally strong. Scores from peaks within a distance of up to 100 bp were assessed to select the peak with the highest score among the cluster for further analysis. These curated peaks were used for all analysis herein (Supplementary file 1).
Total RNA-seq
Request a detailed protocolTotal RNA-seq was performed using the same RNA that was used for the Term-seq library preparations. Total RNA-seq library construction was carried out based on the RNAtag-seq methodology (Shishkin et al., 2015), which was adapted to capture bacterial sRNAs (Melamed et al., 2018). Total RNA-seq RNA libraries were sequenced as for Term-seq. Total RNA-seq data processing followed the same procedures as Term-seq data analysis for QC, adaptor removal and sequencing read mapping.
BCM treatment and DirectRNA-seq
Request a detailed protocolOne culture of E. coli MG1655 cells (GSO989) was grown in LB to an OD600 ~0.5 and the culture was split and half was treated with 100 μg/ml of BCM (gift from Max Gottesman) for 15 min. Total RNA was isolated from 1.5 ml of untreated and BCM-treated cultures using the hot-phenol RNA extraction method followed by ethanol precipitation as described previously (Stringer et al., 2014). Genomic DNA was removed by treating 8 μg of total RNA with 4 U of Turbo DNase (Invitrogen) for 45 min at 37 °C. DNA-free RNA was purified using phenol:chloroform:isoamyl alcohol and ethanol precipitation as described previously (Stringer et al., 2014). rRNA was removed using a Ribo-Zero (Bacteria) kit (Epicenter) according to the manufacturer’s instructions. The RNA libraries were prepared and processed at the Helicos BioSciences facility where poly-A tails and a 3´-dATP block were added to make the RNA suitable for direct sequencing on the HeliScope Single-Molecule Sequencer (Ozsolak and Milos, 2011).
Identification of Rho-dependent 3´ ends
Request a detailed protocolCLC Genomics Workbench (v8.5.1) was used to align DirectRNA-sequencing reads from untreated and BCM-treated samples to the MG1655 NC_000913.3 sense and reverse-complemented genome to properly identify the position of the first mapped 3´ end nucleotide. Mapping parameters were set to default, except for ‘Length fraction’ and ‘Similarity fraction’, which were set to 0.7 and 0.9, respectively. Quality scores were not generated by the HeliScope Sequencer; arbitrary quality scores were added to each read in fasta files to fit import requirements for CLC Genomics Workbench, but they were ignored when mapping. The read count and position of sequenced transcript 3´ ends were used for further analysis. The approximate Rho-dependent transcription termination sites were predicted by identifying the locations of transcriptional readthrough in the BCM-treated sample. The read counts in 800 nt regions upstream (BCM_us) and downstream (BCM_ds) of each position were compared to read counts from the same positions in the untreated sample (Untreated_us, Untreated_ds). We then used a Fisher’s exact test to compare the downstream:upstream read count ratios for untreated and BCM-treated samples, and we calculated a ratio of ratios: (R(BCM/untreated) = ). We refer to the p-value from the Fisher’s exact test as the ‘significance score’, and we refer to the R(BCM/Untreated) ratio as the ‘Rho score’ (Supplementary file 2). Putative Rho termination regions were those genome coordinates with a positive Rho score and a significance score <1e−4. Only the position with the highest Rho significance score within an 800 nt window upstream and downstream is reported in Supplementary file 2. Note that Rho scores and significance scores listed in Supplementary file 3 were calculated for specific positions matching the dominant Term-seq 3´ ends; there may be nearby positions with a lower significance score and/or a higher Rho score.
Classification of 3´ ends
Request a detailed protocolThe intersect function of Bedtools 2.28.0 (Quinlan and Hall, 2010), ran via pybedtools v0.8.0 (Dale et al., 2011), was used to assign each peak to one or more classes: Primary (3´ peaks located on the same strand either within 50 bp downstream of the 3´ end of an annotated mRNA ORF, tRNA, rRNA or sRNA with the highest score), Antisense (3´ peaks located on the opposite strand of an annotated mRNA ORF, tRNA, rRNA or sRNA within 50 bp of its start and end coordinates), Internal (3´ peaks located on the same strand within an annotated mRNA ORF, tRNA, rRNA or sRNA coordinates, excluding the 3´ end coordinate) and Orphan (3´ peak not falling in any of the previous classes).
3´ ends were also categorized according to their position relative to mRNA 5´ UTRs and internal mRNA regions (Supplementary file 3). Any 3´ end (Supplementary file 1) that was located within a region of 200 bp upstream of an annotated start codon to the stop codon were extracted and further analyzed. To remove any 3´ ends that likely belonged to an upstream gene in the same direction, TSS data (Thomason et al., 2015) obtained using the same growth conditions and E. coli strain as Term-seq was considered. All these 3´ ends were examined for the first upstream feature (either a TSS or an ORF stop codon). Any 3´ end where the first upstream feature was a stop codon was eliminated, unless there was also a TSS ≤200 bp upstream the 3´ end or that upstream feature was the stop codon of an annotated ‘leader peptide’ on the EcoCyc E. coli database (mgtL, speFL, hisL, ivbL, ilvL, idlP, leuL, pheL, pheM, pyrL, rhoL, rseD, thrL, tnaC, trpL, uof). Any 3´ end where a TSS was only 20 bp or less upstream was also eliminated. This resulted in the 3´ end coordinates in Supplementary file 3. For the LB 0.4 condition, 3´ ends were given a Rho score from Direct-RNA-seq (as described above) and an intrinsic terminator score (with a custom script as defined in Chen et al., 2013). uORFs for which synthesis was detected by western analysis and/or translational reporter fusions (Hemm et al., 2008; VanOrsdel et al., 2018; Weaver et al., 2019), sRNAs for which synthesis was detected by northern analysis (this study) and other characterized RNA regulators were noted for the LB 0.4 condition.
Comparison of Term-seq 3´ ends and Rho termination regions to equivalent genomic positions identified in other studies
Request a detailed protocolAs detailed in Figure 1—figure supplement 2, we compared the 3´ ends identified by Term-seq with 3´ ends identified in other studies, and we compared Rho termination regions with putative sites of Rho termination identified in other studies. Specifically, we determined how many of the positions in our list of genome coordinates are within a given distance threshold of a genome coordinate from another study. We also performed the reciprocal comparison. Note that the two numbers may differ if two coordinates in one study are close to one coordinate in the other; hence, we include both numbers in the Venn diagrams in Figure 1—figure supplement 2. To determine the extent of overlap between lists we calculated the proportion of the genome that is within the given distance threshold of each genome coordinate in a single list. We then divided this number by double the genome size (doubled to account for the two possible strands), to determine the frequency with which a randomly selected genomic position would overlap with a genome coordinate in that list. We assessed the statistical significance of the overlap between lists of genome coordinates by using a hypergeometric test to compare the number of overlapping positions and the frequency expected by chance. For example, in Figure 1—figure supplements 2C, 19 of the 296 3´ ends in the LB 0.4 dataset are within 10 nt of a 3´ end in the Dar and Sorek dataset. We determined how many genomic positions are within 10 nt of a position in the Dar and Sorek dataset (22899 positions). The input parameters for the hypergeometric test were 219 (number of successes), 296 (sample size), 22899 (number of successes in the population), and 9283304 (population size; twice the genome length, to account for the two possible orientations). The p values are reported as <2.2e−16 since that is the machine epsilon for 64-bit double precision values in R and Python.
β-galactosidase assays
Request a detailed protocolRho transcriptional and MdtU translational reporter assays were performed as previously described (Baniulyte et al., 2017). Briefly, the pMM1constructs (Supplementary file 4) were assayed in MG1655ΔlacZ (AMD054) and MG1655ΔlacZ rhoR66S (GB4) backgrounds. Three separate colonies were grown overnight in LB with 30 μg/ml chloramphenicol, diluted 1:100 in the same medium, and grown to a final OD600 ~ 0.4–0.6 at 37 °C. Cells were lysed in Z buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 0.001 M MgSO4), supplemented with β-mercaptoethanol (50 mM final concentration), sodium dodecyl sulfate (0.001% final concentration), and chloroform. Assays were initiated by adding 2-nitrophenyl β-D-galactopyranoside and stopped by adding Na2CO3. All assays were done at room temperature. The OD600 and A420 of the cultures were measured using a Jenway 6305 spectrophotometer. The translational chiP-lacZ fusions (DJS2979 and DJS2991) were assayed as above, with the following changes. Three separate colonies were grown overnight in LB with 100 μg/ml ampicillin, diluted to an OD600 of 0.05 in the same medium supplemented with 0.2% arabinose and 1 mM IPTG, and grown for 150 min (OD600 ~ 1.5) at 37 °C. Reactions were performed at 28 °C and the OD600 and A420 of the cultures were measured using an Ultrospec 3300 pro spectrophotometer (Amersham Biosciences). For all experiments, β-galactosidase activity was calculated as (1000 x A420)/(OD600 x Vml x timemin).
Northern blot analysis
Request a detailed protocolNorthern blots were performed using total RNA exactly as described previously (Melamed et al., 2020). For small RNAs, 5 μg of RNA were fractionated on 8% polyacrylamide urea gels containing 6 M urea (1:4 mix of Ureagel Complete to Ureagel-8 (National Diagnostics) with 0.08% ammonium persulfate) and transferred to a Zeta-Probe GT membrane (Bio-Rad). For longer RNAs, 10 μg of RNA were fractionated on a 2% NuSieve 3:1 agarose (Lonza), 1X MOPS, 2% formaldehyde gel and transferred to a Zeta-Probe GT membrane (Bio-Rad) via capillary action overnight. For both types of blots, the RNA was crosslinked to the membranes by UV irradiation. RiboRuler High Range and Low Range RNA ladders (Thermo Fisher Scientific) were marked by UV-shadowing. Membranes were blocked in ULTRAhyb-Oligo Hybridization Buffer (Ambion) and hybridized with 5´ 32P-end labeled oligonucleotides probes (listed in Supplementary file 4). After an overnight incubation, the membranes were rinsed with 2X SSC/0.1% SDS and 0.2X SSC/0.1% SDS prior to exposure on film. Blots were stripped by two 7 min incubations in boiling 0.2% SDS followed by two 7 min incubations in boiling water.
Immunoblot analysis
Request a detailed protocolImmunoblot analysis was performed as described previously with minor changes (Zhang et al., 2002). Samples were separated on a Mini-PROTEAN TGX 5–20% Tris-Glycine gel (Bio-Rad) and transferred to a nitrocellulose membrane (Thermo Fisher Scientific). Membranes were blocked in 1X TBST containing 5% milk, probed with a 1:2000 dilution of monoclonal α-FLAG-HRP (Sigma) or a 1:500 dilution of polyclonal α-OmpC follwed by a 1:1000 dilution of peroxidase labeled α-rabbit and developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific) on a Bio-Rad ChemiDoc MP Imaging System.
Genome browsers
Request a detailed protocolThe processed RNA-seq data from this study are available online via UCSC genome browser at the following links:
E. coli Term-seq: https://www.nichd.nih.gov/research/atNICHD/Investigators/storz/ecoli-term-seq
E. coli Rho-dependent 3´ ends (Term-seq LB 0.4 and DirectRNA-seq): https://www.nichd.nih.gov/research/atNICHD/Investigators/storz/ecoli-rho-dependent-3-ends.
Data availability
The raw sequencing data reported in this paper have been deposited in SRA under accession number PRJNA640168. Code for calling 3´ ends in Term-seq sequencing reads can be found at https://github.com/NICHD-BSPC/termseq-peaks (copy archived at https://archive.softwareheritage.org/swh:1:rev:8bb48d2a034b22a312a7e848e0f8694a284a8324/). Code for calling Rho termination regions can be found at https://github.com/gbaniulyte/rhoterm-peaks (copy archived at https://archive.softwareheritage.org/swh:1:rev:831f7cb906d0af5767ffbfbb63ec247579d5e250/). The processed RNA-seq data from this study are available online via UCSC genome browser at the following links: E. coli Term-seq: https://www.nichd.nih.gov/research/atNICHD/Investigators/storz/ecoli-term-seq E. coli Rho-dependent 3´ ends (Term-seq LB 0.4 and DirectRNA-seq): https://www.nichd.nih.gov/research/atNICHD/Investigators/storz/ecoli-rho-dependent-3-ends.
References
-
Prevalence of small base-pairing RNAs derived from diverse genomic lociBiochimica Et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1863:194524.https://doi.org/10.1016/j.bbagrm.2020.194524
-
A role for Rho-dependent polarity in gene regulation by a noncoding small RNAGenes & Development 26:1864–1873.https://doi.org/10.1101/gad.195412.112
-
Riboswitches and translation controlCold Spring Harbor Perspectives in Biology 10:a032797.https://doi.org/10.1101/cshperspect.a032797
-
High-resolution RNA 3'-ends mapping of bacterial Rho-dependent transcriptsNucleic Acids Research 46:6797–6805.https://doi.org/10.1093/nar/gky274
-
The sponge RNAs of Bacteria – How to find them and their role in regulating the post-transcriptional networkBiochimica Et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1863:194565.https://doi.org/10.1016/j.bbagrm.2020.194565
-
Sfold web server for statistical folding and rational design of nucleic acidsNucleic Acids Research 32:W135–W141.https://doi.org/10.1093/nar/gkh449
-
Sponges and predators in the small RNA worldMicrobiology Spectrum 6:2018.https://doi.org/10.1128/microbiolspec.RWR-0021-2018
-
Mimicry, deception and competition: the life of competing endogenous RNAsWiley Interdisciplinary Reviews: RNA 10:e1525.https://doi.org/10.1002/wrna.1525
-
Remodelling of the Escherichia coli outer membrane by two small regulatory RNAsMolecular Microbiology 59:231–247.https://doi.org/10.1111/j.1365-2958.2005.04929.x
-
Small membrane proteins found by comparative genomics and ribosome binding site modelsMolecular Microbiology 70:1487–1501.https://doi.org/10.1111/j.1365-2958.2008.06495.x
-
Identification of a spermidine excretion protein complex (MdtJI) in Escherichia coliJournal of Bacteriology 190:872–878.https://doi.org/10.1128/JB.01505-07
-
Selective structural change by spermidine in the Bulged-out region of Double-stranded RNA and its effect on RNA functionJournal of Biological Chemistry 283:32989–32994.https://doi.org/10.1074/jbc.M806027200
-
Cell Growth of Wall-Free L-Form Bacteria Is Limited by Oxidative DamageCurrent Biology 25:1613–1618.https://doi.org/10.1016/j.cub.2015.04.031
-
Regulation of the Escherichia coli tna operon: nascent leader peptide control at the tnaC stop codonJournal of bacteriology 179:1774–1779.https://doi.org/10.1128/JB.179.5.1774-1779.1997
-
Learning from the Leaders: Gene Regulation by the Transcription Termination Factor RhoTrends in Biochemical Sciences 41:690–699.https://doi.org/10.1016/j.tibs.2016.05.012
-
The Bacterial Transcription Termination Factor Rho Coordinates Mg2+Homeostasis with Translational SignalsJournal of Molecular Biology 427:3834–3849.https://doi.org/10.1016/j.jmb.2015.10.020
-
ChIP-seq guidelines and practices of the ENCODE and modENCODE consortiaGenome Research 22:1813–1831.https://doi.org/10.1101/gr.136184.111
-
Fast gapped-read alignment with Bowtie 2Nature Methods 9:357–359.https://doi.org/10.1038/nmeth.1923
-
The Sequence Alignment/Map format and SAMtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
The Subread aligner: fast, accurate and scalable read mapping by seed-and-voteNucleic Acids Research 41:e108.https://doi.org/10.1093/nar/gkt214
-
RNA thermometers in bacterial pathogensMicrobiology Spectrum 6:RWR-0012-2017.https://doi.org/10.1128/microbiolspec.RWR-0012-2017
-
The EMBL-EBI search and sequence analysis tools APIs in 2019Nucleic Acids Research 47:W636–W641.https://doi.org/10.1093/nar/gkz268
-
Mutational Analysis and Secondary Structure Model of the RNP1-like Sequence Motif of Transcription Termination Factor RhoJournal of Molecular Biology 257:895–908.https://doi.org/10.1006/jmbi.1996.0210
-
Mapping the small RNA interactome in bacteria using RIL-seqNature Protocols 13:1–33.https://doi.org/10.1038/nprot.2017.115
-
Rho Protein: Roles and MechanismsAnnual Review of Microbiology 71:687–709.https://doi.org/10.1146/annurev-micro-030117-020432
-
Alternative ORFs and small ORFs: shedding light on the dark proteomeNucleic Acids Research 48:1029–1042.https://doi.org/10.1093/nar/gkz734
-
Switching off small RNA regulation with trap-mRNAMolecular Microbiology 73:790–800.https://doi.org/10.1111/j.1365-2958.2009.06807.x
-
Transcriptome profiling using single-molecule direct RNA sequencingMethods Mol Biol 733:51–61.https://doi.org/10.3389/fpls.2017.01205
-
Rho and NusG suppress pervasive antisense transcription in Escherichia coliGenes & Development 26:2621–2633.https://doi.org/10.1101/gad.196741.112
-
deepTools2: a next generation web server for deep-sequencing data analysisNucleic Acids Research 44:W160–W165.https://doi.org/10.1093/nar/gkw257
-
Mechanisms of Bacterial Transcription TerminationJournal of Molecular Biology 431:4030–4039.https://doi.org/10.1016/j.jmb.2019.04.003
-
Global regulation by CsrA and its RNA antagonistsMicrobiology Spectrum 6:2017.https://doi.org/10.1128/microbiolspec.RWR-0009-2017
-
Regulation by Small RNAs in Bacteria: Expanding FrontiersMolecular Cell 43:880–891.https://doi.org/10.1016/j.molcel.2011.08.022
-
Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNAGenes & Development 19:2355–2366.https://doi.org/10.1101/gad.354405
-
Identifying New Small Proteins in Escherichia coliPROTEOMICS 18:e1700064.https://doi.org/10.1002/pmic.201700064
-
SMRT-Cappable-seq reveals complex operon variants in bacteriaNature Communications 9:3676.https://doi.org/10.1038/s41467-018-05997-6
-
Diverse mechanisms for regulating ribosomal protein synthesis in Escherichia coliProg Nucleic Acid Res Mol Biol 47:331–370.https://doi.org/10.1016/s0079-6603(08)60256-1
Article and author information
Author details
Gabriele Baniulyte

Funding
National Institutes of Health (1Fi2GM133345-01)
- Philip P Adams
National Institutes of Health (1ZIAHD001608-28)
- Gisela Storz
National Institutes of Health (1DP2OD007188)
- Joseph T Wade
National Science Foundation (DBI 1757170)
- Molly Monge
National Institutes of Health (1ZICHD008986-01)
- Ryan K Dale
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank S Melamed, T Updegrove, A Wagh, L Walling, K Yoo, A Zhang, and H Zhang for experimental and technical assistance, and K Papenfort, T Updegrove and members of the Storz lab for helpful comments on the manuscript. We also thank M Gottesman for the gift of bicyclomycin, the Wadsworth Center Applied Genomic Technologies and Media Core Facilities for technical assistance, and the NICHD Molecular Genomics Core, particularly T Li, for library sequencing. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Copyright
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Metrics
-
- 6,797
- views
-
- 748
- downloads
-
- 89
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Regulatory roles of Escherichia coli 5' UTR and ORF-internal RNAs detected by 3' end mapping
eLife 10:e62438.
https://doi.org/10.7554/eLife.62438
Further reading
-
- Microbiology and Infectious Disease
Chlamydia trachomatis serovar L2 (Ct), an obligate intracellular bacterium that does not encode FtsZ, divides by a polarized budding process. In the absence of FtsZ, we show that FtsK, a chromosomal translocase, is critical for divisome assembly in Ct. Chlamydial FtsK forms discrete foci at the septum and at the base of the progenitor mother cell, and our data indicate that FtsK foci at the base of the mother cell mark the location of nascent divisome complexes that form at the site where a daughter cell will emerge in the next round of division. The divisome in Ct has a hybrid composition, containing elements of the divisome and elongasome from other bacteria, and FtsK is recruited to nascent divisomes prior to the other chlamydial divisome proteins assayed, including the PBP2 and PBP3 transpeptidases, and MreB and MreC. Knocking down FtsK prevents divisome assembly in Ct and inhibits cell division and septal peptidoglycan synthesis. We further show that MreB does not function like FtsZ and serve as a scaffold for the assembly of the Ct divisome. Rather, MreB is one of the last proteins recruited to the chlamydial divisome, and it is necessary for the formation of septal peptidoglycan rings. Our studies illustrate the critical role of chlamydial FtsK in coordinating divisome assembly and peptidoglycan synthesis in this obligate intracellular bacterial pathogen.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}