Repeated origins, widespread gene flow, and allelic interactions of target-site herbicide resistance mutations
- Department of Ecology and Evolutionary Biology, University of Toronto, Canada
- Graduate Group in Computational Biology, University of California, Berkeley, United States
- Department of Crop Sciences, University of Illinois Urbana-Champaign, United States
- Department of Molecular Biology, Max Planck Institute for Biology Tübingen, Germany
Figures
Figure 1
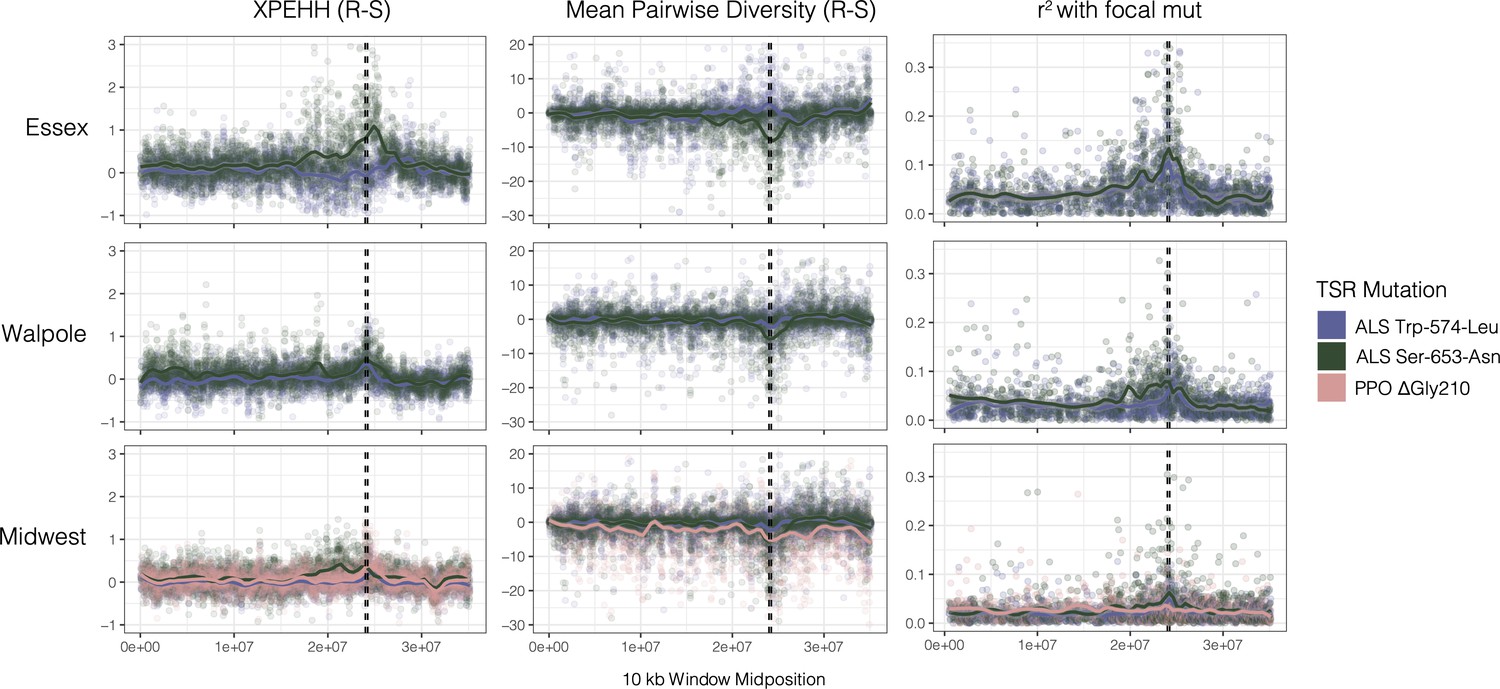
Sweep-scan summary statistics by geographic region.
(Left) Difference in integrated haplotype homozygosities (XPEHH) between haplotypes carrying the focal TSR mutation and susceptible haplotypes. (Middle) Difference in mean pairwise diversity between haplotypes carrying the focal TSR mutation and susceptible haplotypes. (Right) r2 of other missense mutations with focal TSR mutation on genotype, rather than haplotype data. In all columns, dashed vertical lines denote PPO (left) and ALS (right) genes, which are only 250kb apart in the genome.
Figure 2 with 6 supplements
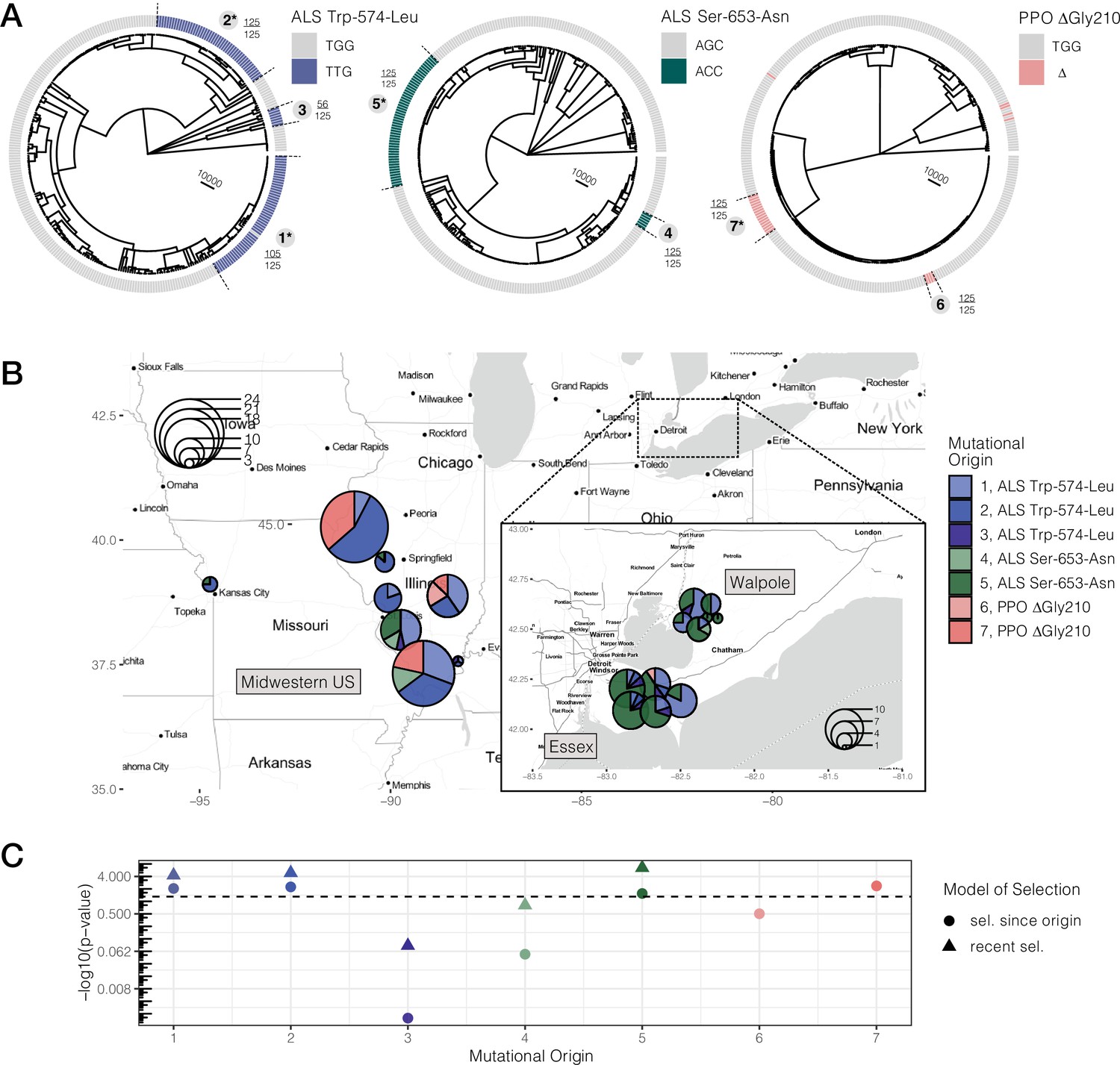
Repeated-independent origins and range-wide distribution of three target-site resistance mutations, along with their associated significance of selection over two different timescales.
(A) Trees at focal TSR loci corresponding to an ARG estimated across 20 kb SNPs. Bold numbers around trees identify clusters of resistant haplotypes consistent with independent origins. The presence of an asterisk at each origin number implies significant evidence of selection since the mutation arose de novo at p < 0.05 against the null distribution, as in C. Support for monophyly for each origin across 125 samples of 1250 MCMC iterations is depicted by the fraction found outside each cluster. (B) Geographic distribution of haplotypes originating from distinct mutational lineages as inferred from A. TSR mutational lineages are found across numerous populations and agricultural regions, although regions show clear differences in the frequency of some mutations. (C) Results of tree-based tests of non-neutral allele frequency change (Speidel et al., 2019) from each mutational origin of TSR under two alternative models of selection; selection on a mutation since its origin versus selection over more recent timescales (on the last 0.01% of the tree). The horizontal dashed line denotes the p-value cutoff of α = 0.05, after false discovery rate correction.
-
Figure 2—source data 1
Tree-based coalescent test for selection under two scenarios; selection on since the mutation first arose and selection even more recent timescales (i.e.the last 1% of the tree).
All bolded significant p-values remain significant after a 5% FDR correction. Values in parentheses in the p-value columns illustrate the ranking of each origin for significance in each test.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig2-data1-v2.txt
-
Figure 2—source data 2
Tree sequence corresponding to ALS Trp-574-Leu, ALS Ser-653-Asn, and PPO ΔGly210 extracted from the most likely iteration of the ARGweaver MCMC.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig2-data2-v2.zip
-
Figure 2—source data 3
Resistance status at ALS Trp-574-Leu, ALS Ser-653-Asn, and PPO ΔGly210 for haplotypes mapped in Figure 2.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig2-data3-v2.txt
Figure 2—figure supplement 1
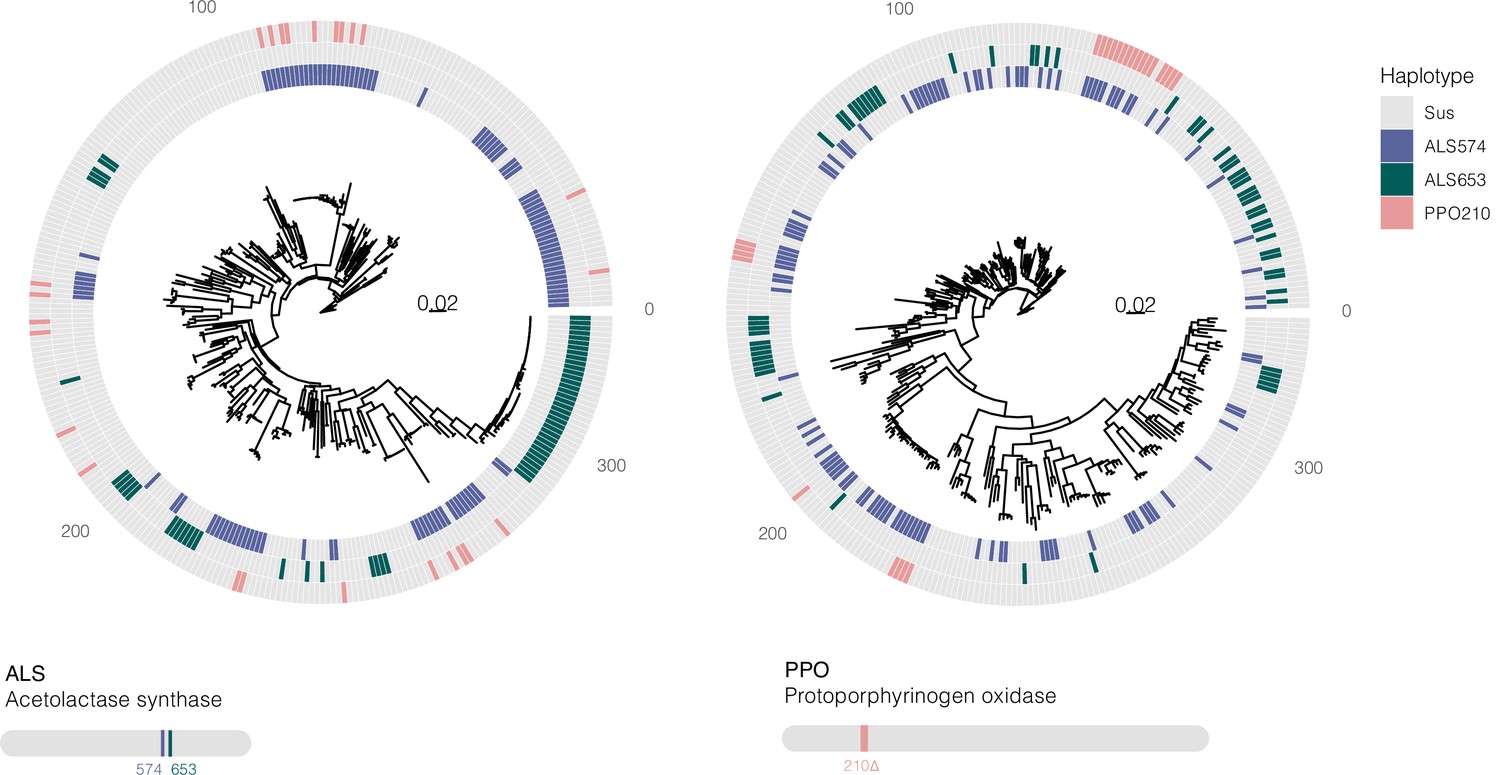
Bootstrapped gene trees of ALS (3 kb) and PPO (10 kb) (coding sequence + 1 kb on either side) alongside TSR mutations across all 162 individuals.
Colored grids indicate a haplotype harboring a focal resistance SNP (ALS Trp-574-Leu mutations in purple, ALS Ser-653-Asn mutations in green, and PPO ΔGly210 deletions in pink). Numerous multiple origins are apparent for all resistance mutations, in excess of the number inferred from ARG-based approaches.
Figure 2—figure supplement 2
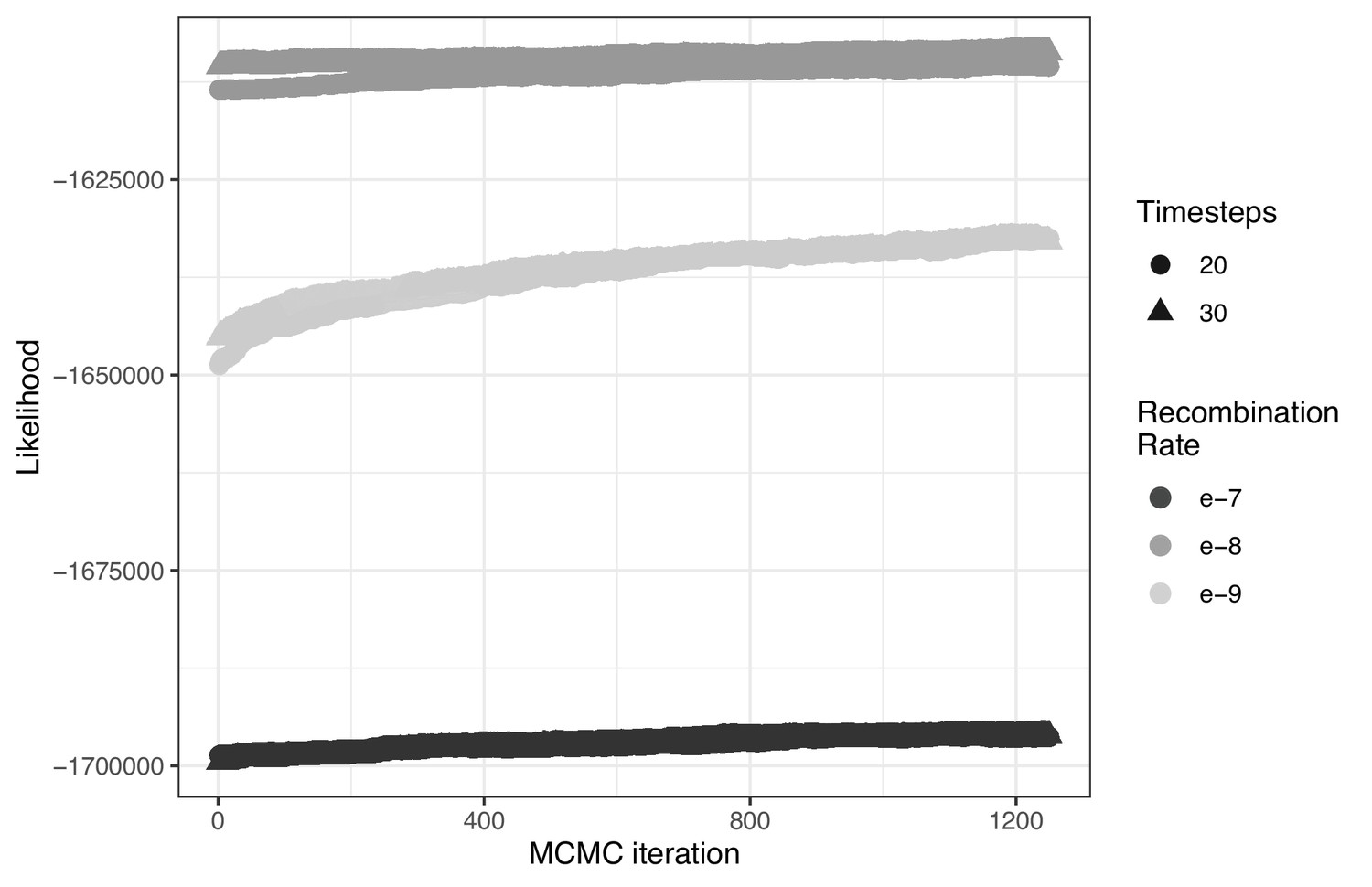
The influence of both the number of timesteps coalescent events are estimated over (t) and constant recombination rate magnitude (r) on ARG likelihood across 1250 MCMC iterations.
Figure 2—figure supplement 3
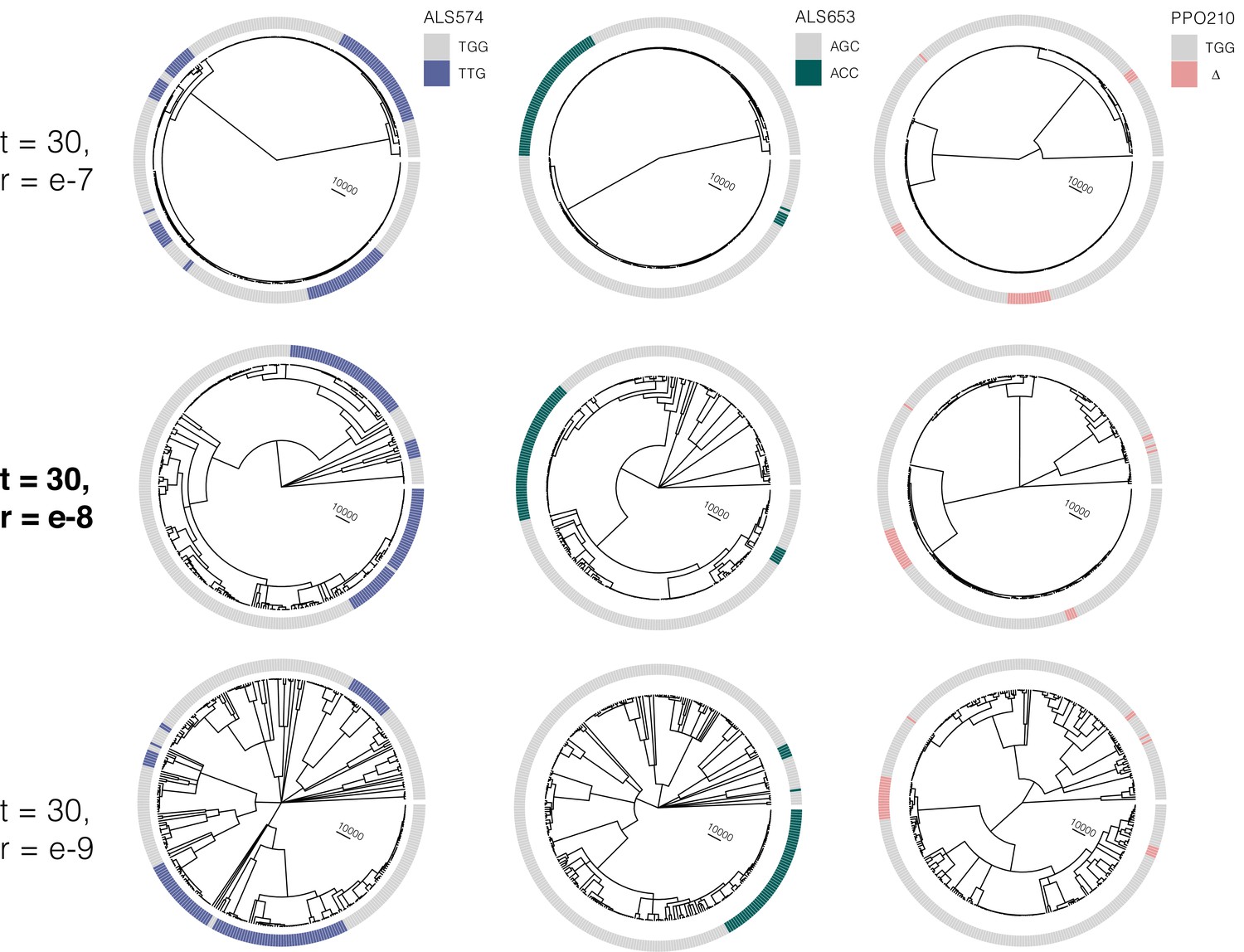
At the most likely number of (timesteps = 30), the influence of increasing the recombination rate parameter constant value from r = e-7 to r = e–9 on the tree sequence inference from the most likely ARG.
Figure 2—figure supplement 4
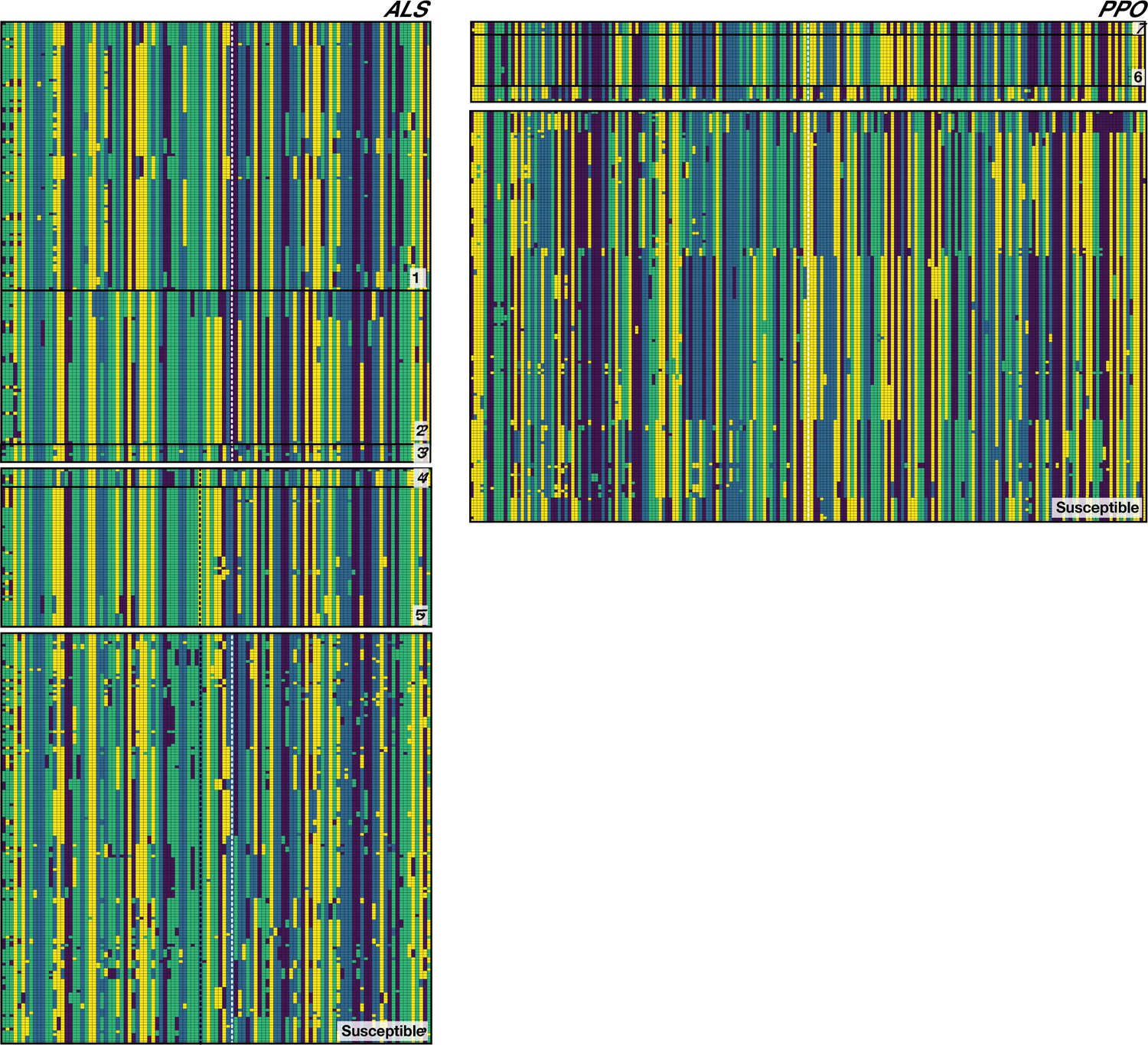
Phased haplotypes corresponding to distinct origins of target-site resistance mutations at the ALS Trp-574-Leu (Left side, grey vertical dashed line), ALS Ser-653-Asn (left side, black vertical dashed line) and PPO ΔGly210 (right side, dashed vertical white line) positions, relative to susceptible lineages.
Haplotypes corresponding to each named origin in Figure 2 depicted by number on the bottom right of each set.
Figure 2—figure supplement 5
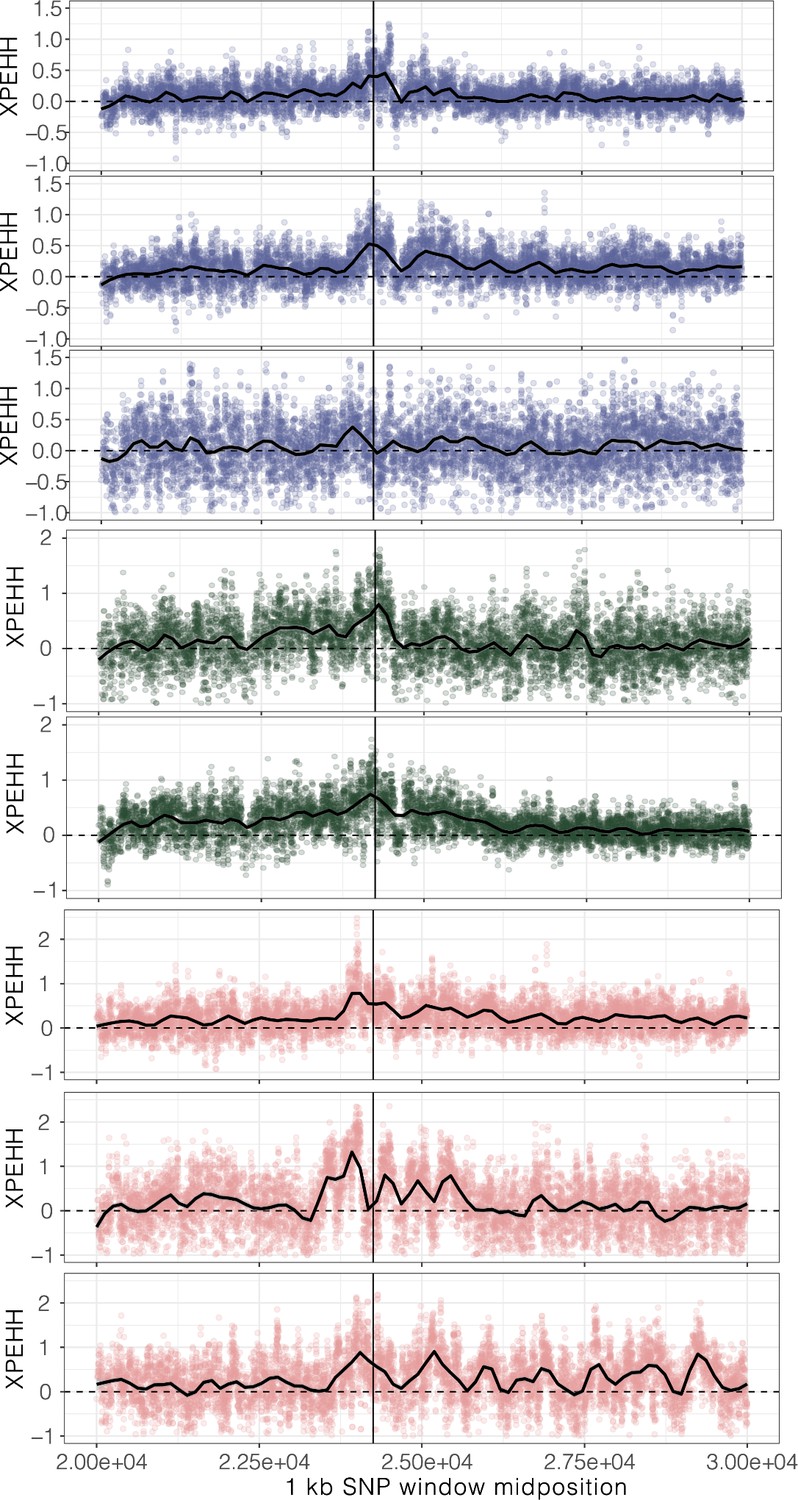
XPEHH, cross-population extended haplotype homozygosity, for haplotypes mapping to each origin of ALS Trp-574-Leu, ALS Ser-653-Asn, and PPO ΔGly210 (1–7, top to bottom, plus unplaced PPO ΔGly210 haplotypes as the last row) in comparison to susceptible haplotypes.
Figure 2—figure supplement 6
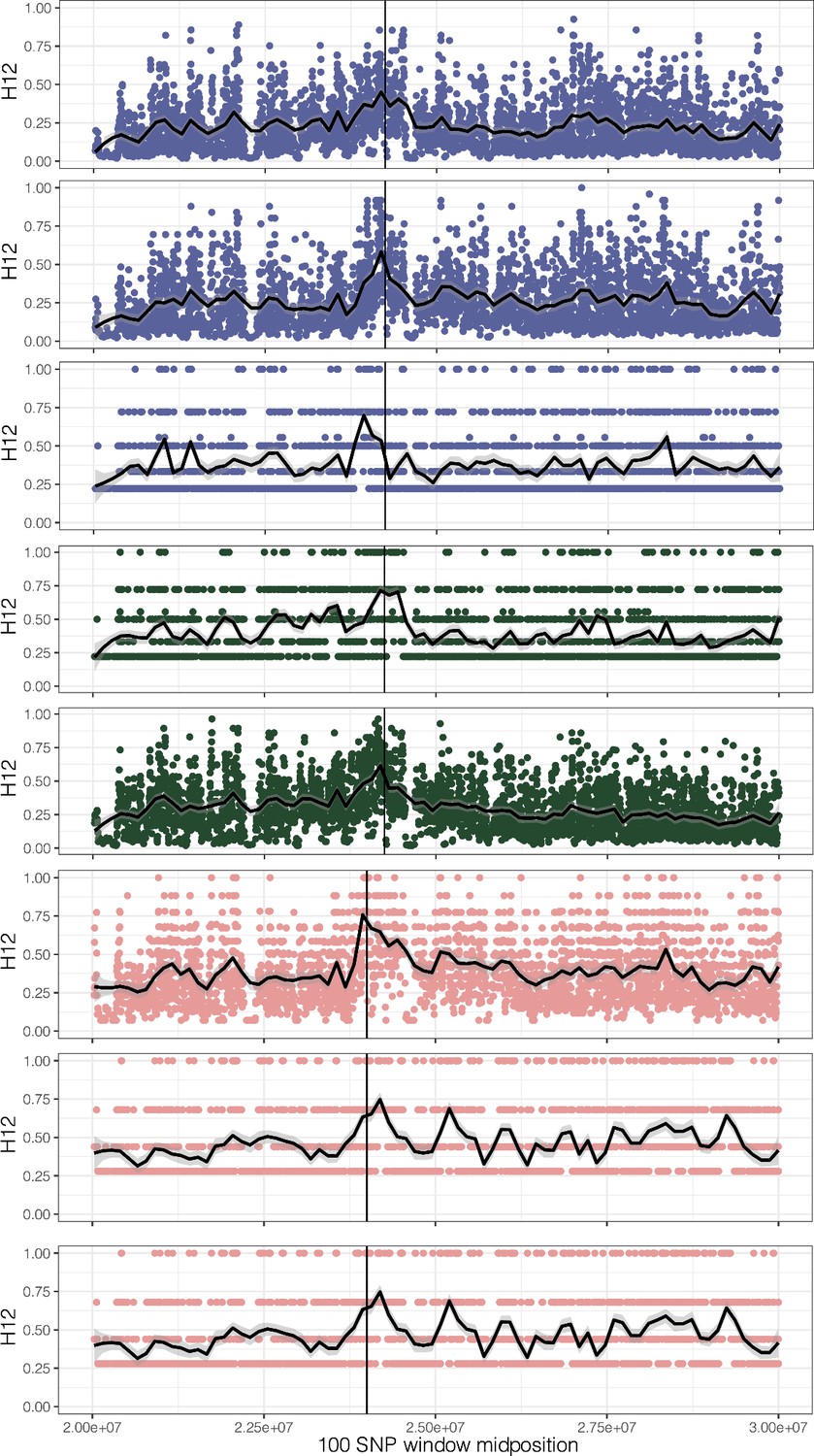
H12, homozygosity of the two most common haplotypes, for each origin of ALS Trp-574-Leu, ALS Ser-653-Asn, and PPO ΔGly210 (1–7, top to bottom, plus unplaced PPO ΔGly210 haplotypes as the last row).
A hamming distance of 5 was used, collapsing haplotypes that have fewer than five differences in each 100 SNP window.
Figure 3
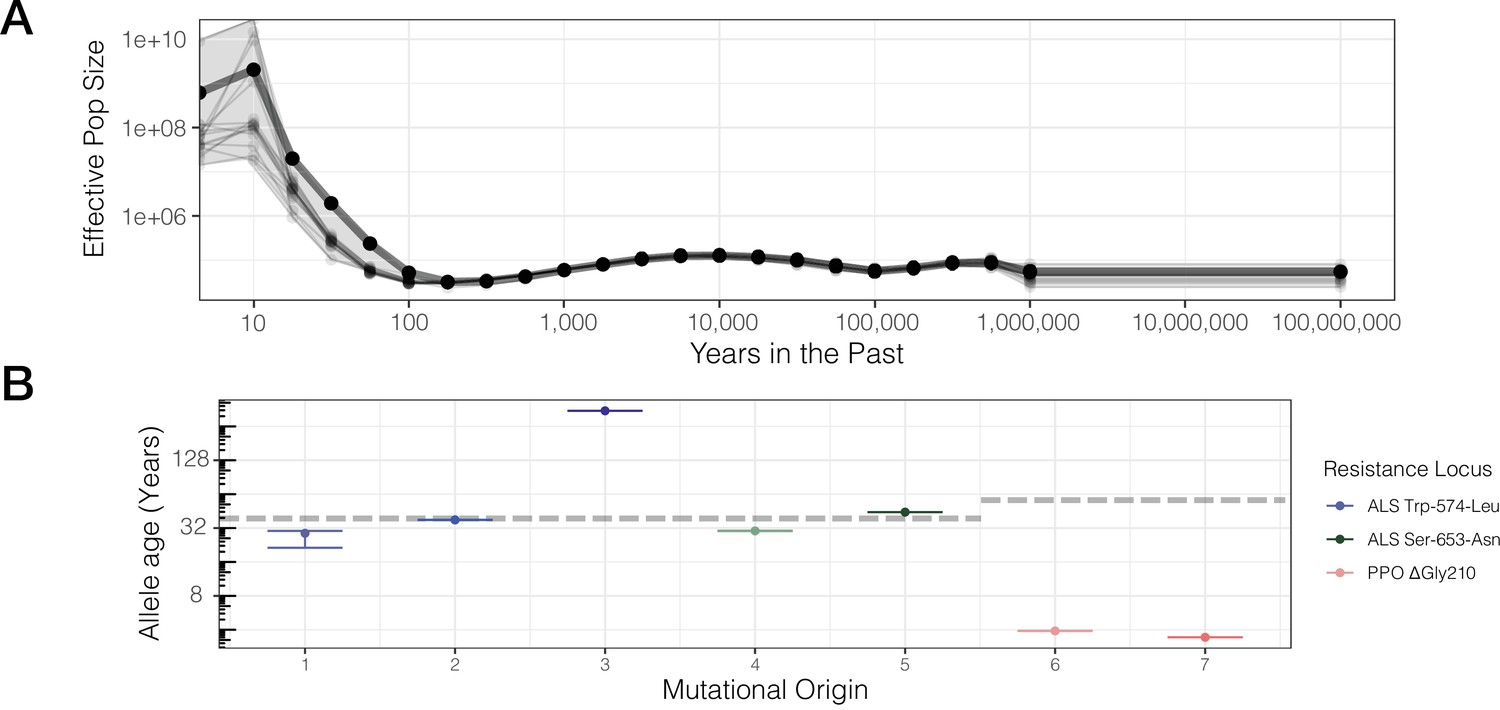
Contemporary population expansion of A. tuberculatus and corresponding ages of TSR variants.
(A) Relate-inferred effective population size through time, illustrating a remarkable population expansion occurring over the last 100 years. The bold line indicates results from genome-wide SNPs, whereas thinner lines represent results from chromosome-by-chromosome analyses, with the shaded area showing the bounds of the variance in the chromosome-by-chromosome data. (B) Allele age inferred from the geometric mean effective population size estimate over the timescale of contemporary herbicide use ( < 50 years ago, GM[Ne] = 83,294,700). Horizontal dashed lines for ALS Trp-574-Leu and ALS Ser-653-Asn, and PPO origins represent the approximate onset of ALS and PPO herbicide use, respectively.
-
Figure 3—source data 1
Effective population size estimates for each chromosome, and genome-wide, from Relate.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig3-data1-v2.txt
-
Figure 3—source data 2
Unscaled allele age estimates corresponding to the seven inferred origins of ALS Trp-574-Leu, ALS Ser-653-Asn, and PPO ΔGly210.
Scaling was performed by dividing the raw age (based on an assumed Ne = 500,000) by 166.5894 (corresponding to the geometric mean contemporary Ne estimate of 83,294,700).
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig3-data2-v2.csv
Figure 4 with 4 supplements
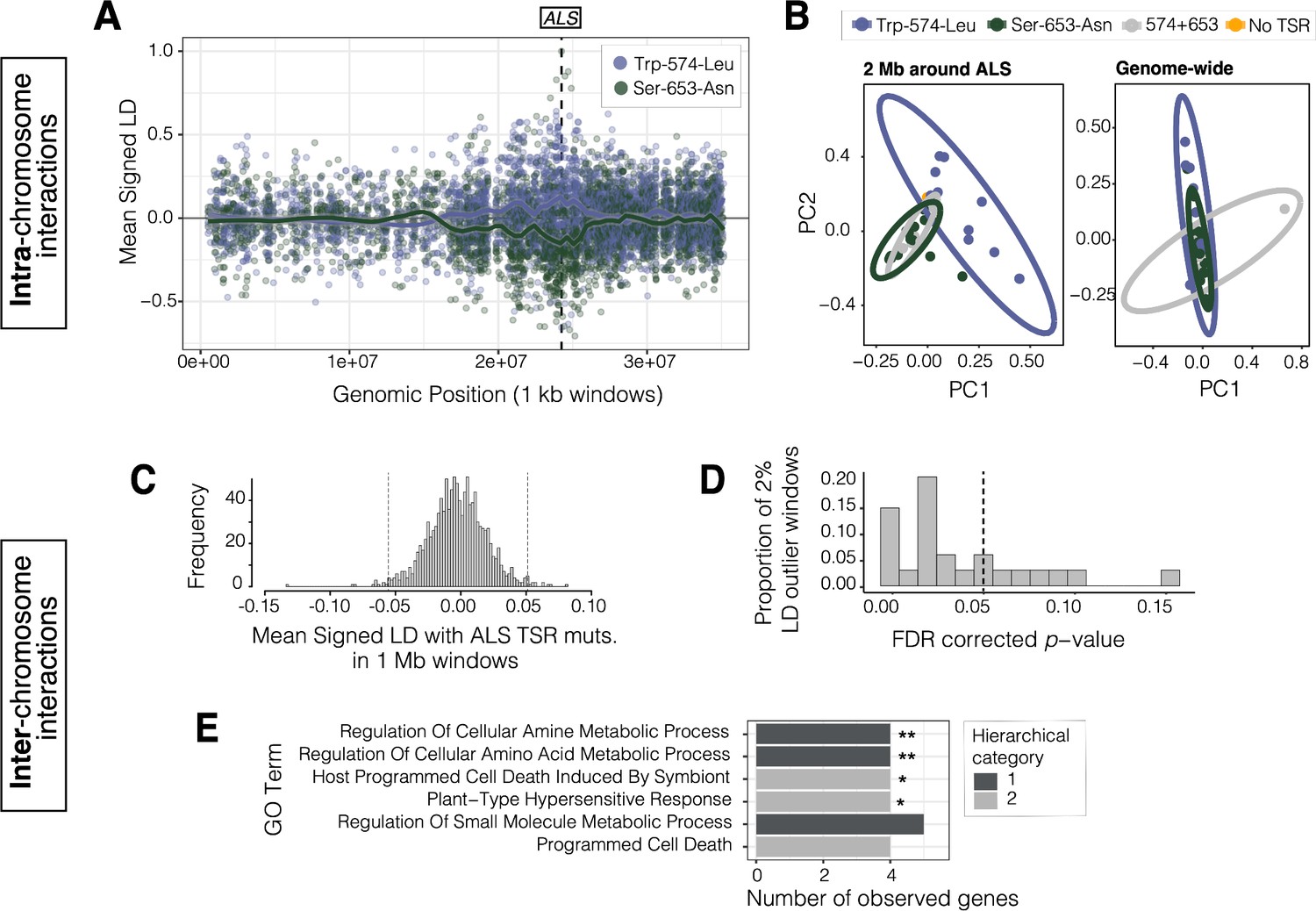
Signals of intra- and inter-chromosomal allelic interactions with target-site resistance mutations.
(A) The breadth of haplotype competition between TSR mutations, illustrated by repulsion linkage disequilibrium (opposite signed LD, r) between two target-site-resistance mutations and bi-allelic missense SNPs surrounding them on Scaffold 11 in Essex. Each point shows mean LD in non-overlapping 10 kb windows. A smoothing spline shows that missense SNPs tend to be in positive LD with ALS Trp-574-Leu but negative LD with ALS Ser-653-Asn in Essex. (B) Signatures of population structure for 2 Mb around ALS compared to genome-wide, based on PCAs of genotypes in Essex. Ellipses represent 95% CIs assuming a multivariate distribution. (C) Distribution of mean signed LD of ALS TSR resistance mutations (ALS 574 or 653) with 1 Mb windows genome-wide in Essex, excluding the ALS containing Scaffold 11. Upper and lower 1% quantile indicated by dashed vertical lines. (D) Distribution of p-values from top 2% of genome-wide windows with high absolute signed LD with ALS TSR mutations, from permuting individual assignment within genomic windows and recalculating LD 1,000 times. (E) GO terms significantly enriched for biological process after FDR correction from the set of 348 genes mapping to the top 13, 1 Mb windows that show significantly extreme LD with ALS TSR mutations in Essex. Number of asterisks represent significance level after bonferroni correction (** = p < 0.01, * = p < 0.05).
-
Figure 4—source data 1
Signed LD in 1 kb windows with two target-site-resistance mutations and bi-allelic missense SNPs surrounding them on Scaffold 11 in Essex.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig4-data1-v2.txt
-
Figure 4—source data 2
Local PCA at 2 Mb around ALS, along with a genome-wide PCA (excluding the ALS containing scaffold 11), in Essex.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig4-data2-v2.zip
-
Figure 4—source data 3
Signed LD of 1 Mb windows across the genome (aside from scaffold 11) with either ALS Trp-574-Leu and ALS Ser-653-Asn.
- https://cdn.elifesciences.org/articles/70242/elife-70242-fig4-data3-v2.txt
Figure 4—figure supplement 1
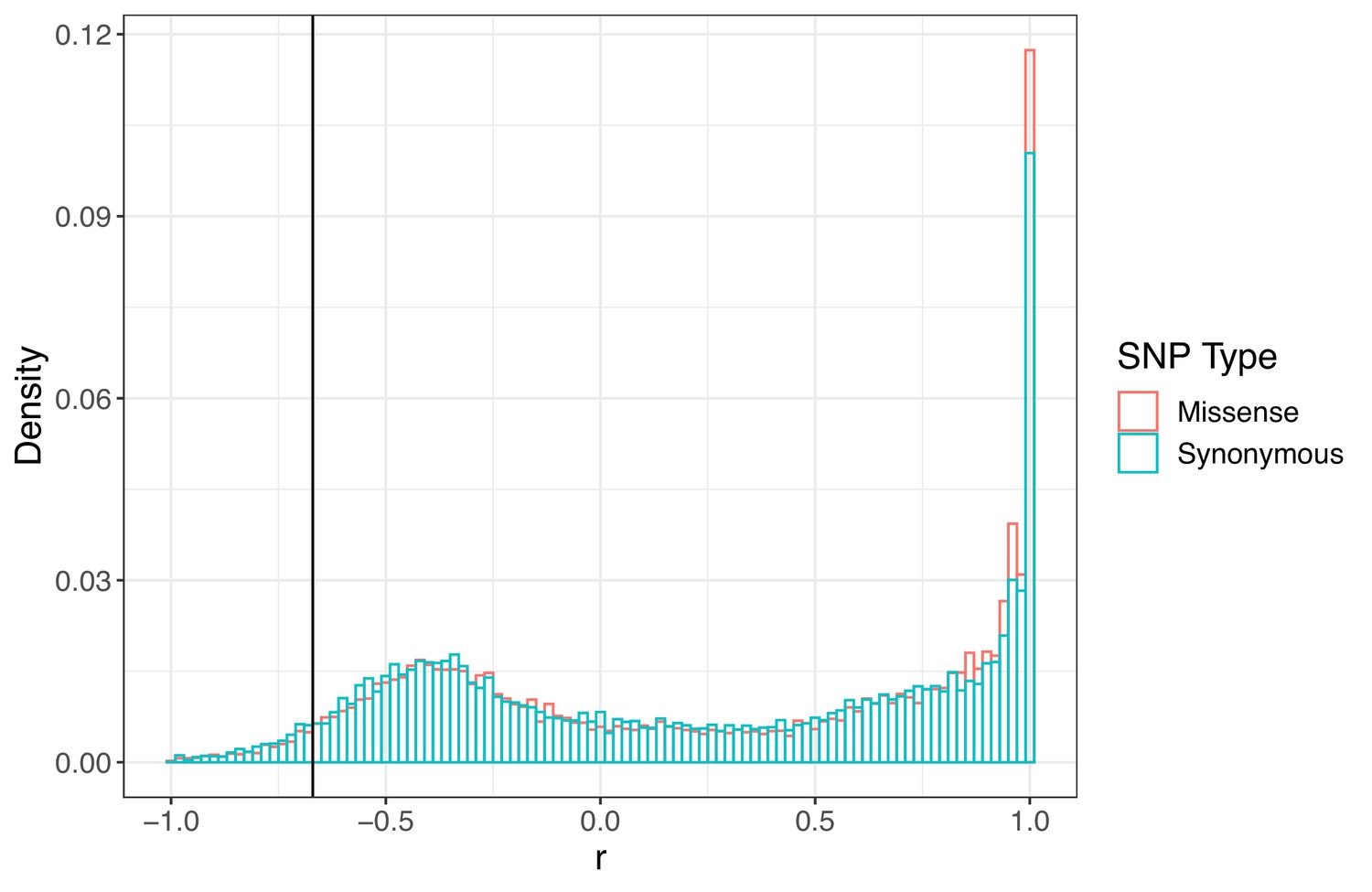
Signed LD (r) between pairs of missense (nonsynonymous) and synonymous SNPs of similar frequency (minor allele frequency > 0.20) and physical distance ( < 500 bp) as ALS Trp-574-Leu and ALS Ser-653-Asn across the genome.
Observed signed LD between ALS Trp-574-Leu and ALS Ser-653-Asn in Essex indicated by the vertical line (r = −0.69).
Figure 4—figure supplement 2
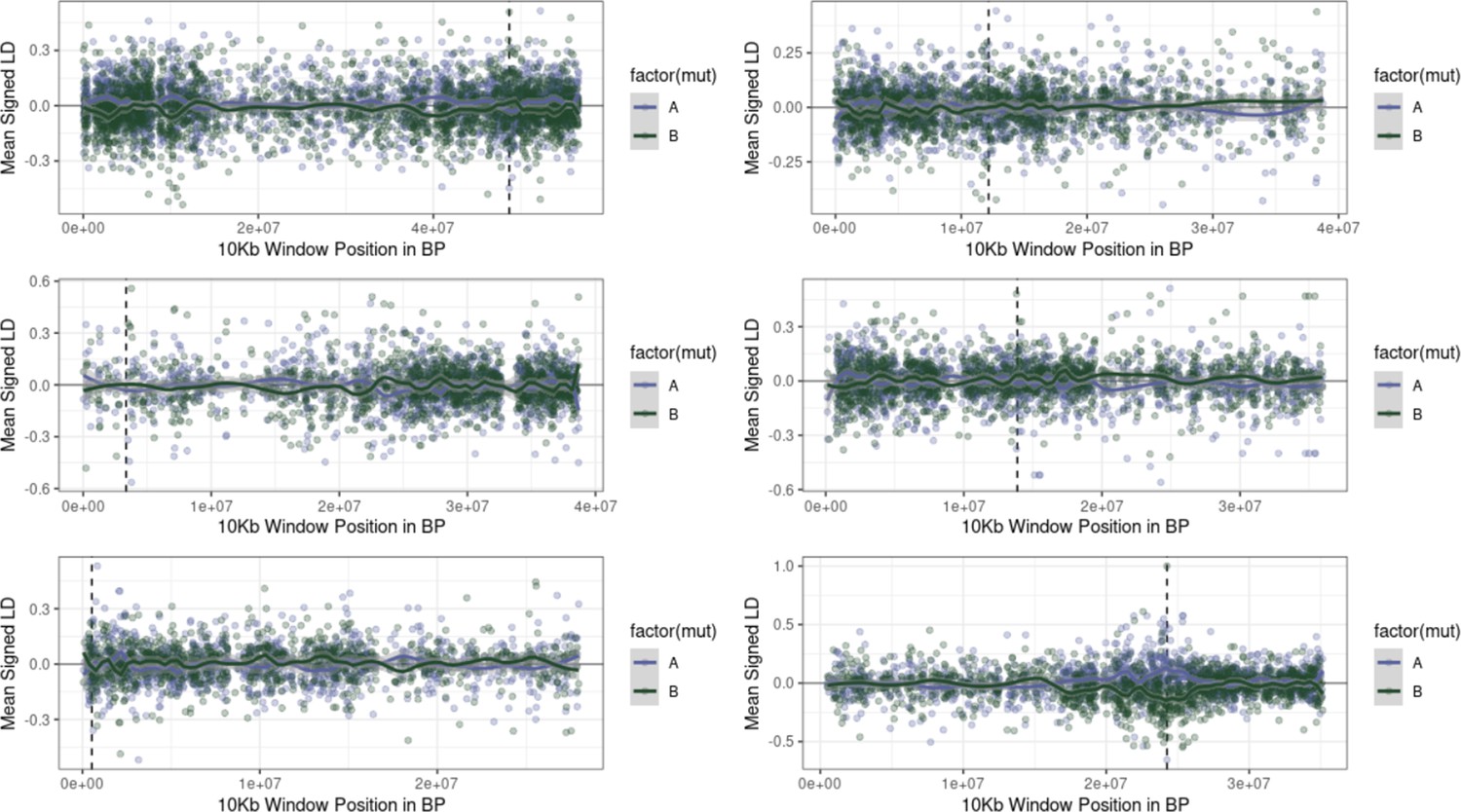
Examples of the extent of extended repulsion elsewhere across the genome, for pairwise comparisons in Figure 4—figure supplement 1 that were more extreme than observed.
The first five Figures show the limited breadth of repulsion associated with these pairwise comparisons, in contrast to the extended repulsion between ALS Trp-574-Leu and ALS Ser-653-Asn (bottom right).
Figure 4—figure supplement 3
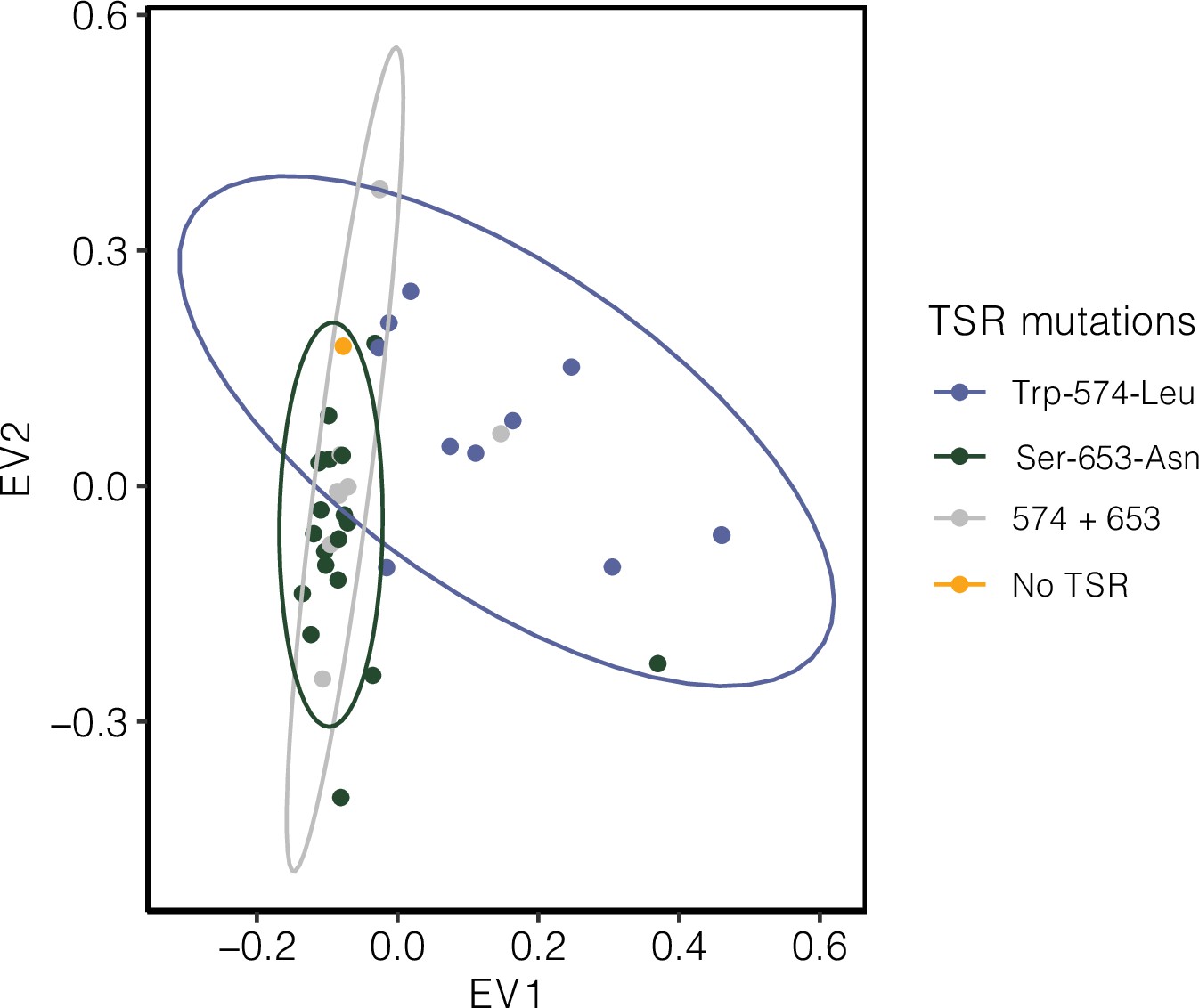
Signature of population structure for 10 Mb around ALS based on PCAs of genotypes in Essex.
Ellipses represent 95% CIs assuming a multivariate distribution.
Figure 4—figure supplement 4
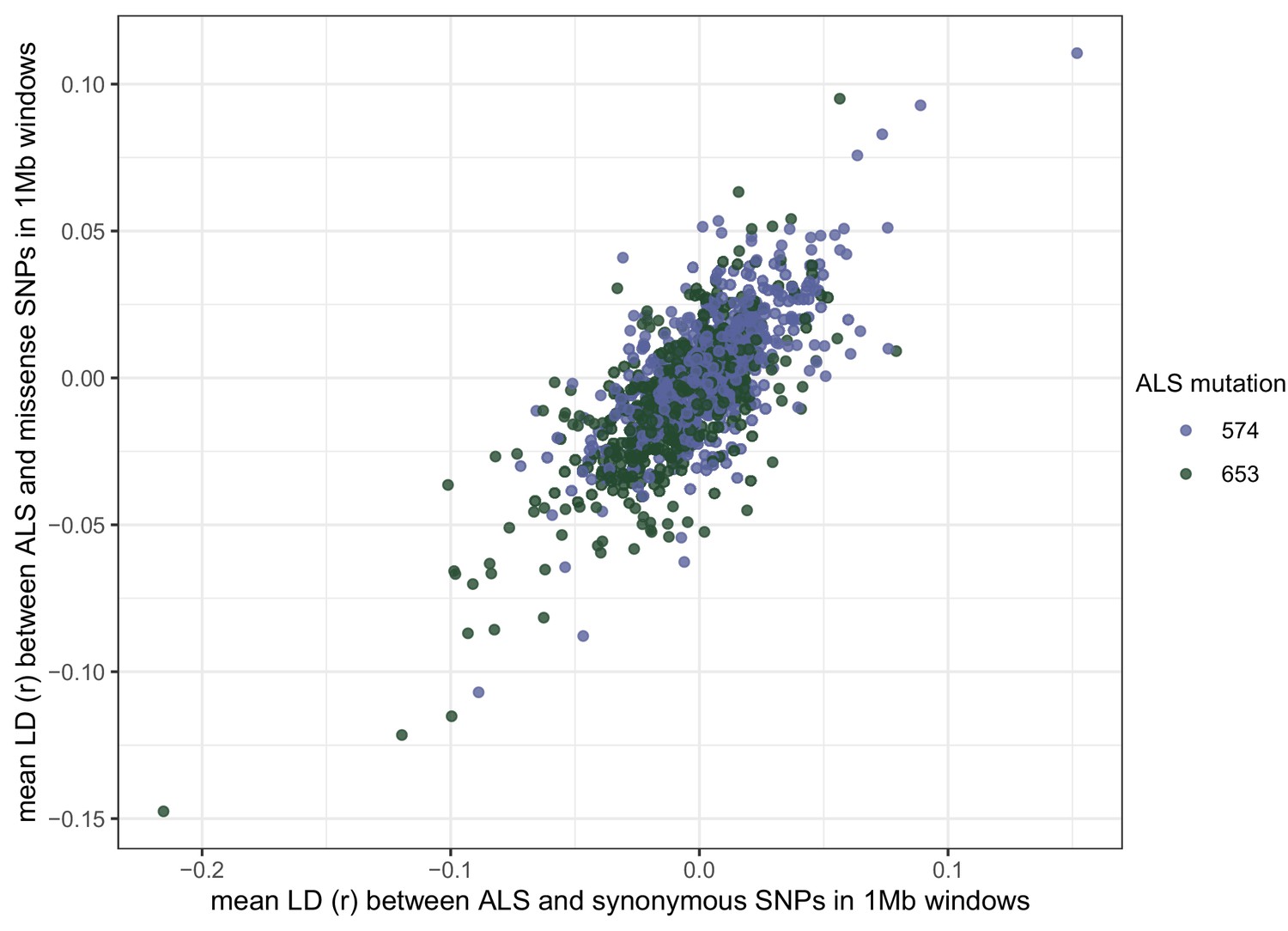
Correlation of signed LD (r) between two target-site resistance mutations, and synonymous or missense mutations across the genome.
Each point represents the mean r between all synonymous or missense mutations and an ALS mutation in a non-overlapping 1 Mb window.
Tables
Table 1
Number and frequency of resistant individuals and mutations at loci known to be causal for resistance to PPO and ALS herbicides, both totals, and within each agricultural region.
Relative frequencies given in parentheses.
| PPO | ALS | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔGly210 | Trp-574-Leu | Ser-653-Asn | Ser-653-Thr | Gly-654-Phe | Pro-197-Leu | Pro-197-His | Ala-122-Ser | Asp-376-Glu | |
| Total number of individuals with TSR mutations | 22 (0.145) | 80(0.526) | 48 (0.316) | 2(0.013) | 1(0.007) | 1(0.007) | 1(0.007) | 2(0.013) | 3(0.020) |
| Total number ofTSR mutations | 25 (0.082) | 106 (0.349) | 59 (0.194) | 2(0.007) | 1(0.003) | 1(0.003) | 1(0.003) | 2(0.007) | 3(0.010) |
| TSR mutations in Walpole, Ontario, Canada | 0 | 12(0.162) | 12(0.162) | 1(0.0135) | 0 | 0 | 0 | 0 | 0 |
| TSR mutations in Essex, Ontario, Canada | 1(0.0125) | 23(0.288) | 35(0.438) | 0 | 1(0.0125) | 0 | 1(0.0125) | 2(0.025) | 3(0.0375) |
| TSR mutations in Midwestern US | 24(0.16) | 71(0.47) | 12(0.08) | 0 | 0 | 1(0.0067) | 0 | 0 | 0 |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Repeated origins, widespread gene flow, and allelic interactions of target-site herbicide resistance mutations
eLife 11:e70242.
https://doi.org/10.7554/eLife.70242




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}