The transcription factor Xrp1 is required for PERK-mediated antioxidant gene induction in Drosophila
- NYU Grossman School of Medicine, United States
Abstract
PERK is an endoplasmic reticulum (ER) transmembrane sensor that phosphorylates eIF2α to initiate the Unfolded Protein Response (UPR). eIF2α phosphorylation promotes stress-responsive gene expression most notably through the transcription factor ATF4 that contains a regulatory 5’ leader. Possible PERK effectors other than ATF4 remain poorly understood. Here, we report that the bZIP transcription factor Xrp1 is required for ATF4-independent PERK signaling. Cell-type-specific gene expression profiling in Drosophila indicated that delta-family glutathione-S-transferases (gstD) are prominently induced by the UPR-activating transgene Rh1G69D. Perk was necessary and sufficient for such gstD induction, but ATF4 was not required. Instead, Perk and other regulators of eIF2α phosphorylation regulated Xrp1 protein levels to induce gstDs. The Xrp1 5’ leader has a conserved upstream Open Reading Frame (uORF) analogous to those that regulate ATF4 translation. The gstD-GFP reporter induction required putative Xrp1 binding sites. These results indicate that antioxidant genes are highly induced by a previously unrecognized UPR signaling axis consisting of PERK and Xrp1.
Introduction
The endoplasmic reticulum (ER) is the site where most membrane and secretory proteins undergo folding and maturation. This organelle contains an elaborate network of chaperones, redox buffers, and signaling mediators, which work together to maintain ER homeostasis. When the amount of misfolded or nascent proteins exceeds the folding capacity of a given cell, the ER initiates a gene expression regulatory program that is referred to as the Unfolded Protein Response (UPR) (Karagöz et al., 2019; Walter and Ron, 2011).
The ER also represents an important nexus between protein folding and oxidative stress. The ER maintains an oxidizing environment for the formation of intra- and intermolecular disulfide bonds that contribute to the oxidative folding of client proteins. A product of this reaction is hydrogen peroxide (Gross et al., 2006; Tu and Weissman, 2004; Tu and Weissman, 2002), and excessive protein misfolding in the ER can cause the accumulation of reactive oxygen species (ROS). Consistently, genes involved in redox homeostasis are induced in response to ER stress (Cullinan et al., 2003; Harding et al., 2003; Malhotra et al., 2008; Mukaigasa et al., 2018).
In metazoans, there are three evolutionarily conserved branches of the UPR initiated by the ER transmembrane proteins IRE1, PERK (PKR-like ER Kinase, also known as Pancreatic ER Kinase (PEK)), and ATF6 (Walter and Ron, 2011). The best studied downstream effectors of IRE1 and PERK signaling are the bZIP family transcription factors XBP1 and ATF4, respectively. Once activated in response to ER stress, these transcription factors induce the expression of genes involved in ER quality control, antioxidant response, and amino acid transport (Han et al., 2013; Harding et al., 2003; Walter and Ron, 2011). The Drosophila genome encodes mediators of all three branches of the UPR, and the roles of the IRE1-XBP1 and PERK-ATF4 branches in Drosophila development and tissue homeostasis have been established (Mitra and Ryoo, 2019; Ryoo, 2015).
The PERK branch of UPR draws considerable interest in part because its abnormal regulation underlies many metabolic and neurodegenerative diseases (Delépine et al., 2000; Ma et al., 2013; Pennuto et al., 2008). Stress-activated PERK is best known to initiate downstream signaling by phospho-inhibiting the translation initiation factor eIF2α (Harding et al., 1999; Shi et al., 1998). While most mRNA translation becomes attenuated under these conditions, ATF4 protein synthesis increases to mediate a signaling response. Such ATF4 induction requires ATF4’s regulatory 5’ leader sequence that has an upstream Open Reading Frame (uORF) that overlaps with the main ORF in a different reading frame. This overlapping uORF interferes with the main ORF translation in unstressed cells. (Harding et al., 2000; Kang et al., 2015; Vattem and Wek, 2004). But eIF2α phosphorylation causes the scanning ribosomes to bypass this uORF, ultimately allowing the translation of the main ORF assisted by the noncanonical translation initiation factors eIF2D and DENR (Bohlen et al., 2020; Vasudevan et al., 2020). The literature also reports PERK effectors that may be independent of ATF4. These include a small number of factors that are translationally induced in parallel to ATF4 in stressed mammalian cells (Andreev et al., 2015; Baird et al., 2014; Palam et al., 2011; Zhou et al., 2008). Compared to the ATF4 axis, the roles of these ATF4-independent PERK effectors remain poorly understood.
Here, we report that a previously uncharacterized UPR signaling axis is required for the expression of the most significantly induced UPR targets in the larval eye disc of Drosophila melanogaster. Specifically, glutathione-S-transferases (gstDs) were among the most significantly induced UPR target genes in Drosophila. We further show that such gstD induction was dependent on Perk, but did not require crc, the Drosophila ortholog of ATF4. Instead, this response required Xrp1, which encodes a bZIP transcription factor with no previously established connections to the UPR. Together, these findings suggest that PERK-Xrp1 forms a previously unrecognized signaling axis that mediates the induction of the most highly upregulated UPR targets in Drosophila.
Results
RNA-sequencing reveals upregulation of antioxidant genes in response to ER stress
The Drosophila eye imaginal disc has been used as a platform to study ER stress responses (Kang and Ryoo, 2009; Kang et al., 2012; Ryoo et al., 2007), but the global transcriptional changes associated with ER stress have not been thoroughly investigated in this context. To probe these changes in an unbiased approach, we performed RNA-seq on control and ER-stressed third instar eye imaginal discs. To genetically impose ER stress in this tissue, we misexpressed a mutant allele of Rhodopsin-1 (Rh1), a multipass membrane protein that is synthesized in the ER (O’Tousa et al., 1985; Zuker et al., 1985). The mutant allele Rh1G69D has been used as a potent genetic inducer of ER stress and UPR signaling (Colley et al., 1995; Kang and Ryoo, 2009; Kurada and O’Tousa, 1995; Ryoo et al., 2007). We chose to drive the expression of Rh1G69D in eye discs posterior to the morphogenetic furrow using Gal4 driven by the synthetic Glass Multiple Repeat promoter (henceforth referred to as GMR > Rh1G69D) (Brand and Perrimon, 1993; Freeman, 1996). To profile gene expression specifically in these cells, we simultaneously drove the expression of nuclear envelope-localized EGFP::Msp300KASH (Hall et al., 2017; Ma and Weake, 2014), enabling us to isolate only EGFP-labeled nuclei for further analysis (Figure 1A) (see Materials and methods). We confirmed successful enrichment of EGFP expressing nuclei through qRT-PCR and imaging (Figure 1B and C), and prepared total RNA from them for RNA-seq. The resulting RNA-seq data was subjected to differential expression analysis to identify genes induced by ER stress (Supplementary file 1). The full results of our RNA-seq experiment are available through the Gene Expression Omnibus (GEO; accession number GSE150058).
Figure 1 with 1 supplement see all
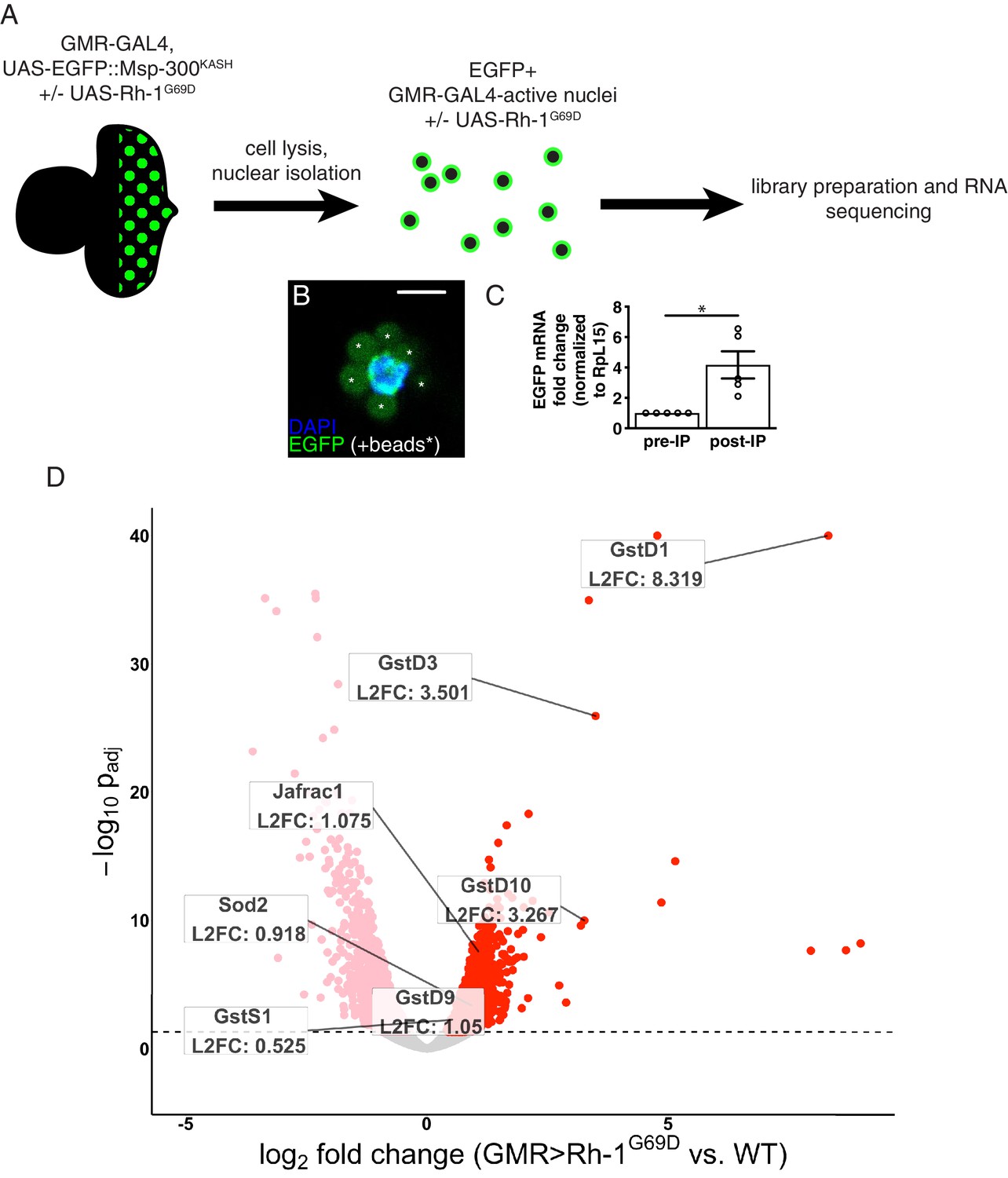
RNA-sequencing reveals upregulation of antioxidant genes in response to ER stress.
(A) Workflow for isolation of tagged nuclei from eye imaginal discs and preparation of total RNA for transcriptional profiling. (B) A representative image of an isolated, EGFP-tagged nucleus. Asterisks label auto-fluorescent anti-EGFP-coupled beads. Scale bar = 5 µm. (C) Representative qRT-PCR of pre- and post-isolation disc nuclei showing a significant enrichment of EGFP mRNA following isolation. The error bar represents standard error (SE). Statistical significance was assessed through a two tailed t-test. * = p < 0.05. (D) Volcano plot of genes differentially expressed in the presence or absence of Rh1G69D expression. Genes significantly up/downregulated in response to Rh1G69D are shown in red and pink, respectively. A select set of antioxidant genes induced by Rh1G69D are labeled with their log2 fold change values. Note: -log10 padj of gstD1 exceeded the bounds of the graph (180) and was constrained to the maximum displayed value of 40 for readability.
Gene Ontology (GO) term analysis of the RNA-seq data indicated that genes related to glutathione metabolism (p = 2.08*10–4) and cell redox homeostasis (p = 3.29*10–6) were noticeably enriched in GMR > Rh1G69D samples. Among this group, the most significantly upregulated gene was the delta family glutathione-S-transferase gstD1 (Figure 1D). Other notable genes induced by ER stress included the superoxide dismutase Sod2, the peroxiredoxin Jafrac1 as well as multiple other glutathione-S-transferase genes. These findings were suggestive of an antioxidant response. Consistently, we found that GMR > Rh1G69D discs showed signs of ROS accumulation. Specifically, the ROS sensitive reporter cyto-roGFP2-Orp1 that gains fluorescence upon ROS-initiated Orp1 oxidation (Albrecht et al., 2011) was activated in GMR > Rh1G69D but not in control eye imaginal discs (Figure 1—figure supplement 1). These observations prompted us to investigate the link between ER stress and antioxidant gene expression.
ER stress induces gstD-GFP expression in Drosophila imaginal discs
To validate our RNA-seq findings, we utilized a transgenic reporter of gstD1 expression, gstD-GFP, in which a regulatory DNA sequence between the gstD1 and gstD2 genes drives the expression of GFP (Figure 2A; Sykiotis and Bohmann, 2008). While control discs expressing lacZ (GMR > lacZ) showed no discernible gstD-GFP expression in the posterior eye disc (Figure 2B and D), we observed marked induction of gstD-GFP in GMR > Rh1G69D discs (Figure 2C and D), in agreement with our RNA-seq results.
Figure 2 with 3 supplements see all
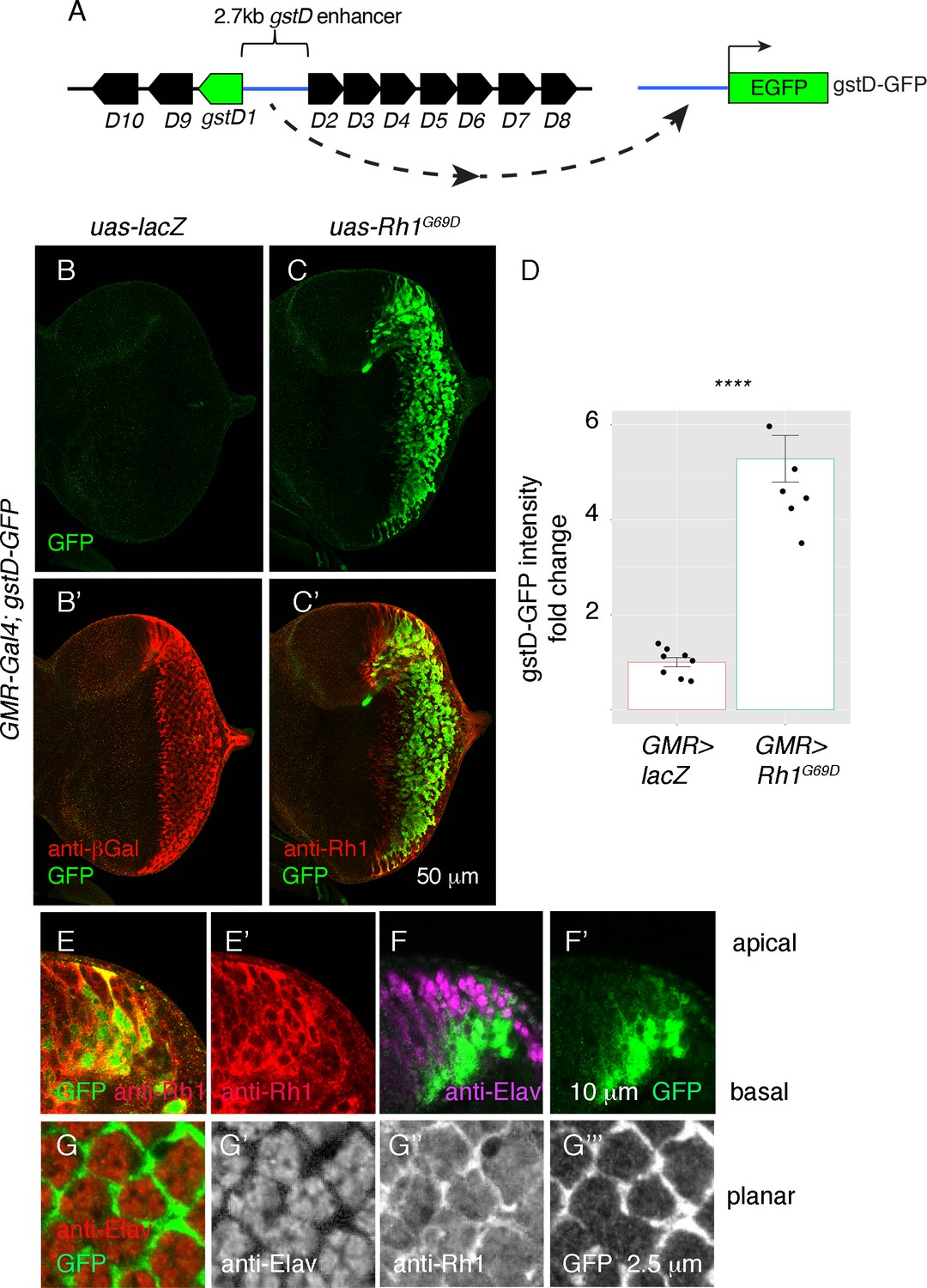
ER stress induces gstD-GFP expression in the eye imaginal disc.
(A) Schematic of the gstD-GFP reporter and the gstD locus, which encodes multiple gstD isozymes in close proximity to the 2.7 kb gstD1/gstD2 intergenic enhancer (scale is approximate). (B–C) Planar views of gstD-GFP larval eye discs also expressing either lacZ or Rh1G69D driven by GMR-Gal4. (B’, C’) show merged images of discs with gstD-GFP expression in green, and Rh1 or lacZ in red. (B, C) show only the green channel. Anterior is to the left and posterior to the right. Scale bar = 50 μm. (D) Quantification of gstD-GFP pixel intensity fold change from eye discs with the indicated genotypes. Statistical significance based on t test (two tailed). **** = p < 0.0001. (E, F) A magnified view of GMR > Rh1G69D eye discs in apico-basal orientation. gstD-GFP signals are marked in green. (E) Posterior eye disc double labeled with anti-Rh1 (red). (F) An equivalent region labeled with anti-Elav antibody that marks photoreceptors (magenta). Scale bar = 10 μm (G) A magnified view of GMR > Rh1G69D eye discs in planar orientation with gstD-GFP (green) and anti-Elav labeling (red). Individual channels are shown separately in (G’ – G’’’). Scale bar = 2.5 µm.
Examination of the gstD-GFP reporter also allowed us to visualize the pattern of gene expression in response to Rh1G69D expression. Expression of GMR-Gal4 driven Rh1G69D expression was detected in all cell types of the posterior eye disc (Figure 2C and E), but among this population of cells, gstD-GFP expression occurred primarily in non-photoreceptor cells. This was evident in eye discs regions with an apical-to-basal orientation, where gstD-GFP signal was primarily detected in the basal support cell layer. GFP was not present in the apical photoreceptor layer marked by anti-Elav antibody labeling, even though those apical cells expressed Rh1G69D (Figure 2E and F). Confocal sections on the apical layer with photoreceptor cell bodies further confirmed that the gstD-GFP-positive signal was coming from non-photoreceptor cells (Figure 2G). We therefore conclude that gstD-GFP induction by ER stress occurs in a cell-type-specific manner.
To further validate that gstD-GFP is induced by ER stress, we treated imaginal discs with culture media containing dithiothreitol (DTT), which causes ER stress by interfering with oxidative protein folding. Most cells of the larval eye and wing imaginal discs did not express gstD-GFP under control conditions. However, 2 mM DTT treatment for 4 hr induced gstD-GFP expression in a large population of cells in both the eye and wing imaginal discs (Figure 2—figure supplement 1). Similar to the outcome of Rh1G69D overexpression, DTT treatment induced gstD-GFP primarily in the non-neuronal cell layer (Figure 2—figure supplement 1 A-D). gstD-GFP expression was also induced by a different ER stress-imposing chemical, the N-linked glycosylation inhibitor tunicamycin (Figure 2—figure supplement 1 G-H). These results further support the notion that gstDs are significant UPR targets in Drosophila tissues, with some degree of cell-type specificity.
ER stress-induced gstD-GFP expression does not require the antioxidant response factor cncC
The gstD-GFP reporter responds to known oxidative stressors, including hydrogen peroxide and the oxygen radical generator N,N′-dimethyl-4,4′-bipyridinium dichloride (paraquat) (Sykiotis and Bohmann, 2008). The transcription factor Cap’n’collar-C (cncC), an Nrf2 homolog, mediates this response in Drosophila (Hochmuth et al., 2011; Sykiotis and Bohmann, 2008). This led us to test whether cncC is required for ER stress-mediated gstD-GFP expression. Specifically, we generated mutant mitotic clones negatively marked with armadillo-lacZ in GMR > Rh1G69D eye discs. We examined two different mutant alleles of cnc; the vl110 allele which has a deletion that spans significant parts of the coding sequence (Mohler et al., 1995), and the k6 allele which has a nonsense mutation that specifically truncates longer ROS-responsive Cnc isoforms including CncC (Veraksa et al., 2000). Mutant mosaic clones of these cncC alleles failed to block the induction of gstD-GFP by GMR > Rh1G69D. Contrary to our expectations, cncC mutant clones showed enhanced gstD-GFP expression as compared to the neighboring wild-type cells (Figure 2—figure supplement 2 A-C). This enhanced gstD-GFP expression in the mutant clones could reflect increased proteostatic stress associated with cncC loss. Based on these data, we conclude that cncC is not required for gstD-GFP induction in response to ER stress.
Consistent with these genetic experiments, we also observed that transcriptional induction of gstDs in cultured Drosophila S2 cells was cncC-independent. When ER stress was pharmacologically induced in Drosophila S2 cells by tunicamycin, we observed robust transcriptional induction of gstD2, which shares the same 2.7 kb enhancer as gstD1 (Tang and Tu, 1994; Toung et al., 1993). Such induction of gstD2 was unaffected when cncC was knocked down in S2 cells prior to tunicamycin treatment (Figure 2—figure supplement 2 D, E). The knockdown efficiency of cnc as estimated through q-RT-PCR from these cells was 92.46%. As a control, we validated the known roles of cncC in antioxidant response by utilizing the oxidative stressor paraquat, which leads to transcriptional induction of gstD2 in S2 cells as well (Figure 2—figure supplement 2 E). Consistent with previous reports, induction of gstD2 in response to paraquat was blocked after cncC knockdown in S2 cells (Figure 2—figure supplement 2 E). These results further support the idea that cncC is not required for tunicamycin-induced gstD gene expression.
gstD-GFP induction by ER stress requires PERK that phosphorylates eIF2α
We next considered the three canonical branches of the UPR as candidate mediators of gstD-GFP induction by GMR > Rh1G69D. We found no evidence for the requirement of Ire1 or Atf6. Specifically, GMR > Rh1G69D eye discs with negatively marked Ire1 mutant mosaic clones still induced gstD-GFP. If anything, there appeared to be a general increase in gstD-GFP expression in Ire1 mutant clones (Figure 2—figure supplement 3 A), consistent with previous observations that impairment of the IRE1 axis of the UPR could cause a concomitant increase in the activity of the remaining UPR branches (Huang et al., 2017). Likewise, Atf6 mutant discs bearing a piggyBac insertion in the Atf6 coding sequence, Atf6LL07432, did not impair gstD-GFP induction by GMR > Rh1G69D (Figure 2—figure supplement 3 B, C).
We next tested whether gstD-GFP induction was reliant on Perk using the previously described Perke01744 mutant allele (Wang et al., 2015). In contrast to Atf6 mutant discs and Ire1 mutant clones, Perk loss-of-function mutant eye discs expressing GMR > Rh1G69D were unable to induce gstD-GFP (Figure 3A–C and E). Conversely, we overexpressed Perk in eye discs. We had previously shown that such overexpression of Perk is sufficient for its autoactivation, possibly by overwhelming the repressive capacity of BiP (Malzer et al., 2010). Accordingly, we found that overexpression of Perk was sufficient to induce gstD-GFP expression (Figure 3D and E).
Figure 3 with 1 supplement see all

Perk is necessary and sufficient to induce gstD-GFP expression.
(A–D) Representative eye imaginal discs with gstD-GFP (green) alone channels (A–D), and gstD-GFP (green) channels merged with anti-Rh1 (red) (A’-D’). The genotypes of the discs are as follows: (A) GMR-Gal4; gstD-GFP/+, (B) GMR-Gal4; gstD-GFP/UAS-Rh1G69D, (C) GMR-Gal4; gstD-GFP/UAS-Rh1G69D; Perke01744 (D) GMR-Gal4; gstD-GFP/UAS-Perk. (E) Quantification of gstD-GFP pixel intensity fold change from posterior eye discs of the indicated genotypes from A-D. (F) qRT-PCR of gstD2 normalized to RpL15 in S2 cells treated with 10 ug/mL tunicamycin for 0, 4, or 8 hr. Cells were either pre-treated with control dsRNA (GFP) or those targeting Perk. (G–I) gstD-GFP (green) containing GMR > Rh1G69D eye discs with either control mosaic clones (G), or those with Perk loss-of-function clones (H, I). Homozygous Perk e01744 clones in H’ and I are marked by the absence of β-galactosidase (red) expression. Note that gstD-GFP expression occurs broadly in the background of control mosaic clones, but is largely absent in Perk homozygous mutant clones. (I, I’) A magnified view of the dotted inset in (H’). (I’) β-galactosidase only channel (marking Perk-positive cells; in white). Scale bar = 50 μm. * = p < 0.05, ** = p < 0.005, *** = p < 0.001.
PERK is best known to phosphorylate eIF2α in response to ER stress (Figure 3—figure supplement 1 A). To test if such kinase activity is required for gstD-GFP induction, we expressed a Perk transgene with a mutation that disrupts its kinase activity (Malzer et al., 2010). While wild-type Perk expression robustly induced gstD-GFP, the kinase dead Perk (PerkKD) transgene failed to induce the reporter under otherwise equivalent conditions (Figure 3—figure supplement 1 B, C). To further test if eIF2α phosphorylation causes the induction of gstD-GFP, we employed an RNAi line against gadd34 (Malzer et al., 2010). gadd34 encodes a phosphatase subunit that helps to dephosphorylate eIF2α, and therefore, the loss of gadd34 increases phospho-eIF2α levels (Figure 3—figure supplement 1 A). We found that knockdown of gadd34 using the eye specific GMR-Gal4 and ey-Gal4 drivers induces gstD-GFP in posterior eye discs (Figure 3—figure supplement 1 D, E). A striking induction of gstD-GFP was also observed when gadd34 was knocked down in the wing disc posterior compartment using the hh-Gal4 driver (Figure 3—figure supplement 1 F, G). These results support the idea that PERK-mediated phosphorylation of eIF2α promotes gstD-GFP induction across many tissue types.
To independently validate the role of Perk, we turned to S2 culture cells where tunicamycin (Tm) treatment induces gstD2 transcripts as detected through q-RT PCR. Perk dsRNA treatment resulted in an 82.7% reduction in Perk transcripts as assessed by q-RT-PCR. Knockdown of Perk in this setup blunted the induction of gstD2 (Figure 3F).
To further test if Perk has a cell-autonomous role in gstD-GFP induction, we examined eye discs with Perk loss-of-function mosaic clones. Whereas GMR> Rh1G69D robustly induced gstD-GFP in control mosaic clones without the Perk mutation (Figure 3G), the gstD-GFP signal was largely lost in eye discs with Perk homozygous mutant clones (Figure 3H, I). The small number of the residual GFP signals came from Perk + mosaic clones, indicating that Perk’s role in gstD-GFP expression is cell-autonomous (Figure 3I). Together, these results independently support Perk’s role in gstD induction in ER-stressed cells.
gstD-GFP induction does not require crc
As ATF4 is the best-characterized downstream effector of PERK in Drosophila and mammals (Mitra and Ryoo, 2019; Ryoo, 2015; Walter and Ron, 2011; Figure 4A), we examined whether the Drosophila ATF4 ortholog crc is required for gstD-GFP induction in GMR > Rh1G69D discs. For this, we employed the crc1 allele, a strong hypomorph bearing a missense mutation in the crc coding sequence (Hewes et al., 2000). To our surprise, loss of crc did not impair gstD-GFP induction in the eye discs (Figure 4B’ and C’). Rh1G69D expression levels were similar in discs with or without crc (Figure 4B’’ and C’’). As a positive control to validate crc activation, we used Thor-lacZ, a lacZ-based enhancer trap inserted upstream of the crc target gene Thor (Bernal and Kimbrell, 2000; Kang et al., 2017; Figure 4A). Consistent with previously published results, Thor-lacZ was induced in response to GMR > Rh1G69D and such induction was suppressed in crc1 homozygotes (Figure 4B and C). In an S2 cell-based assay, knockdown of crc did not impair gstD2 induction in response to tunicamycin treatment (Figure 4D), but did block the induction of Thor (Figure 4E). Consistently, overexpression of crc using UAS-crcleaderless (Vasudevan et al., 2020) also did not induce gstD-GFP expression (Figure 4G). We confirmed that the uas-crcleaderless transgene is functional, as its expression was sufficient to induce Thor-lacZ reporter expression in eye discs (Figure 4H, I). These data strongly indicate that the induction of gstDs in response to ER stress is dependent on Perk, but not crc.
Figure 4
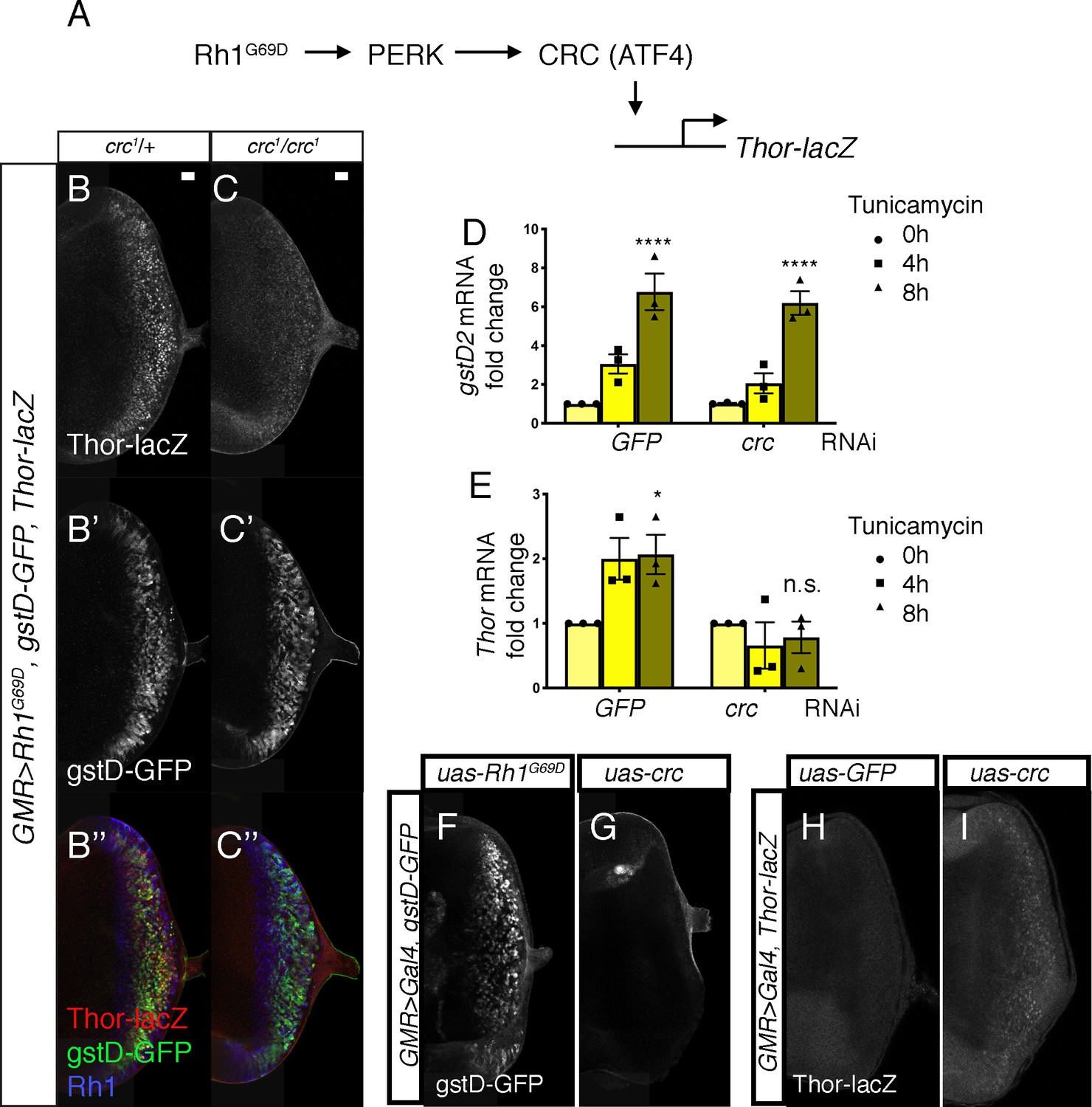
PERK-mediated gstD expression is ATF4 (crc)-independent.
(A) Schematic diagram of the Drosophila PERK-ATF4 signaling pathway. CRC is the Drosophila ATF4 ortholog, and Thor-lacZ is a CRC transcriptional target. (B, C) Eye imaginal discs expressing GMR > Rh1G69D in either the crc1/+ control (B) or in the crc1 homozygous backgrounds (C). These discs also have Thor-lacZ that reports CRC activity (B, C, in white), and gstD-GFP (B’, C’, in white). (B’’, C’’) Images of discs with Thor-lacZ (red), gstD-GFP (green) and anti-Rh1 (blue) channels merged. Note that crc1 homozygous mutants block Thor-lacZ expression (C), but still allows gstD-GFP expression in Rh1G69D expressing cells (C’, C’’). (D, E) qRT-PCR analysis of gstD2 and Thor normalized to Rpl15 in S2 cells challenged with 10 µg/mL tunicamycin for 0, 4, or 8 hr. Cells were either treated with control dsRNA (GFP) or that target crc. (F, G) gstD-GFP expression (white) in eye discs that are overexpressing either Rh1G69D (F) or crcleaderless (G) through the GMR-Gal4 driver. (H, I) Thor-lacZ expression (white) in eye discs overexpressing either a control GFP transgene (H), or crcleaderless (I) through the GMR-Gal4 driver. Scale bar = 20 μm. * = p < 0.05, **** = p < 0.0001. Genotypes: (B) GMR-Gal4; gstD-GFP, crc1/UAS-Rh1G69D, Thor-lacZ. (C) GMR-Gal4; gstD-GFP, crc1/crc1, UAS-Rh1G69D, Thor-lacZ. (F) GMR-Gal4; gstD-GFP/UAS-Rh1G69D. (G) GMR-Gal4; gstD-GFP/UAS-crcleaderless. (H) GMR-Gal4; Thor-lacZ/UAS-GFP. (I) GMR-Gal4; Thor-lacZ/UAS-crcleaderless.
The induction of gstD genes and other antioxidants in response to ER stress require the bZIP transcription factor Xrp1
We next sought to determine how PERK activation induced the expression of gstD-GFP independently of crc, and turned our focus to another stress response transcription factor, Xrp1. Although Xrp1 has no known association with the UPR, Xrp1 drew our attention as it reportedly mediates the induction of the gstD1-lacZ reporter in response to ribosome mutations that cause cell competition (Ji et al., 2019).
To assess a possible role of Xrp1 in UPR signaling, we performed clonal analysis with an Xrp1 mutant allele, Xrp1M2-73, which bears a nonsense mutation truncating all Xrp1 isoforms prior to both the AT-hook motif and bZIP domain (Lee et al., 2018). Loss of Xrp1 had no effect on basal gstD-GFP expression levels (Figure 5A). GMR > Rh1G69D discs prominently expressed gstD-GFP, but otherwise equivalent eye discs with Xrp1 mutant clones had marked reduction of the gstD-GFP signal (Figure 5B and C). The small patches of GFP-positive cells in these discs were all within the Xrp1+ mosaic cells (Figure 5C’ D).
Figure 5
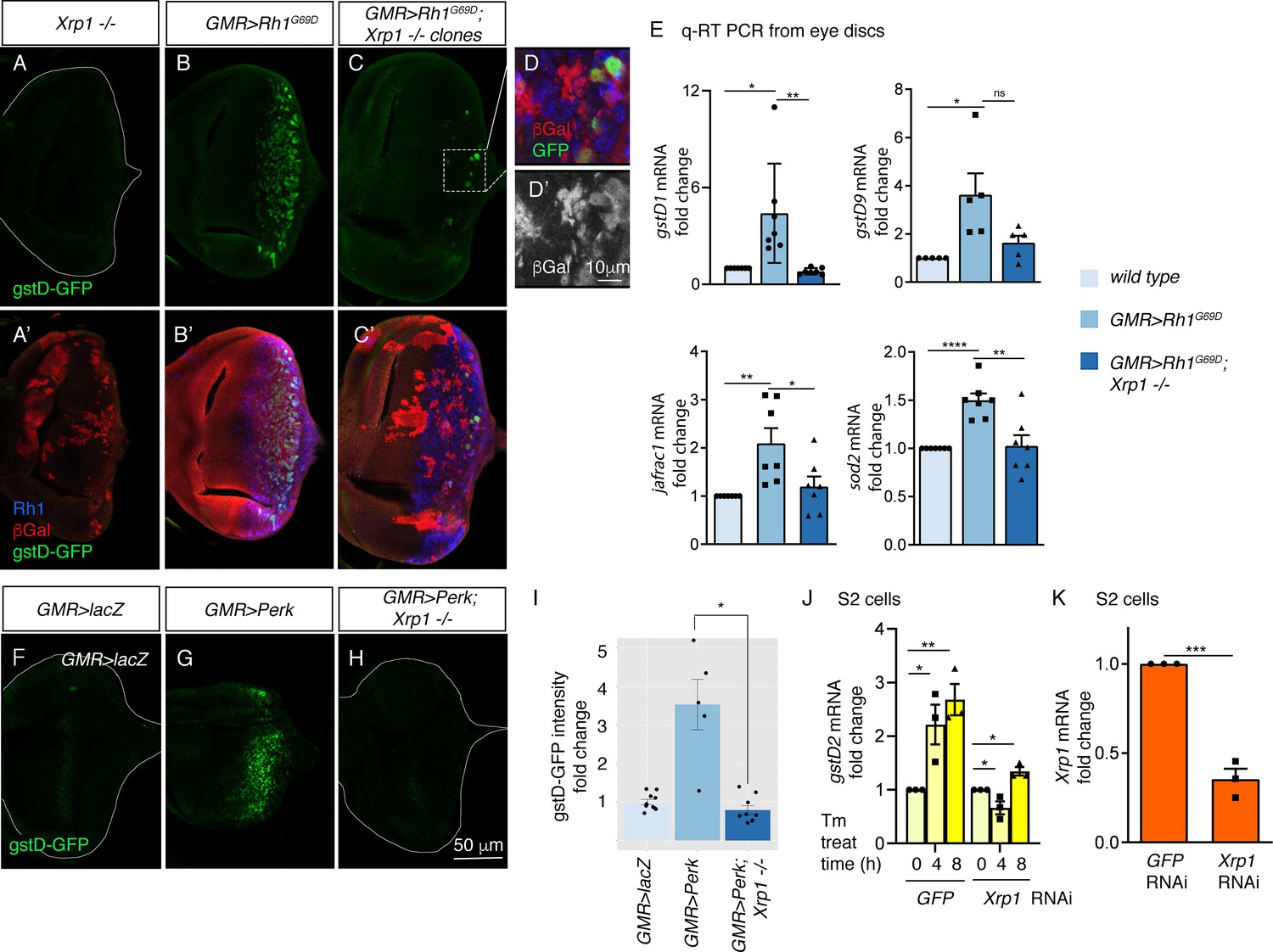
PERK regulates gstD gene expression through the bZIP transcription factor Xrp1.
(A–G) gstD-GFP expression (green) in eye discs. (A, A’) An eye disc with homozygous Xrp1M2-73 mutant clones that are marked by the absence of βGal (red). (B, B’) A GMR> Rh1G69D disc in a control genetic background. (C, C’) A GMR> Rh1G69D disc with Xrp1M2-73 loss-of-function mosaic clones (non-red). (A–C) gstD-GFP only channels. (A’-C’) Merged images with Rh1 is stained in blue and βgal is in red. Note the absence of gstD-GFP in Xrp1 mutant clones. (D, D’) Magnified view of the inset marked in C. The scale bar here represents 10 μm. (D’) is the βGal only channel of the image in (D). (E) Quantitative(q) RT-PCR results of indicated genes from dissected eye discs. The genotypes are color labeled. (F–H) Eye discs expressing control lacZ (E), Perk (F), or expressing Perk in the background of Xrp1M2-73 mosaic clones (G). (I) Quantification of gstD-GFP intensity fold change in the indicated genotypes. (J, K) q RT-PCR results from S2 cells. (J) gstD2 mRNA levels from S2 cells challenged with 10 µg/mL tunicamycin for 0, 4 or 8 hr. The cells were pretreated with dsRNA against either GFP (lanes 1–3) or Xrp1 (lanes 4–6). (K) Xrp1 mRNA levels in cells pretreated with dsRNA against GFP or Xrp1. All qRT-PCR results were normalized with Rpl15. Two tailed t tests were used to assess statistical significance. The scale bar of 50 μm applies to all images except for (D). * = p < 0.05, ** = p < 0.005, *** = p < 0.001. Genotypes: (A) GMR-Gal4, ey-FLP; gstD-GFP/+; FRT82, Xrp1M2-73/FRT82, arm-lacZ. (B) GMR-Gal4, ey-FLP; gstD-GFP/UAS-Rh1G69D. (C) GMR-Gal4, ey-FLP; gstD-GFP/UAS-Rh1G69D; FRT82, Xrp1M2-73/FRT82, arm-lacZ. (E) GMR-Gal4, ey-FLP; gstD-GFP/UAS-lacZ. (F) GMR-Gal4, ey-FLP; gstD-GFP/UAS-Rh1G69D. (G) GMR-Gal4, ey-FLP; gstD-GFP/UAS-Rh1G69D; FRT82, Xrp1M2-73/ FRT82, arm-lacZ.
To validate the findings with the gstD-GFP reporter, we assessed the mRNA levels of gstD1 and other select antioxidant genes from dissected eye imaginal discs through qRT-PCR. A subset of the analyzed transcripts showed induction in GMR > Rh1G69D disc samples dependent on Xrp1 (Figure 5E). These included gstD1, jafrac1 that encodes a cytoplasmic peroxidase and superoxide dismutase 2 (sod2) that detoxifies superoxides in the mitochondria.
The requirement of Xrp1 in gstD-GFP expression in response to GMR > Rh1G69D prompted us to further examine the epistatic relationship between Perk and Xrp1. We found that gstD-GFP induction caused by GMR > Perk was completely abolished in Xrp1 mutant eye discs (Figure 5F–I). Together, these data indicate that Xrp1 is epistatic to Perk in mediating gstD-GFP induction as part of UPR signaling.
We further confirmed the role of Xrp1 in cultured S2 cells. Tunicamycin treatment induced gstD2 significantly within 4 and 8 hr, but cells with Xrp1 knockdown did not show a statistically significant induction (Figure 5J and K). Together, these results independently support Xrp1’s role in gstD induction in ER-stressed cells.
Xrp1 protein is induced by Perk-dependent UPR
We next examined if ER stress induces Xrp1 expression through anti-Xrp1 immunohistochemistry. Control eye imaginal discs did not show obvious anti-Xrp1 signals beyond background (Figure 6A and A’), but GMR > Rh1G69D eye discs displayed nuclear anti-Xrp1 immunoreactivity in cells that expressed Rh1G69D (Figure 6B, B’ and D). Consistent with the role of Perk and eIF2α phosphorylation, Xrp1 protein induction by GMR > Rh1G69D was significantly reduced in the Perke01744 mutant background (Figure 6C, C’ and D). While the reduction was statistically significant (Figure 6D), the residual level of Xrp1 in the Perk mutant discs indicated a partial dependency of Xrp1 induction on Perk. Interestingly, we found that Xrp1 transcript levels did not change significantly in GMR > Rh1G69D discs as compared to controls, as assessed from our RNA-seq analysis (Supplementary file 1) and by qRT-PCR analysis (Figure 6E). These results indicate that Perk mediates the induction of Xrp1 protein through a post-transcriptional mechanism.
Figure 6
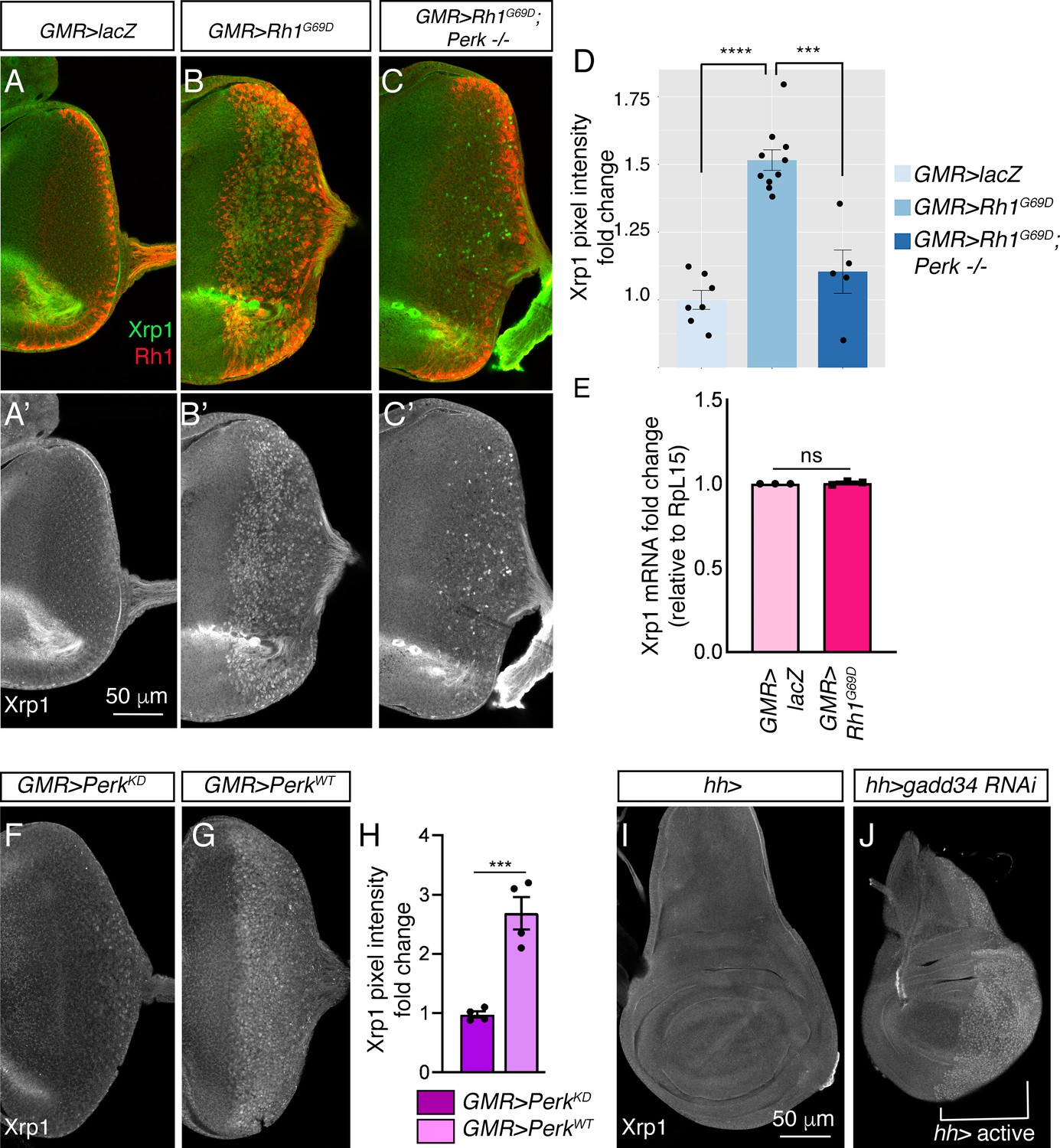
Xrp1 induction is regulated by Perk and eIF2α phosphorylation.
(A–C) Eye discs labeled with anti-Xrp1 (green) and anti-Rh1 (red). (A’-C’) Anti-Xrp1 single channels of images in (A–C). Genotypes: (A, A’) GMR-Gal4; UAS-lacZ/+. (B, B’) GMR-Gal4; UAS-Rh1G69D/+. (C, C’) GMR-Gal4; UAS-Rh1G69D/+; Perke01744. The scale bar represents 50 µm. (D) Quantification of anti-Xrp1 pixel intensities from eye discs in (A–D). (E) qRT-PCR analysis of Xrp1 from GMR > LacZ (control) and GMR > Rh1G69D eye discs. Xrp1 qRT-PCR results were normalized with that of Rpl15. (F, G) Anti-Xrp1 immunolabeling (white) in eye imaginal discs. Genotypes: (F) GMR-Gal4; uas-PerkKD. (G) GMR-Gal4; uas-PerkWT. (H) Quantification of anti-Xrp1 pixel intensities in (F, G). (I, J) Anti-Xrp1 immunolabeling in wing discs. Genotypes: (I) hh-Gal4/+. (J) uas-gadd34 RNAi/+; hh-Gal4. The white bracket indicates the posterior compartment where hh-Gal4 drives the transgene expression. In all graphs, two tailed t tests were used to assess statistical significance. *** = p < 0.0005, **** = p < 0.00005 and ns = non-significant, respectively.
To test if Perk’s kinase activity is involved in Xrp1 protein induction, we compared the effects of overexpressing PerkWT versus PerkKD (Kinase Dead). Xrp1 protein levels increased in GMR > PerkWT eye discs, but not in GMR > PerkKD discs (Figure 6F, G and H). To further test if PERK’s phosphorylation target eIF2α controls gstD-GFP expression, we knocked down gadd34 in wing discs using the posterior compartment-specific hh-Gal4 driver. While control wing discs showed a low basal anti-Xrp1 signal throughout the tissue, depletion of gadd34 by RNAi resulted in distinct nuclear anti-Xrp1 signals throughout the posterior compartment (Figure 6I and J). These results indicate that Xrp1 induction is regulated by eIF2α phosphorylation.
PERK’s well-established downstream effector, ATF4, has a 5’ regulatory leader sequence with multiple upstream Open Reading Frames (uORFs) that allows its translational induction in response to stress. To examine if there is an analogous 5’ leader in Xrp1, we used a bioinformatic program that predicts initiation codons (https://atgpr.dbcls.jp). This approach did not detect initiation codons in the 5’ leader of Xrp1’s shorter splice isoforms (e.g. isoform D and E). By contrast, two putative uORFs in the 5’ leader of the long splice isoforms of Xrp1 (e.g. isoform F and G) were identified, with uORF1 predicted to encode a 124 a.a. length peptide, and the uORF2 with a 288 a.a. peptide. The uORF2 overlapped with the main ORF, but was in a different reading frame (Figure 7A). Such an arrangement is similar to that of ATF4’s last uORF.
Figure 7

Predicted upstream Open Reading Frames (ORFs) in Xrp1.
(A) A schematic diagram of the predicted ORFs in D. melanogaster Xrp1. uORF1 and uORF2 in the 5’ leader have initiation start codons with Kozak sequences. uORF1 encodes a peptide of 124 amino acid residues. uORF2 encodes a peptide of 288 amino acid residues. uORF2 overlaps with the main Xrp1 ORF, but is in a different reading frame. (B) Amino acid sequence alignment between D. melanogaster Xrp1 uORF2, and the Xrp1 main ORFs from the species, D. persimili, D. navojoa, D. grimshawi. BoxShade was used for visualizing alignments, with those in black indicating sequence identity. The first 219 residues of the D. melanogaster Xrp1 uORF2 show high-sequence conservation with the main Xrp1 ORFs from other species. (C) Amino acid sequence alignment between the main ORFs of D. melanogaster and other Drosophila species. D. melanogaster’s Xrp1 main ORF shows high-sequence conservation with other Drosophila species beginning from the 140th amino acid residue.
To assess the likelihood that Xrp1 uORFs are peptide coding sequences, we performed Protein BLAST searches with the encoded peptide sequences. Xrp1’s uORF1 did not have any homologous hits in other species. However, sequences homologous to D. melanogaster Xrp1 uORF2 were identified in D. kikkawai (Percent identity = 71.85%), D. persimilis (Percent identity = 48.67%), D. navojoa (Percent identity = 50.44%), D. grimshawi (Percent identity = 49.48%), and D. busckii (Percent identity = 38.69%). The homologous sequences in these other species were part of their Xrp1 main ORF N-terminal regions (Figure 7B). The C-terminal regions of these main ORFs all encoded the AT-hook and bZIP DNA-binding domains homologous to that in the D. melanogaster’s Xrp1 main ORF (Figure 7C). The phylogenetic conservation of D. melanogaster uORF2 at the peptide level supports the idea that this is a functional uORF that has been under selective pressure during evolution. The similarities in the arrangements of the D. melanogaster Xrp1 and ATF4 5’ leaders, together with the observation that Xrp1 is induced in response to eIF2α phosphorylation, suggest that Xrp1 and ATF4 share similar mechanisms for their translational induction in response to stress.
Xrp1 binding sites within the gstD enhancer are essential for gstD-GFP induction
To determine if Xrp1 regulates gstDs directly, we examined the gstD 2.7 kb enhancer for putative Xrp1 and ATF4 binding sites (Figure 8A). To do so, we used the Xrp1 position frequency matrix derived from a previous Xrp1 ChIP-seq analysis (Baillon et al., 2018) and a publicly available ATF4 position frequency matrix (see Materials and methods). Our binding score analysis predicted three putative binding sites each for Xrp1 and ATF4 in the enhancer (Figure 8B and C). Two of the three highest scoring Xrp1 binding sites overlapped with two of the ATF4 sites, while one was predicted to be a unique Xrp1 site.
Figure 8

Xrp1 binding sites in the gstD enhancer are required for Rh1G69D -induced gstD-GFP expression.
(A) Sequence logos representing Xrp1 and ATF4 position frequency matrices. (B) Binding score analysis for Xrp1 and ATF4 sites in the gstD 2.7 kb enhancer region. Those that score high in the forward strand are depicted in green, whereas those in the reverse strand are shown in blue. The four putative binding sites are numbered below the graphs. (C) Putative binding site sequences. The site numbers match those whose positions are indicated in (B). (D) A schematic diagram of the CUT&RUN approach to assess Xrp1 binding to target DNA. When HA-tagged Xrp1 (light blue) binds to a specific DNA locus, that DNA can be recovered by adding anti-HA antibody (red) and pAG-MNase (dark blue) that cleaves adjacent DNA prior to immunoprecipitation. (E) Putative Xrp1 target gene enrichment after Xrp1HA protein pull down as assessed through q-PCR (using CUT&RUN). The y axis shows fold change increases in target gene DNA recovery in response to DTT treatment (normalized to q-PCR values from controls without DTT treatment). (F - J) Representative eye discs either expressing control lacZ (F) or those expressing GMR> Rh1G69D (G–I), with variants of the gstD-GFP reporter in green: gstD WT-GFP (F, G) gstD ΔATF4-GFP (H) and gstD Xrp1m-GFP (I). (G) shows a merged image with anti-Rh1 (red) and the gstD-GFP reporter (green). (G’, H, I) are gstD-GFP single channel images. (J) Quantification of gstD-GFP intensity fold change. Two tailed t tests were used to assess statistical significance. ** = p < 0.005, *** = p < 0.0005, **** = p < 0.00001, n.s. = not significant.
To test if Xrp1 binds to this locus, we used the CUT&RUN approach (Skene et al., 2018) to pull down HA-tagged Xrp1 proteins from Drosophila larval tissues to examine if putative Xrp1 target gene DNAs co-purify (Figure 8D, see Materials and methods). We specifically employed an Xrp1HA transgenic fly line that has an HA-tagged transgene inserted in the endogenous Xrp1 locus (Blanco et al., 2020). Larval tissues incubated with DTT gave strong q-RT-PCR signals for gstD regulatory DNA, while control tissues without DTT treatment had minimal DNA recovered. Upd3 was previously reported as an Xrp1 target gene, and as expected, increased Upd3 DNA was recovered in response to DTT treatment. Tubulin DNA was used as a negative control, which had minimal DNA recovery even after DTT treatment (Figure 8E). These results support the idea that Xrp1 binds to the gstD locus in larval cells under ER stress.
We then reconstructed new gstD-GFP reporters with (gstDWT-GFP) or without (gstD∆ATF4-GFP) the predicted ATF4-binding sites. To generate an Xrp1 binding site-deficient gstD-GFP reporter (gstDXrp1m-GFP), we introduced mutations in the remaining Xrp1 binding site within the gstD∆ATF4-GFP reporter. To control for genetic backgrounds, we targeted these reporters to a specific attP landing site to generate transgenic flies. While GMR > Rh1G69D effectively induced both the wild type and gstD∆ATF4-GFP reporters, gstDXrp1m-GFP was not induced under otherwise identical conditions (Figure 8F–J). Together, these results indicate that gstD genes are direct targets of Xrp1 in the context of ER stress.
Discussion
Here, we report that ER stress activates a previously unrecognized UPR axis mediated by PERK and Xrp1. Specifically, we showed that gstD family genes are among the most highly induced UPR targets in Drosophila, and that such induction requires Perk, one of the three established ER stress sensors in metazoans. Surprisingly, the induction of gstD genes in this context did not require crc, the ATF4 ortholog. Instead, we found that a poorly characterized transcription factor Xrp1 is induced downstream of Perk to promote the expression of gstDs and other antioxidant genes.
Our findings are surprising given that ATF4 is considered a major effector of PERK-mediated transcription response (Karagöz et al., 2019; Walter and Ron, 2011). ATF4 was the first PERK downstream transcription factor to be identified in part based on the similarity of its regulatory mechanisms with that of yeast GCN4 (Dever et al., 1992; Harding et al., 2000). But more recent studies have shown there could be parallel effectors downstream of PERK activation (Andreev et al., 2015; Baird et al., 2014; Palam et al., 2011; Zhou et al., 2008). The functional significance of these alternative factors had remained poorly understood. Our study here has led us to conclude that an ATF4-independent branch of PERK signaling is required for the expression of the most highly induced UPR target in Drosophila.
As a potential mediator of this ATF4-independent PERK signaling, we first considered cncC as a prime candidate for a few reasons: cncC is an established regulator of gstD-GFP induction (Sykiotis and Bohmann, 2008), and previous studies had reported that Nrf2 is activated by PERK in cultured mammalian cells and in zebrafish (Cullinan et al., 2003; Cullinan and Diehl, 2004; Mukaigasa et al., 2018). However, our results reported here do not support the simple idea that gstD-GFP is induced by CncC, which in turn is activated by PERK. Specifically, we found that the loss of Perk blocked gstD-GFP induction in this experimental setup, but the loss of cncC did not. While Nrf2/CncC clearly regulates antioxidant gene expression in response to paraquat, our results indicate that PERK mediates an independent antioxidant response in Drosophila.
Our data indicates that this ATF4-independent PERK signaling response requires the AT-hook bZIP transcription factor Xrp1. Several pieces of evidence support the idea that Xrp1 is translationally induced, analogous to the mechanism reported for ATF4 induction. First, our RNA-seq and qRT-PCR results indicate that Xrp1 transcript levels do not change significantly in Rh1G69D expressing eye discs. These results argue against the idea that Xrp1 is induced at the transcriptional level. Second, we find that PERK’s kinase domain is required for Xrp1 protein induction. Third, knockdown of gadd34, which increases phospho-eIF2α levels downstream of Perk, is sufficient to induce Xrp1 protein and gstD-GFP expression. Finally, we find that Xrp1’s 5’ leader has a uORF that overlaps with the main ORF, similar to what is found in ATF4’s regulatory 5’ leader sequence. Moreover, Xrp1’s uORF2 encodes a peptide sequence that is phylogenetically conserved in other Drosophila species. High-sequence conservation at the peptide level enhances confidence that uORF2 is a peptide coding sequence.
Xrp1 is known to respond to ionizing radiation, motor neuron-degeneration in a Drosophila model for amyotrophic lateral sclerosis (ALS), and in cell competition caused by Minute mutations that cause haplo-insufficiency of ribosomal protein genes (Akdemir et al., 2007; Baillon et al., 2018; Lee et al., 2018; Mallik et al., 2018). Interestingly, two recent studies reported that these Minute cells induce gstD-GFP, and also show signs of proteotoxic stress as evidenced by enhanced eIF2α phosphorylation (Baumgartner et al., 2021; Recasens-Alvarez et al., 2021). Although these studies did not examine the relationships between Xrp1, gstD-GFP and eIF2α kinases such as Perk, our findings make it plausible that the PERK-Xrp1 signaling axis regulates cell competition caused by Minute mutations.
Despite the rising levels of interest in Xrp1 as a stress response factor, the identity of its mammalian equivalent remains unresolved. Xrp1 is well conserved in the Dipteran insects, but neither NCBI Blast searches nor Hidden Markov Model-based analyses identify clear orthologs in other orders (Akdemir et al., 2007; Blanco et al., 2020; Mallik et al., 2018; Baillon et al., 2018). Such evolutionary divergence is not unprecedented in UPR signaling: GCN4 is considered a yeast equivalent of ATF4, but they are not the closest homologs in terms of their peptide sequences (Harding et al., 2000). Likewise, the yeast equivalent of XBP1 (IRE1 effector, not to be confused with Xrp1 in this study) is Hac1, but there is little sequence conservation between the two genes (Yoshida et al., 2001; Shen et al., 2001; Calfon et al., 2002). Yet, the UPR signaling mechanisms are considered to be conserved due to the shared regulatory mechanisms. Along these lines, mammalian cells may have functional equivalents of Xrp1. We consider among the candidate equivalent factors those with regulatory 5’ leader sequences that respond to eIF2α phosphorylation (Palam et al., 2011; Zhou et al., 2008; Andreev et al., 2015). Based on the emerging roles of Xrp1 in Drosophila models of human diseases, we speculate that those ATF4-independent PERK signaling effectors may play more significant roles in diseases associated with UPR than had been generally assumed.
We note in our study that genes encoding cytoplasmic glutathione S-transferases (GSTs) such as gstD1 and gstD9 are among the most prominently induced UPR targets in our eye imaginal disc-based gene expression profiling analysis. Previous studies also reported these as ER stress-inducible genes in Drosophila S2 cells (Malzer et al., 2018). GSTs are cytoplasmic proteins that participate in the detoxification of harmful, often lipophilic intracellular compounds damaged by ROS. These enzymes catalyze the formation of water-soluble glutathione conjugates that can be more easily eliminated from the cell (Low et al., 2010; Low et al., 2007; Sharma et al., 2004; Wilce and Parker, 1994). It is noteworthy that ROS is generated as a byproduct of Ero-1-mediated oxidative protein folding, and such ROS generation increases when mutant proteins undergo repeated futile cycles of protein oxidation (Gross et al., 2006; Tang and Tu, 1994; Tu and Weissman, 2004). Therefore, we speculate that cytoplasmic GSTs evolved as UPR targets as they have the ability to detoxify lipid peroxides or oxidized ER proteins that increase in response to ER stress.
In conclusion, our findings support the idea that an ATF4-independent branch of PERK signaling mediates the expression of the most highly induced UPR targets in eye disc cells. This axis of the UPR requires Xrp1, a gene that had not previously been associated with ER stress response. The identification of this new axis of UPR signaling may pave the way for a better mechanistic understanding of various physiological and pathological processes associated with abnormal UPR signaling in metazoans.
Materials and methods
Fly stocks and husbandry
Request a detailed protocolWe reared flies at ambient temperature on a standard cornmeal-agar diet supplemented with molasses, and carried out crosses at 25°C. We performed all gene overexpression experiments using the Gal4/UAS binary expression system (Brand and Perrimon, 1993). We used the following fly stocks: w1118, gstD-GFP (Sykiotis and Bohmann, 2008), GMR-Gal4 (Freeman, 1996), Rh1-Gal4 (Pichaud and Desplan, 2001), hh-Gal4 (Calleja et al., 1996), UAS-EGFP::Msp300KASH (Hall et al., 2017; Ma and Weake, 2014; RRID: BDSC_92584), UAS-Rh1G69D (Ryoo et al., 2007), UAS-PerkWT (RRID: BDSC_76248), UAS-PerkKD (Malzer et al., 2010; RRID: BDSC_76249), UAS-cyto-roGFP-Orp1 (Albrecht et al., 2011; RRID: BDSC_67670), ey-FLP (RRID: BDSC_5576), FRT82B,arm-lacZ (RRID: BDSC_7369), FRT82B,p(w+)[90E] (RRID: BDSC_2050), UAS-lacZ (RRID: BDSC_1776), FRT82B,cncK6/TM6B (Veraksa et al., 2000), FRT82B, cncvl110 (Mohler et al., 1995), FRT82B,Ire1f02170/TM6B (Ryoo et al., 2013; RRID: BDSC_18520), FRT82B,Perke01744 (Wang et al., 2015; RRID: BDSC_85557), FRT82B, Xrp1M2-73 (Lee et al., 2018; RRID: BDSC_81270), Xrp1HA (Blanco et al., 2020), Atf6LL07432 (Kyoto DGGR, #142049), crc1 (Hewes et al., 2000; RRID: DGGR_105823), Thork13517 (Thor-lacZ)(Bernal and Kimbrell, 2000; RRID: BDSC_9558), uas-crcleaderless (Vasudevan et al., 2020), uas-gadd34 RNAi (VDRC #107545). The gadd34 RNAi line was expressed together with uas-dicer2 on the 3rd chromosome to enhance knockdown efficiency.
Identification of ATF4 and Xrp1 binding sites in the gstD1/gstD2 intergenic enhancer
Request a detailed protocolWe modified an existing Python code that that uses TBP position frequency matrix to predict TATA boxes (Stevens and Boucher, 2015) to calculate transcription factor binding scores. We deposited this modified Python code in Github (https://github.com/finnroach/transcription-factor-binding; copy archived at swh:1:rev:32ecddcc5cd7c8a22b2bb3db073162fc62d79447; Finnegan, 2021).
To identify putative ATF4 binding sites in the gstD1 upstream enhancer, we utilized the position frequency matrix (PFM) information of human ATF4 available on JASPAR (jaspar.genereg.net, Matrix ID:MA0833.1). The code outputs only binding sites with scores that are greater than 76% of the optimal ATF4 binding sequence. This cutoff both picks up two known ATF4 binding sites within the Drosophila Thor intron sequence (Kang et al., 2017) and allows leeway to find slightly lower affinity binding sites. Inputting the gstD1 enhancer sequence into this code revealed three unique putative ATF4 binding half-sites with the following sequences: cgttccctcatac (77% of optimal binding score), aatttcatcattt (83% of optimal binding score), and tatttcatcaccc (86% of optimal binding score).
To score putative Xrp1 binding sites, we used information available from a previous Xrp1 ChIP-seq study. The sequence logo of Xrp1 position frequency matrix was reported previously (Baillon et al., 2018). The Xrp1 position frequency matrix itself is available from the link (https://www.biorxiv.org/content/10.1101/467894v1). We show in Figure 8 the outputs of sites with scores greater than 95 % of the optimal Xrp1 binding sequence.
Generation of transgenic lines: gstDWT-GFP, gstD∆ATF4-GFP, and gstDXrp1m-GFP
Request a detailed protocolTo construct the gstDWT-GFP reporter, we amplified the entire genomic region of gstD-GFP flies containing the gstD-GFP reporter, then cloned the amplicon into BglII/NotI-digested pattB using InFusion cloning (ClonTech). To make gstD∆ATF4-GFP, we used commercial gene synthesis (General BioSystems, Inc) to reconstruct the entire reporter region previously amplified by PCR for the creation of gstDWT-GFP, but with the predicted ATF4-binding sites deleted. The three deleted sequences are (in the order of from that closest to gstD2 towards the gstD1 coding sequence): ggtgatgaaata, aatgatgaaatt, atgagggaa.
To generate the gstDXrp1m-GFP reporter, we performed directed mutagenesis on the gstD∆ATF4-GFP to mutate the remaining Xrp1 binding site. Specifically, the sequence ttgtgaaatc was mutated to ttcccgggtc. The wild type and mutant gstD-GFP DNA were then subcloned into the pattB vector, and sent to BestGene.Inc (Chino Hills, CA) for embryo injection. Standard phiC31 integrase-mediated germline recombination approach (Groth et al., 2004) was used to target the plasmids to an attP landing site at cytological position 51 C (Bloomington Stock Center, #24482).
Affinity-based isolation of EGFP-labeled nuclear membranes and RNA extraction
Request a detailed protocolWe followed published protocols to isolate GMR > EGFP::Msp-300KASH-positive nuclei (Hall et al., 2018; Ma and Weake, 2014). We dissected eye-antennal discs from approximately 100 larvae per sample in phosphate buffered saline (PBS), pH 7.4 with 0.1% Tween-20 (Sigma-Aldrich, cat. #P7949). We then washed dissected discs in ice-cold nuclear isolation buffer (10 mM HEPES-KOH, pH 7.5; 2.5 mM MgCl2; 10 mM KCl), then lysed cells in 1 mL of ice-cold nuclear isolation buffer in a 2 mL Dounce homogenizer (VWR, cat. #62400–595). We next filtered the homogenate through a 40 µm Flowmi cell strainer (WVR, cat. #BAH136800040), after which we incubated a 20 µL pre-isolation aliquot of the filtrate with 0.1 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Millipore-Sigma, cat. #D9542) and mounted on a slide for imaging (Fisher, cat. #12-550-433). We incubated the remaining filtrate with anti-EGFP-coupled protein G Dynabeads (Invitrogen, cat. #10,003D) for 1 hr at 4°C with gentle end-over-end rotation. Next, we collected the beads using a magnetic microcentrifuge tube holder (Sigma, cat. #Z740155) and washed the collected beads with wash buffer (PBS, pH 7.4; 2.5 mM MgCl2), then resuspended the beads in a final volume of 150 µL of wash buffer. We incubated an aliquot of the post-isolation sample with 0.1 µg/mL DAPI and mounted the sample on a slide for imaging. Finally, we suspended the post-isolation nuclei in 1 mL of Trizol reagent (Life Technologies, cat. #15596018) for RNA extraction following standard procedures. Prior to RNA precipitation with isopropanol, we added 0.3 M sodium acetate and glycogen (Invitrogen, cat. #AM9510) to facilitate visualization of the RNA pellet. We then suspended the pellet in RNAse-free water and purified it using a Qiagen RNeasy MinElute cleanup kit (Qiagen, cat. #74204) following standard protocols.
Preparation of cDNA libraries, RNA-Seq, and data processing
Request a detailed protocolThe NYU Genome Technology Center performed library preparation and RNA sequencing. We quantified RNA on an Agilent 2100 BioAnalyzer (Agilent, cat. #G2939BA). For cDNA library preparation and ribodepletion, we utilized a custom Drosophila Nugen Ovation Trio low-input library preparation kit (Tecan Genomics), using approximately 1.5 ng total RNA per sample. For sequencing, we performed paired-end 50 bp sequencing of samples on an Illumina NovaSeq 6000 platform (Illumina, cat. #20012850) using half of a 100 cycle SP flow cell (Illumina, cat. #20027464). We used the bcl2fastq2 Conversion software (v2.20) to convert per-cycle BCL base call files outputted by the sequencing instrument (RTA v3.4.4) into the fastq format in order to generate per-read per-sample fastq files. For subsequent data processing steps, we used the Seq-N-Slide automated workflow developed by Igor Dolgalev (https://github.com/igordot/sns; Dolgalev, 2021). For read mapping, we used the alignment program STAR (v2.6.1d) (Dobin et al., 2013; Dobin and Gingeras, 2015) to map reads of each sample to the Drosophila melanogaster reference genome dm6, and for quality control we used the application Fastq Screen (v0.13.0) (Wingett and Andrews, 2018) to check for contaminating sequences. We employed featureCounts (Subread package v1.6.3) (Liao et al., 2014) to generate matrices of read counts for annotated genomic features. For differential gene statistical comparisons between groups of samples contrasted by genotype, we used the DESeq2 package (R v3.6.1) (Love et al., 2014) in the R statistical programming environment. For filtering of stably induced genes, we calculated the coefficient of variation (CV) of FPKM-normalized counts for all genes across all three GMR > Rh1G69D samples, and excluded genes with a CV greater than 0.3 from downstream analyses (Liang et al., 2020).
Antibodies, and immunofluorescence and confocal microscopy
Request a detailed protocolTo generate a guinea pig anti-Xrp1 antibody, we expressed the Xrp1 long isoform (isoform F) cloned in pSV272 (Francis et al., 2016) in BL21 pLys E. coli strain by IPTG induction. After cells lysis and sonication in pre-chilled binding buffer (20 mM Tris pH 8.0, 0.5 M NaCl, 5 mM imidazole), we centrifuged the lysate and washed the insoluble pellet containing His-tag fused recombinant Xrp1 in wash buffer (20 mM Tris pH 8.0, 0.5 M NaCl, 5 mM imidazole), and then solubilized the pellet in denaturing buffer (8 M Urea with 20 mM Tris pH 8.0 and 100 mM NaCl). We ran the solubilized fraction containing His-Xrp1 through a Ni2+-NTA column and washed the column with reducing concentrations of Urea in the denaturing buffer for purification and re-folding. We eluted His-Xrp1 from the column with Tris buffer containing 400 mM imidazole, and sent the purified protein for custom polyclonal antibody production (Covance Inc). The crude antiserum was affinity purified and used at 1:10 dilution for immunolabeling.
We immunoprecipitated EGFP-labeled imaginal disc nuclei with mouse anti-EGFP (Roche, cat. #11814460001). For immunofluorescence, we used the following primary and secondary antibodies at the indicated dilutions: rabbit anti-β-Gal (1:500, MP Biomedicals, cat. #55976), Rabbit anti-GFP (1:500 Invitrogen #A6455), chicken anti-EGFP (1:500, Aves lab, cat. #GFP-1020), mouse anti-Rh1 (1:500, Developmental Studies Hybridoma Bank (DSHB), 4C5 concentrate), rat anti-Elav (1:50, DSHB, 7E8A10 concentrate), mouse anti-Wingless (1:25, DSHB, 4D4), mouse-anti-HA (1:500, Cell Signaling, 2,367 S), Alexa Fluor 647 goat anti-mouse (1:500, Life Technologies, cat. #A-21235), Alexa Fluor 488 goat anti-chicken (1:500, Life Technologies, cat. #A-11039), Alexa Fluor 546 goat anti-rabbit (1:500, Life Technologies, cat. #A-11035).
We followed standard protocols for whole-mount immunofluorescence. We fixed discs in 1 X PBS with 0.2% Triton X-100 (Millipore-Sigma, cat. #T8787) (PBTx 0.2%) and 4% paraformaldehyde (Alfa Aesar, cat. #43368) for 20 min at ambient temperature with gentle rocking. Next, following three short rinses with PBTx 0.2%, we incubated discs with primary antibodies in PBTx 0.2 % for 1 hr at ambient temperature with gentle rocking. Following primary antibody incubation, we washed discs for 3*10 min with PBTx 0.2%, then covered from light and incubated with secondary antibody in PBTx 0.2% for 1 hr at ambient temperature with gentle rocking. Following secondary antibody incubation, we washed discs 3*10 min with PBTx 0.2%, then mounted in 50% glycerol (Millipore-Sigma, cat. #G5516) with 0.1 µg/mL DAPI. For all confocal micrographs, we captured images on a Zeiss LSM 700 confocal microscope (Carl Zeiss). For image acquisition, we scanned all imaginal discs under a 40 X water objective, and all isolated nuclei under a 100 X oil objective.
S2 cell culture, RNAi, and drug treatments
Request a detailed protocolWe cultured Drosophila S2 cells in ambient conditions in S2 cell medium (Fisher, cat. #21720024) supplemented with 1% penicillin/streptomycin (Life Technologies, cat. #15-140-122) and 10% heat-inactivated FBS (Fisher, cat. #10082147). For maintenance of cell lines, we passaged cells every 3–4 days and utilized them for experiments between passages 6 and 15.
We performed RNAi using a modified dsRNA bathing protocol (Ryoo et al., 2007). We generated dsRNA against cncC, Perk, Xrp1 and crc mRNAs following an established T7 in vitro transcription protocol (ThermoFisher, cat. #AM1334) (Armknecht et al., 2005). On day 0, we added approximately 20 µg of the indicated dsRNA to 1 mL containing 106 cells in serum-free S2 cell medium in a six-well dish. After 30 min of incubation at ambient temperature, we added 3 mL of S2 cell medium with serum and incubated the cells at ambient temperature. On day 3, we harvested the cells, resuspended them at 1*106 cells/mL in serum-free S2 cell medium, and subjected them to another round of dsRNA bathing as described above. On day 6, we re-plated cells at a concentration of ~2*106 cells/mL for drug treatments. For drug treatments, we exposed cells to either 20 mM paraquat (Sigma, cat. #856177) or 10 µg/mL tunicamycin (Fisher, cat. #351610) for the indicated times, then harvested and resuspended the treated cells in 500 µL Trizol reagent. Following RNA isolation, we then treated the samples with Turbo DNAse (Invitrogen, cat. #AM1907) for 25 min at 37°C to remove traces of contaminating genomic DNA. We utilized the following oligos to generate T7 double-stranded RNA:
EGFP-F: TAATACGACTCACTATAGGGAACAGCCACAACGTCTATATC
EGFP-R: TAATACGACTCACTATAGGGTTGGACAAACCACAACTAGAA
PERK-F: TAATACGACTCACTATAGGGTGGCACAAGGAGGGGAAC
PERK-R: TAATACGACTCACTATAGGGGCACCACTGGACCTAGTAAA
CncC-F: TAATACGACTCACTATAGGGGGGCTGCAAGCTTCCG
CncC-R: TAATACGACTCACTATAGGGGCGGTGCTGAGGGGTG
ATF4-F: TAATACGACTCACTATAGGGGCGGTGTAGAGGATCGAAAG
ATF4-R: TAATACGACTCACTATAGGGCACTGTCCGATTTGCAGAAA
Xrp1-F: TAATACGACTCACTATAGGGAGGACGAAGAGGAGACTACCACCG
Xrp1-R: TAATACGACTCACTATAGGGAGGAGTAAGTGCTCTTCTGCCGCT
Molecular biology and qRT-PCR
Request a detailed protocolWe amplified the gstD-GFP reporter from transgenic fly genomic DNA for InFusion cloning into BglII/NotI-digested pattB using the following oligos: gstD-GFP-F: attcgttaacagatcattgcaactggttgttaacct gstD-GFP-R: cctcgagccgcggcccgccttaagatacattgatgagt.
For qRT-PCRs, we reverse transcribed approximately 500 ng of total RNA from S2 cells in a 20 µL reaction with random primers using Maxima H reverse transcriptase (Thermo Scientific, cat. #EP0752) following manufacturer protocols. From this, we used 1 µL of cDNA per well for qPCR on a BioRad CFX96 Touch Real-Time PCR Detection System (BioRad, cat. #1855196) using Power SYBR Green PCR master mix (Life Technologies, cat. #4367659). We determined cycle threshold (Ct) values with BioRad CFX Manager software. For mRNA fold change calculations, we used the ∆∆Ct method, normalized to the housekeeping gene RpL15 (Livak and Schmittgen, 2001). We used the following oligos for qRT-PCR:
RpL15-F: AGGATGCACTTATGGCAAGC
RpL15-R: GCGCAATCCAATACGAGTTC
Thor-F: TAAGATGTCCGCTTCACCCA
Thor-R: CGTAGATAAGTTTGGTGCCTCC gstD2-F: CCGTCTATCTGGTGGAGAAGTA gstD2-R: GAGTTCCCATGTCGAAGTACAG
EGFP::Msp-300KASH-F: GAGGGATACGTGCAGGAGAG
EGFP::Msp-300KASH-R: GATCCTGTTGACGAGGGTGT
Gstd1-F: CATCGCGAGTTTCACAACAG
Gstd1-R: GTTGAGCAGCTTCTTGTTCAG
Gstd9-F: TTGCCGTTCCATCCTGATGAC
Gstd9-R: GCTTAAGATGCTCGCCGGCAT
Gstd10-F: TGCCGCTCTGTTCTGATG
Gstd10-R: CTAGCTCGGGTGTTGATAAT
SOD2-F: TCGCAAACTGCAAGCCTG
SOD2-R: CATGATCTCCCGGCAGAT
JAFRAC1-F: ATCATTGCGTTCTCGGAG
JAFRAC1-R: GCGTGTTGATCCAGGCCAA
Quantification of gstD-GFP pixel intensity changes
Request a detailed protocolWe used ImageJ (https://imagej.nih.gov/ij/index.html) to calculate average pixel intensities of gstD-GFP signals from eye disc microscopic images. We specifically measured pixel intensities of the GMR-Gal4 expressing region in posterior eye discs. To calculate fold change of the gstD-GFP signal, we divided the average pixel intensities of interest with that from control discs. Statistical significance was assessed through two tailed t tests unless otherwise stated. We plotted graphs using the ggplot2 package in the R programming environment.
Sample preparation for CUT and RUN
Request a detailed protocolApproximately, 100 tissues including eye, wing, leg discs and brains for each experimental condition were isolated from third instar wandering larvae (Genotype: hspflp; FRT42/Cyo;Xrp1[HA-1]/TM6B). The isolated tissues were soaked in 2 mM DTT for 6 hr in S2 media at RT in shaking condition. After incubation, samples were centrifuged at 1200 rpm for 5 min at 4° and 1 X trypsin was added to the pellet to isolate single-cell suspension. Trypsin digestion was continued for 4 hr at RT in a nutator. After digestion, the reaction mixture was centrifuged at 1200 rpm for 5 min and the isolated cells were re-suspended in cold wash buffer (2% FBS in 1 X DPBS without Ca/Mg and EDTA). The cells were kept on ice from this step. The cell suspension was filtered using a 40 µm cell strainer. Isolated cells were counted in haemocytometer and approximately 250,000 cells/sample were used for further processing using CUTANA ChIC/ CUT & RUN kit (Epicypher, 14–1048) according to the manufacture’s protocol. For IP reaction of HA-tagged Xrp1, ChIP grade anti-HA antibody (Abcam, ab9110) was used. As a control isotype, ChIP grade control IgG was used (Abcam, ab171870).
Validation of Xrp1 binding to gstD locus by qPCR assay from CUT and RUN sample
Request a detailed protocolA total of 500 picogram of eluted DNA was used as a template for qPCR analysis in a 20 µL reaction with the following primers on a BioRad CFX96 Touch Real-Time PCR Detection System (BioRad, cat. #1855196) using Power SYBR Green PCR master mix (Life Technologies, cat. #4367659). We determined cycle threshold (Ct) values with BioRad CFX Manager software. For relative fold change in binding event calculations, ∆Ct value was evaluated in comparison to the control IgG for each condition, while Δ∆Ct was generated by normalizing to unstressed condition (Without DTT). Here, UPD3 was used as a known binding partner of Xrp1 (Baillon et al., 2018) and Tubulin was used as an Xrp1 unbound chromatin control. We used the following oligos for qRT-PCR:
GstD1 F: TTGCGCTCTTAACGTCGAGAAC
GstD1 R: CAGCTGGATTTCGGCATTATGT
UPD3 F: TCGATACTTCTGAACCCGC
UPD3 R: CCTAAACCCATCACACCT
Tubulin F: AGCTCGATAACTCCGCATTGGC
Tubulin R: AGAGCGAGACGGCCGAT
Data availability
Sequencing data have been deposited in GEO under the accession code GSE150058. Source Data files have been provided for Figures 2-6 and 8.
-
NCBI Gene Expression OmnibusID GSE150058. Transcriptional changes associated with endoplasmic reticulum stress in the eye imaginal disc of Drosophila melanogaster.
References
-
Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKαMolecular Biology of the Cell 25:1686–1697.https://doi.org/10.1091/mbc.E14-02-0704
-
Proteotoxic stress is a driver of the loser status and cell competitionNature Cell Biology 23:136–146.https://doi.org/10.1038/s41556-020-00627-0
-
Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survivalMolecular and Cellular Biology 23:7198–7209.https://doi.org/10.1128/MCB.23.20.7198-7209.2003
-
PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stressThe Journal of Biological Chemistry 279:20108–20117.https://doi.org/10.1074/jbc.M314219200
-
STAR: ultrafast universal RNA-seq alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Mapping RNA-seq Reads with STARCurrent Protocols in Bioinformatics 51:11.14.1–11.14.19.https://doi.org/10.1002/0471250953.bi1114s51
-
SoftwareTranscription Factor Binding Analysis, version swh:1:rev:32ecddcc5cd7c8a22b2bb3db073162fc62d79447Software Heritage.
-
The requirement of IRE1 and XBP1 in resolving physiological stress during Drosophila developmentJournal of Cell Science 130:3040–3049.https://doi.org/10.1242/jcs.203612
-
4E-BP is a target of the GCN2-ATF4 pathway during Drosophila development and agingThe Journal of Cell Biology 216:115–129.https://doi.org/10.1083/jcb.201511073
-
The unfolded protein response: Detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulumCold Spring Harbor Perspectives in Biology 11:a033886.https://doi.org/10.1101/cshperspect.a033886
-
Recognition and detoxification of the insecticide DDT by Drosophila melanogaster glutathione S-transferase D1Journal of Molecular Biology 399:358–366.https://doi.org/10.1016/j.jmb.2010.04.020
-
Affinity-based isolation of tagged nuclei from Drosophila tissues for gene expression analysisJournal of Visualized Experiments 10:51418.https://doi.org/10.3791/51418
-
Xrp1 genetically interacts with the ALS-associated FUS orthologue caz and mediates its toxicityThe Journal of Cell Biology 217:3947–3964.https://doi.org/10.1083/jcb.201802151
-
Impaired tissue growth is mediated by checkpoint kinase 1 (CHK1) in the integrated stress responseJournal of Cell Science 123:2892–2900.https://doi.org/10.1242/jcs.070078
-
The unfolded protein response in metazoan developmentJournal of Cell Science 132:jcs217216.https://doi.org/10.1242/jcs.217216
-
Phosphorylation of EIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translationThe Journal of Biological Chemistry 286:10939–10949.https://doi.org/10.1074/jbc.M110.216093
-
BookBiological sequencesIn: Stevens T, editors. Python Programming for Biology. Cambridge University Press. pp. 181–207.
-
Biochemical characterization of Drosophila glutathione S-transferases D1 and D21The Journal of Biological Chemistry 269:27876–27884.
-
The glutathione S-transferase D genes A divergently organized, intronless gene family in Drosophila melanogasterThe Journal of Biological Chemistry 268:9737–9746.https://doi.org/10.1016/S0021-9258(18)98410-3
-
Oxidative protein folding in eukaryotes: Mechanisms and consequencesThe Journal of Cell Biology 164:341–346.https://doi.org/10.1083/jcb.200311055
-
Cap “n” collar B cooperates with a small Maf subunit to specify pharyngeal development and suppress deformed homeotic function in the Drosophila headDevelopment 127:4023–4037.
-
Structure and function of glutathione S-transferasesBiochimica et Biophysica Acta 1205:1–18.https://doi.org/10.1016/0167-4838(94)90086-8
-
Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditionsThe Journal of Biological Chemistry 283:7064–7073.https://doi.org/10.1074/jbc.M708530200
Article and author information
Author details
Funding
National Eye Institute (R01 EY020866)
- Hyung Don Ryoo
National Institute of General Medical Sciences (R01 GM125954)
- Hyung Don Ryoo
National Institute of General Medical Sciences (T32 GM136573)
- Brian Brown
Eunice Kennedy Shriver National Institute of Child Health and Human Development (T32 HD007520)
- Brian Brown
National Eye Institute (K99 EY029013)
- Deepika Vasudevan
NYU School of Medicine (P30 CA061087)
- Deepika Vasudevan
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Nicholas Baker, Claude Desplan, Michael Garabedian, Moses Chao, Erika Bach and Jessica Treisman for helpful comments. We also thank Heinrich Jasper, Nicholas Baker, Konrad Basler, Donald Rio, Thomas Hurd, Temesgen Fufa, Robert Hufnagel, Arjita Sarkar and Vikki Weake for fly strains, plasmids and technical advice. This project was supported by NIH R01 EY020866 and GM125954 to HDR, T32 HD007520 and T32 GM136573 training grants support for BB, K99 EY029013 to DV, and the Cancer Center Support Grant P30 CA061087 at the Perlmutter Cancer Center to the Genome Technology Center.
Copyright
© 2021, Brown et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The transcription factor Xrp1 is required for PERK-mediated antioxidant gene induction in Drosophila
eLife 10:e74047.
https://doi.org/10.7554/eLife.74047
Further reading
-
- Genetics and Genomics
- Microbiology and Infectious Disease
Polyamines are biologically ubiquitous cations that bind to nucleic acids, ribosomes, and phospholipids and, thereby, modulate numerous processes, including surface motility in Escherichia coli. We characterized the metabolic pathways that contribute to polyamine-dependent control of surface motility in the commonly used strain W3110 and the transcriptome of a mutant lacking a putrescine synthetic pathway that was required for surface motility. Genetic analysis showed that surface motility required type 1 pili, the simultaneous presence of two independent putrescine anabolic pathways, and modulation by putrescine transport and catabolism. An immunological assay for FimA—the major pili subunit, reverse transcription quantitative PCR of fimA, and transmission electron microscopy confirmed that pili synthesis required putrescine. Comparative RNAseq analysis of a wild type and ΔspeB mutant which exhibits impaired pili synthesis showed that the latter had fewer transcripts for pili structural genes and for fimB which codes for the phase variation recombinase that orients the fim operon promoter in the ON phase, although loss of speB did not affect the promoter orientation. Results from the RNAseq analysis also suggested (a) changes in transcripts for several transcription factor genes that affect fim operon expression, (b) compensatory mechanisms for low putrescine which implies a putrescine homeostatic network, and (c) decreased transcripts of genes for oxidative energy metabolism and iron transport which a previous genetic analysis suggests may be sufficient to account for the pili defect in putrescine synthesis mutants. We conclude that pili synthesis requires putrescine and putrescine concentration is controlled by a complex homeostatic network that includes the genes of oxidative energy metabolism.
-
- Genetics and Genomics
Resistance to anthelmintics, particularly the macrocyclic lactone ivermectin (IVM), presents a substantial global challenge for parasite control. We found that the functional loss of an evolutionarily conserved E3 ubiquitin ligase, UBR-1, leads to IVM resistance in Caenorhabditis elegans. Multiple IVM-inhibiting activities, including viability, body size, pharyngeal pumping, and locomotion, were significantly ameliorated in various ubr-1 mutants. Interestingly, exogenous application of glutamate induces IVM resistance in wild-type animals. The sensitivity of all IVM-affected phenotypes of ubr-1 is restored by eliminating proteins associated with glutamate metabolism or signaling: GOT-1, a transaminase that converts aspartate to glutamate, and EAT-4, a vesicular glutamate transporter. We demonstrated that IVM-targeted GluCls (glutamate-gated chloride channels) are downregulated and that the IVM-mediated inhibition of serotonin-activated pharynx Ca2+ activity is diminished in ubr-1. Additionally, enhancing glutamate uptake in ubr-1 mutants through ceftriaxone completely restored their IVM sensitivity. Therefore, UBR-1 deficiency-mediated aberrant glutamate signaling leads to ivermectin resistance in C. elegans.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}