Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs
- Institute for Research in Immunology and Cancer (IRIC), and Department of Biochemistry and Molecular Medicine, Université de Montréal, Canada
- Domain Therapeutics North America, Canada
- Department of Drug Design and Pharmacology, University of Copenhagen, Denmark
- Internal Medicine Research Unit, Pfizer Worldwide Research, Development and Medical, United States
- Pfizer Global R&D, United States
- Domain Therapeutics, France
Abstract
The recognition that individual GPCRs can activate multiple signaling pathways has raised the possibility of developing drugs selectively targeting therapeutically relevant ones. This requires tools to determine which G proteins and βarrestins are activated by a given receptor. Here, we present a set of BRET sensors monitoring the activation of the 12 G protein subtypes based on the translocation of their effectors to the plasma membrane (EMTA). Unlike most of the existing detection systems, EMTA does not require modification of receptors or G proteins (except for Gs). EMTA was found to be suitable for the detection of constitutive activity, inverse agonism, biased signaling and polypharmacology. Profiling of 100 therapeutically relevant human GPCRs resulted in 1500 pathway-specific concentration-response curves and revealed a great diversity of coupling profiles ranging from exquisite selectivity to broad promiscuity. Overall, this work describes unique resources for studying the complexities underlying GPCR signaling and pharmacology.
Editor's evaluation
The authors describe a novel set of biosensors to assess the coupling specificity of 100 therapeutically relevant G proteins-coupled receptors (GPCRs) to various G proteins. The utility of the assay system is well-supported by the data. These tools are likely to be useful for many specific studies of individual receptors, including efforts to discover ligands that display functional selectivity (bias) between G protein pathways or between G proteins and arrestins. The work provides a rich repository of data informing on the possible effector coupling of 100 GPCRs and a set of analytical tools that could guide the development of new drugs, including efforts to discover ligands that display functional selectivity (bias) between G protein pathways or between G proteins and arrestins.
https://doi.org/10.7554/eLife.74101.sa0Introduction
G protein-coupled receptors (GPCRs) play crucial roles in the regulation of a wide variety of physiological processes and represent one-third of clinically prescribed drugs (Hauser et al., 2017). Classically, GPCR-mediated signal transduction was believed to rely on linear signaling pathways whereby a given GPCR selectively activates a single G protein family, defined by the nature of its Gα subunit (Oldham and Hamm, 2008). Gα proteins are divided into four major families (Gs, Gi/o, Gq/11, and G12/13) encoded by 16 human genes. Once activated, these proteins each trigger different downstream effectors yielding different biological outcomes. It has now become evident that many GPCRs can couple to more than one G protein family and that ligands can selectively promote the activation of different subsets of these pathways (Namkung et al., 2018; Quoyer et al., 2013). These observations extended the concept of ligand-biased signaling, which was first established for ligand-directed selectivity between βarrestin and G protein (Azzi et al., 2003; Wei et al., 2003), to functional selectivity between G proteins. Ligand-directed functional selectivity represents a promising avenue for GPCRs drug discovery since it offers the opportunity of activating pathways important for therapeutic efficacy while minimizing activation of pathways responsible for undesirable side effects (Galandrin et al., 2007; Kenakin, 2019).
To fully explore the potential of functional selectivity, it is essential to have an exhaustive description of the signaling partners that can be activated by a given receptor, providing receptor- and ligand-specific signaling signatures. Currently, few assays allow for an exhaustive pathway-specific analysis of GPCR signaling; these include BRET-based G protein activation sensors platforms (Galés et al., 2005; Masuho et al., 2015; Maziarz et al., 2020; Mende et al., 2018; Olsen et al., 2020) and the TGF-α shedding assay (Inoue et al., 2019). However, several of these platforms require modification of G protein subunits that may create functional distortions. Moreover, these assays may detect non-productive conformational rearrangements of the G protein heterotrimer as was recently reported for G12 (Okashah et al., 2020).
Here, we describe unique sensors that do not require modification of receptors or G proteins (except for Gs) for interrogating the signaling profiles of GPCRs. The platform includes 15 pathway-selective enhanced bystander bioluminescence resonance energy transfer (ebBRET) biosensors monitoring the translocation of downstream effectors to the plasma membrane for Gi/o, Gq/11, and G12/13, the dissociation of the Gα subunit from the plasma membrane for Gs and the recruitment of βarrestin to the plasma membrane. Overall, the new ebBRET-based Effector Membrane Translocation Assays, named EMTA, provide a readily accessible large scale and comprehensive platform to study constitutive and ligand-directed GPCR signaling. The signaling signatures of 100 GPCRs using the EMTA platform also provides a rich source of information to explore the principles underlying receptor/G protein/βarrestin coupling selectivity relationships. It thus provides a unique set of tools that is complementary to previously described platforms and existing datasets, and offers a map of the coupling potentials for individual GPCR that will stimulate future studies investigating the relevance of these couplings in different physiological systems.
Results
ebBRET-based G protein effector membrane translocation assay (EMTA) allows detection of each Gα protein subunit activation
To detect the activation of Gα subtypes, we created an EMTA biosensor platform based on ebBRET (Namkung et al., 2016; Figure 1A). The biosensors at the heart of EMTA consist of sub-domains of the G protein-effector proteins p63-RhoGEF, Rap1GAP and PDZ-RhoGEF that selectively interact with activated Gq/11, Gi/o, or G12/13, respectively. These domains were fused at their C-terminus to Renilla luciferase (RlucII) and co-expressed with different unmodified receptor and Gα protein subtypes. Upon GPCR activation, the energy donor-fused effectors translocate to the plasma membrane to bind activated Gα proteins, bringing RlucII in close proximity to the energy acceptor, Renilla green fluorescent protein, targeted to the plasma membrane through a CAAX motif (rGFP-CAAX), thus leading to an increase in ebBRET. The same plasma membrane translocation principle is used to measure βarrestin recruitment (Namkung et al., 2016; Figure 1B, top). Because no selective soluble downstream effector of Gs exists, the assay was modified taking advantage of Gαs dissociation from the plasma membrane following its activation (Wedegaertner et al., 1996). In this configuration, the RlucII is directly fused to Gαs (Carr et al., 2014). Its activation upon GPCR stimulation leads to its dissociation from the plasma membrane (Martin and Lambert, 2016), resulting in a reduction in ebBRET (Figure 1B, bottom).
Figure 1
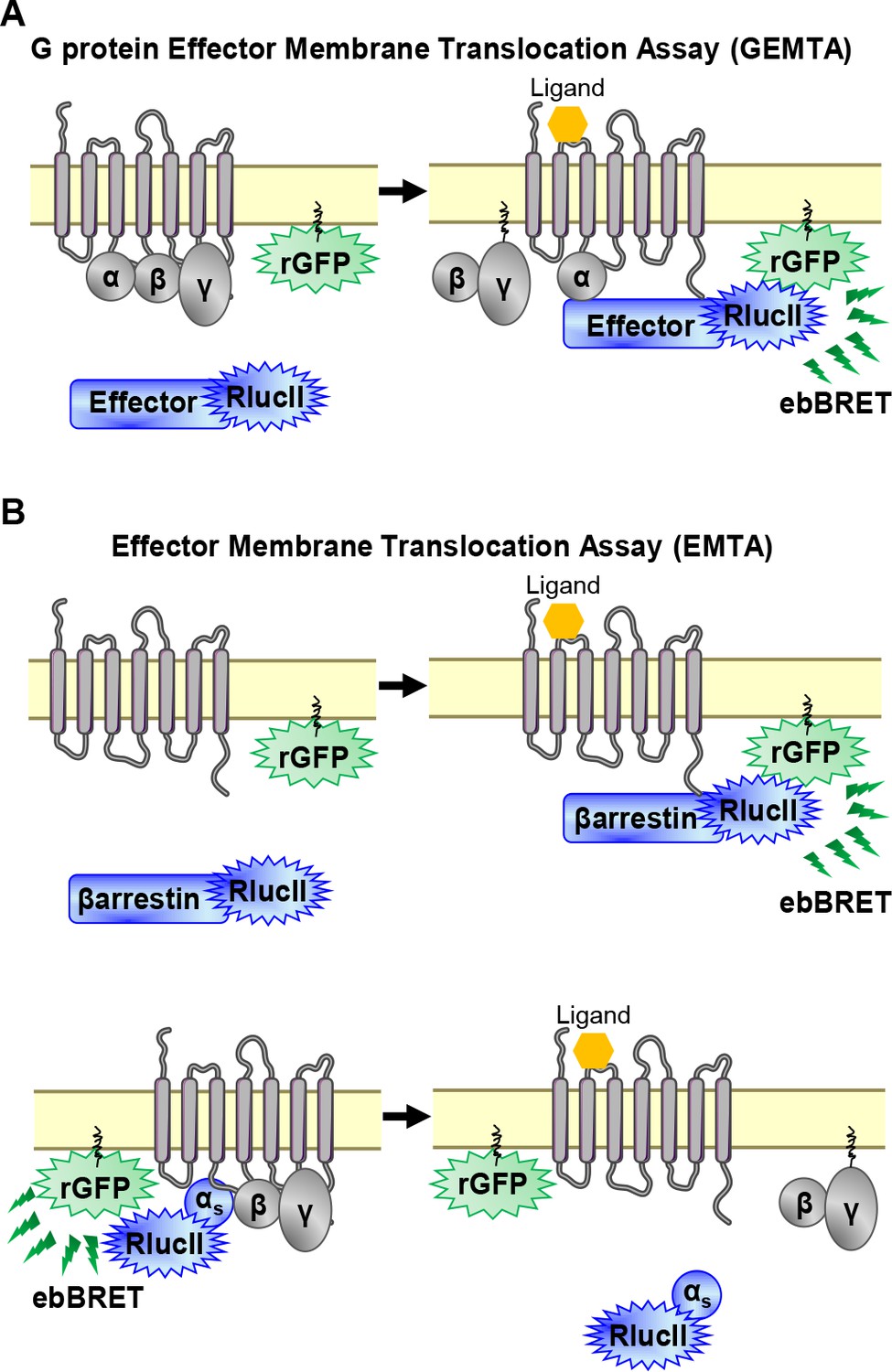
EMTA ebBRET platform to monitor G protein activation and βarrestin recruitment.
(A) Schematic of the G protein Effector Membrane Translocation Assay (GEMTA) to monitor Gα protein activation. Upon receptor activation, RlucII-tagged effector proteins (Effector-RlucII) translocate towards and interact with active Gα subunits from each G protein family, leading to increased ebBRET. (B) Principle of the Effector Membrane Translocation Assay (EMTA) monitoring βarrestin recruitment to the plasma membrane (top) and Gαs activation (bottom). Top; upon receptor activation, RlucII-tagged βarrestins (βarrestin-RlucII) translocate to the plasma membrane, thus increasing ebBRET with rGFP-CAAX. Bottom; Internalization of activated RlucII-tagged Gαs (Gαs-RlucII) following receptor stimulation decreases ebBRET with the membrane-anchored rGFP-CAAX.
The sensitivity and selectivity of the newly created G protein EMTA biosensors, were validated using prototypical GPCRs known to activate specific Gα subtypes. The responses were monitored upon heterologous expression of specific Gα subunits belonging to Gi/o, Gq/11, or G12/13 families in the absence or presence of pharmacological inhibitors and using engineered cells lacking selected Gα subtypes. The dopamine D2 receptor was used to validate the ability of the Gi/o binding domain of Rap1GAP (Jordan et al., 1999; Meng et al., 1999) to selectively detect Gi/o activation. The dopamine-promoted increase in ebBRET between Rap1GAP-RlucII and rGFP-CAAX in the presence of Gαi/o subunits was not affected by the Gq/11-selective inhibitor UBO-QIC (a.k.a., FR900359; Schrage et al., 2015; Figure 2A, left), whereas the Gαi/o family inhibitor, pertussis toxin (PTX), completely blocked the response for all members of Gαi/o family except for Gαz, known to be insensitive to PTX (Casey et al., 1990; Figure 2A, right). Gonadotropin-releasing hormone (GnRH) stimulation of the GnRH receptor (GnRHR), used as a prototypical Gq/11-coupled receptor, promoted ebBRET between the RlucII-fused Gq/11 binding domain of p63-RhoGEF (p63-RhoGEF-RlucII; Lutz et al., 2007; Rojas et al., 2007) and rGFP-CAAX. The ebBRET increase observed in the presence of different Gαq/11 subunits was not significantly (p = 0.077, 0.0636 and 0.073 for Gq, G11, and G14, respectively) affected by PTX (Figure 2B, right), whereas UBO-QIC completely blocked the response for all members of Gαq/11 family except for Gα15, known to be insensitive to UBO-QIC (Schrage et al., 2015; Figure 2B, left). These two G protein-specific EMTA were sensitive enough to detect responses elicited by endogenous G proteins since deletion of Gi/o (ΔGi/o) or Gq/11 (ΔGq/11) subtypes completely abolished the responses induced by D2 or GnRHR activation in the absence of heterologously expressed G proteins (Figure 2—figure supplement 1I). It should however be noted that relying on endogenous proteins does not allow the identification of specific members of Gi/o (i.e.: Gi1, Gi2, Gi3, GoA, GoB, or Gz) or Gq/11 (i.e.: Gq, G11, G14, or G15) families.
Figure 2 with 6 supplements see all
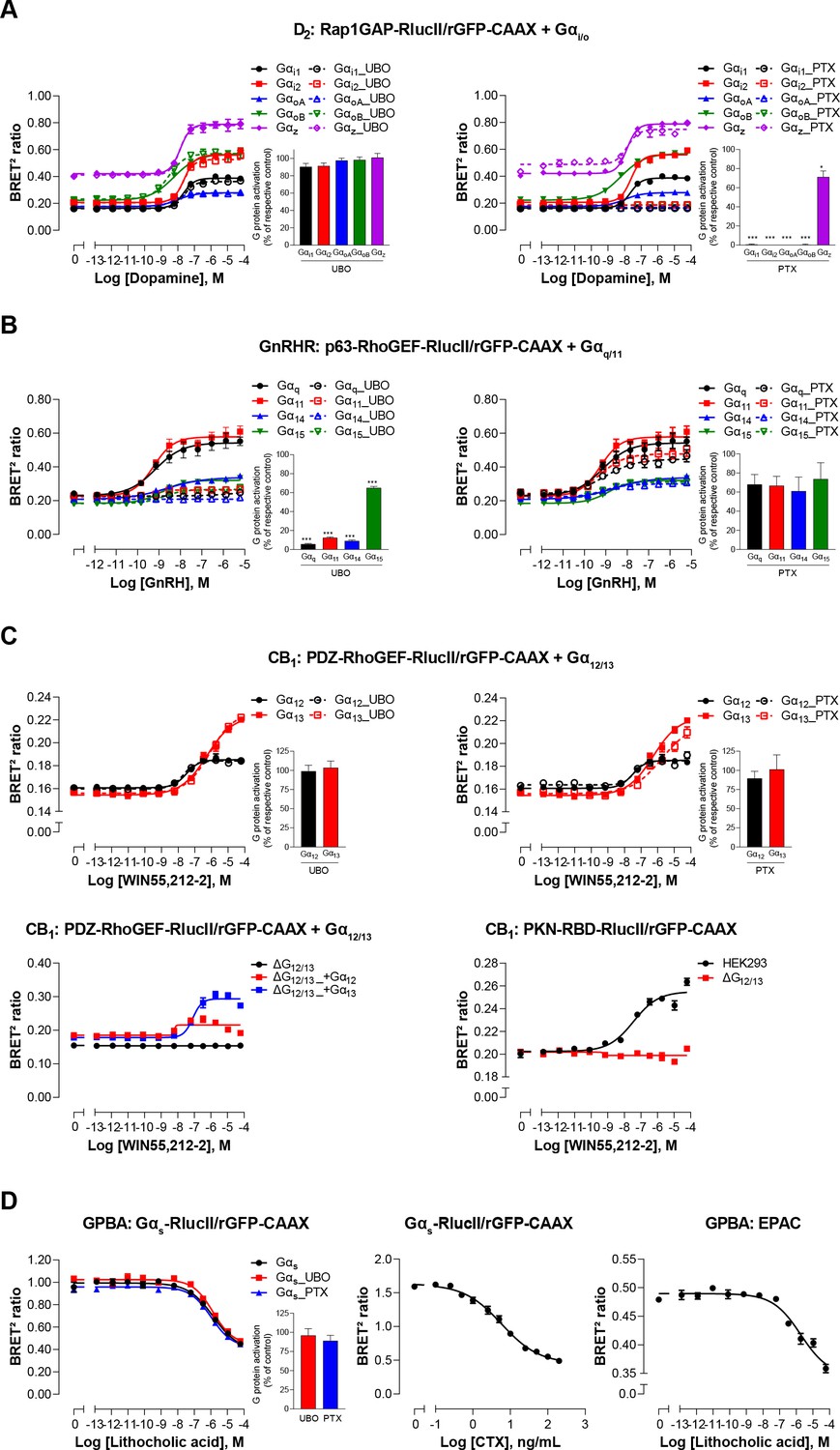
Validation of EMTA ebBRET-based platform to monitor Gα protein activation.
(A) Pharmacological validation of the Gαi/o activation sensor. HEK293 cells were transfected with the D2 receptor and the Gαi/o family-specific sensor, along with each Gαi/o subunit. Concentration-response curve using the Gαi/o activation sensor, in the presence or absence of UBO-QIC (left) or PTX (right) inhibitors. Insets; Emax values determined from concentration-response curves of inhibitor-pretreated cells. (B) Pharmacological validation of the Gαq/11 activation sensor. HEK293 cells were transfected with the GnRH receptor and the Gαq/11 family-specific sensor, along with each Gαq/11 subunit. Concentration-response curve using Gαq/11 activation sensor, in the presence or absence of UBO-QIC (left) or PTX (right) inhibitors. Insets; Emax values determined from dose-response curves of inhibitor-pretreated cells. (C) Validation of the Gα12/13 activation sensor. Cells were transfected with the CB1 receptor and one of the Gα12/13 activation sensors, along with the Gα12 or Gα13 subunits. Concentration-response curves of HEK293 cells (top) or the parental and devoid of G12/13 (ΔG12/13) HEK293 cells (bottom) using the PDZ-RhoGEF-RlucII/rGFP-CAAX (top and bottom left) or PKN-RBD-RlucII/rGFP-CAAX (bottom right) sensors, pretreated or not with UBO-QIC or PTX (top). (D) Pharmacological validation of the Gαs activation sensor. HEK293 cells were transfected with the GPBA receptor and the Gαs activation (left and central) or the EPAC (right) sensors. Left: Concentration-response curves using the Gαs activation sensor in the presence or absence of UBO-QIC or PTX, inhibitors of Gαq or Gαi/o, respectively. Central: Concentration-response activation of the Gαs sensor using CTX, a Gαs activator. Right: Concentration-response curve using the EPAC sensor. Inset; Emax values determined from dose-response curves of inhibitors-pretreated cells. Data are expressed as BRET ratio for the concentration-response curves or expressed in % of respective control cells (Emax graphs) and are the mean ± SEM of 3 (A–C) or 4 (D) independent experiments performed in one replicate. Unpaired t-test (A–D): *p < 0.05 and ***p < 0.001 compared to control cells.
-
Figure 2—source data 1
Raw data of Figure 2.
- https://cdn.elifesciences.org/articles/74101/elife-74101-fig2-data1-v2.xlsx
The selectivity of the G12/13 binding domain of PDZ-RhoGEF (Fukuhara et al., 2001) was confirmed using the cannabinoid receptor type 1 (CB1). The ebBRET between PDZ-RhoGEF-RlucII and rGFP-CAAX in the presence of Gα12 or Gα13 promoted by the cannabinoid agonist WIN-55,212–2 was not affected by UBO-QIC (Figure 2C, top left), nor PTX (Figure 2C, top right). Given the lack of selective G12/13 pharmacological inhibitor, we used HEK293 cells genetically deleted for Gα12 and Gα13 proteins (ΔG12/13) to further confirm the response selectivity. As expected, PDZ-RhoGEF-RlucII/rGFP-CAAX ebBRET was observed only following reintroduction of either Gα12 (ΔG12/13_+G12) or Gα13 (ΔG12/13_+G13) (Figure 2C, bottom left). The G12/13 coupling of CB1 was further confirmed by monitoring the recruitment of PKN to the plasma membrane (Figure 2C, bottom right) in agreement with previous reports (Inoue et al., 2019).
To further assess the selectivity of each EMTA biosensor, we took advantage of the fact that the endothelin-1 receptor (ETA) can activate Gq/11, Gi/o, and G12/13 family members. As shown in Figure 2—figure supplement 2, only over-expression of the Gα family members corresponding to their selective effectors (Rap1GAP for Gi/o, p63-RhoGEF for Gq/11, and PDZ-RhoGEF for G12/13) significantly increased the recruitment of the effector-RlucII to the plasma membrane. A recent study (Chandan et al., 2021) showed that Gi/o can also activate full length PDZ-RhoGEF. Although the domain of PDZ-RhoGEF required for this activation has not been identified yet, the selectivity of our PDZ-RhoGEF sensor for G12/13 vs. all other G protein families most likely results from the fact that we used a truncated version of PDZ-RhoGEF that only contains the G12/13 binding domain and lacks the PDZ domain involved in protein-protein interaction, the actin-binding domain and the DH/PH domains involved in GEF activity and RhoA activation (Aittaleb et al., 2010).
It should be noted that in the heterologous expression configuration, competition with endogenous G proteins did not occur to a significant extent since the potencies of the responses to a given G protein subtype were not affected by genetic deletion of the different G protein family members (Figure 2—figure supplement 1 and Supplementary file 1A). Similarly, overexpression of G proteins, GPCRs or effectors-RlucII did not affect the potencies of the responses observed (Figure 2—figure supplement 3 and Supplementary file 1B-D), indicating that, in our experimental conditions, overexpression of the different components of EMTA sensors must likely not bias the coupling response. In addition to spectrometric assessment of coupling selectivity (above) and activation kinetics (Figure 2—figure supplement 4), EMTA allows to image the real-time recruitment of the G protein effectors to the plasma membrane (Videos 1–3) thus providing spatiotemporal resolution for the imaging detection of Gαi/o, Gαq/11, and Gα12/13 activation.
Video 1
BRET-based imagery of p63-RhoGEF-RlucII recruitment to the plasma membrane upon AT1 activation.
HEK293 cells expressing the p63-RhoGEF-RlucII/rGFP-CAAX sensors with Gαq and AT1 were stimulated with Angiotensin II. BRET levels (the ratio of the acceptor photon count to the total photon count) are expressed as a color code (lowest being black and purple, and highest being red and white).
Video 2
BRET-based imagery of Rap1GAP-RlucII recruitment to the plasma membrane upon D2 activation.
HEK293 cells expressing the Rap1GAP-RlucII/rGFP-CAAX sensors with Gαi2 and D2 were stimulated with dopamine. BRET levels (the ratio of the acceptor photon count to the total photon count) are expressed as a color code (lowest being black and purple, and highest being red and white).
Video 3
BRET-based imagery of PDZ-RhoGEF-RlucII recruitment to the plasma membrane upon TPαR activation.
HEK293 cells expressing the PDZ-RhoGEF-RlucII/rGFP-CAAX + Gα13 and TPαR were stimulated with U46619. BRET levels (the ratio of the acceptor photon count to the total photon count) are expressed as a color code (lowest being black and purple, and highest being red and white).
The sensitivity of the EMTA platform is illustrated by a direct side-by-side comparison of the signals detected with EMTA vs. BRET assays based on Gαβγ dissociation (Gαβγ) (Galés et al., 2005; Galés et al., 2006; Olsen et al., 2020), that reveals a significantly larger assay windows for EMTA for the 6 Gα subunits tested for eight selected receptors, (Figure 2—figure supplement 5).
For the Gαs translocation biosensor, the bile acid receptor (GPBA) was chosen for validation (Kawamata et al., 2003). As expected, lithocholic acid stimulation resulted in a concentration-dependent decrease in ebBRET between Gαs-RlucII and rGFP-CAAX (Figure 2D, left). Cholera toxin (CTX), which directly activates Gαs (De Haan and Hirst, 2004), led to a decrease in ebBRET (Figure 2D, center), confirming that loss of Gαs plasma membrane localization results from its activation. The potency of lithocholic acid to promote Gs dissociation from the plasma membrane was well in line with its potency to increase cAMP production as assessed using a BRET²-based EPAC biosensor (Leduc et al., 2009; Figure 2D, right). The Gs-plasma membrane dissociation ebBRET signal was not affected by UBO-QIC or PTX (Figure 2D, left), confirming the selectivity of the biosensor.
Signaling signatures of one hundred therapeutically relevant receptors reveals distinct G protein and βarrestin selectivity profiles
We used EMTA to assess the signaling signature of a panel of 100 human GPCRs that are either already the target of clinically used drugs (74 receptors), considered for pre- or clinical drug development (6 receptors), or pathophysiologically relevant (Supplementary file 2A). To establish the coupling potentials for each receptor, we quantified its ability to activate 15 pathways: Gαs, Gαi1, Gαi2, GαoA, GαoB, Gαz, Gα12, Gα13, Gαq, Gα11, Gα14, Gα15 and βarrestin 2 as well as βarrestin 1 and 2 in the presence of GRK2 (Supplementary file 3). Emax and pEC50 values were determined (Supplementary file 2) and, based on the pre-determined threshold criteria (Emax ≥mean of vehicle-stimulated +2*SD; see Materials and methods), a ‘yes or no’ agonist-dependent activation was assigned to each signaling pathway and summarized using radial graph representations (Figure 3—figure supplement 1). To assess whether endogenous receptors could contribute to the observed responses, assays were also carried out in cells not transfected with the studied receptor (Figure 3—figure supplement 2). When an agonist-promoted response was observed in non-transfected parental HEK293 cells, this response was not considered as a receptor-specific response (see Materials and methods).
To compare the signaling profiles across all receptors and pathways and to overcome differences in receptor expression levels and individual biosensor dynamic windows, we first min-max normalized Emax and pEC50 values (between 0 and 1) across receptors as a function of a reference receptor yielding the largest response for a given pathway (Figure 3A, left). Then, these values were again min-max normalized (between 0 and 1) for the same receptor across pathways, using the pathway with the largest response for this receptor as the reference (Figure 3A, right; see description in Materials and methods). Such double normalization allows direct comparison of the coupling efficiency to different G proteins for a given receptor and across receptors for a given G protein. This coupling efficiency is summarized as heatmaps (Figure 3B) that reveals a high diversity of signaling profiles. The selectivity toward the different G protein families varies considerably among GPCRs (Figure 4). In our dataset, which is the first using unmodified GPCRs and Gα proteins (except for Gs), 29% of the receptors coupled to only one family, whereas others displayed more promiscuity by coupling to 2, 3, or 4 families (36%, 25%, and 10%, respectively). Receptors coupling to a single G protein family favored the members of the Gi/o family. Indeed, 27% of the receptors coupling to Gi/o only activated this subtype family in comparison to 0, 2.4 and 9.1% for receptors activating G12/13, Gq/11, and Gs, respectively, thus displaying more promiscuous coupling. A detailed comparative analysis of the selectivity profiles that we observed using the EMTA sensors with that of the chimeric G protein-based assay developed by Inoue et al., 2019 and the IUPHAR/BPS Guide to Pharmacology database (GtP; https://www.guidetopharmacology.org/) is presented in the accompanying paper (Hauser et al., 2022). Supplementary file 2C allows a direct comparison of the relative potency determined using EMTA for both the new and the already known (i.e.: identified in GtP database) couplings. As can be seen in the table, although in many cases the potency for the novel couplings is lower, this is not a universal finding since for some receptors, the pEC50s for the new couplings are similar (ex: G12 for CB1; G13 for serotonin 5-HT2C; G12/13 for adenosine 2A (A2A) and prostaglandin E1 (EP1) receptors; Gi/o for corticotropin-releasing hormone receptor 1 (CRFR1), ETA and G protein-coupled receptor 39 (GPR39)) or higher (ex: Gz for serotonin 5-HT2B; G15 for adenosine 3 (A3) and melanocortin 3 (MC3R) receptors; G12 for bradykinin 2 (B2), cholecystokinin A (CCK1), chemokine receptor 6 (CCR6) and ETA receptors; G12/13 for CRFR1 and GPR68) than those for the canonical ones. Interestingly, in many instances the potency for the newly uncovered couplings are similar to those for βarrestins, which is generally lower than for their canonical G proteins, a finding consistent with the role of βarrestins in signaling arrest at the plasma membrane. The potency differences observed for the activation of different G protein subtypes by a given receptor may lead to preferential activation of some pathways over others. This relative selectivity is likely to be influenced by tissue-dependent G protein subtype expression levels. The physiological consequences of such selectivity remain to be investigated.
Figure 3 with 3 supplements see all

Heatmaps illustrating the diversity of receptor-specific signaling signatures detected with the EMTA ebBRET platform.
(A) First, values within each pathway were normalized relative to the maximal response observed across all receptors (max = 1; left). These values were then normalized across pathways for the same receptor, with the highest-ranking pathway serving as the reference (max = 1; right). (B) Heatmap representation of double normalized Emax (left) and pEC50 (right) data. Empty cells (grey) indicate no detected coupling. IUPHAR receptor names are displayed.
Figure 4 with 1 supplement see all
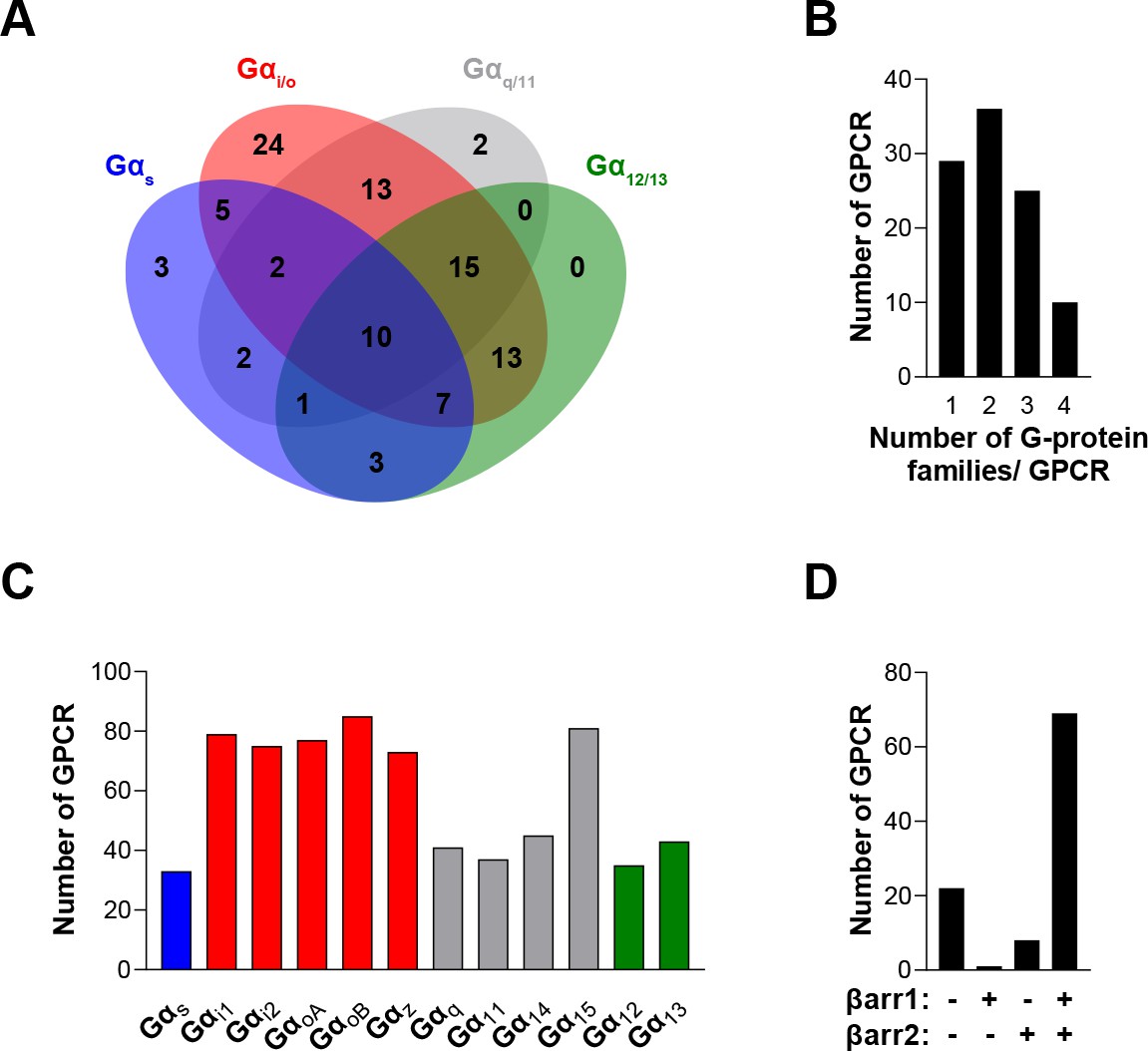
The EMTA ebBRET platform has a unique ability to uncover coupling selectivity between G protein families.
(A) Venn diagram showing the numbers of receptors coupled to each G protein family in the EMTA ebBRET biosensor assay. (B) Evaluation of receptors coupling promiscuity: number of receptors that couple to members of 1, 2, 3, or 4 G protein families. (C) Determination of G protein subunit coupling frequency: number of receptors that activate each Gα subunit. (D) Proportion of receptors recruiting βarrestins: number of receptors that do not recruit (-/-) or that recruit either (+/- or -/+) or both (+/+) βarrestin isotypes. All data are based on double normalized Emax values from Figure 3.
-
Figure 4—source data 1
Raw data of Figure 4.
- https://cdn.elifesciences.org/articles/74101/elife-74101-fig4-data1-v2.xlsx
When examining the frequency of coupling for each Gα subunit family (Figure 4C), the Gi/o family members were the most commonly activated, with 89% of the tested receptors activating a Gi/o family member. In contrast, only 33%, 49%, and 45% of the receptors activate Gs, G12/13, or Gq/11 (excluding Gα15) family members, respectively. Not surprisingly, and consistent with its reported promiscuous coupling, Gα15 was found to be activated by 81% of the receptors. For some receptors, we also observed preferential coupling of distinct members within a subtype family (Figure 3—figure supplement 1). For instance, 33% of Gi/o-coupled receptors can couple to only a subpopulation of the family (Figure 4—figure supplement 1A). For the Gq/11 family, only 44% activate all family members with 45% activating only Gα15 and 11% engaging only two or three members of the family. A matrix expressing the % of receptors engaging a specific Gα subtype that also activated another subtype, is illustrated in Figure 4—figure supplement 1B. When considering individual families, considerable variation within the Gi/o family was observed. The greatest similarities were observed between GαoB and either GαoA or Gαz, and the lowest between Gαi1 and Gαz. A striking example of intra-family coupling selectivity is the serotonin 5-HT2B that activates only GαoB and Gαz and GPR65 that selectively activates GαoB. Similarly, when considering the ligand-promoted responses above our threshold criteria (see Materials and methods), histamine H2 and MC3R receptors show preferred activation of GαoB and Gαz, whereas the prostaglandin F (FP) and neuropeptide Y5 (Y5) receptors preferentially activate GαoB, GαoA, and Gαz. Even when all members of a given family are found to be activated, some receptors activate specific family members with greater potencies (Supplementary file 2C).
When considering βarrestin recruitment, our analysis shows that 22% of receptors did not recruit βarrestin 1 or 2, even in the presence of overexpressed GRK2 (Figure 4D). Among the receptors able to recruit βarrestins, only a very small number selectively recruited βarrestin1 (1.3%) or βarrestin2 (6.4%), most of them recruiting both βarrestins in the presence of GRK2 (92.3%) (Figure 4D). Overexpression of GRK2 potentiated the recruitment of βarrestin2 for 68% of receptors highlighting the importance of GRK2 expression level in determining βarrestin activation (Supplementary files 3 and 2).
Comparison with previous datasets reveals commonalities and crucial differences
We compared the signaling profiles obtained here with those presented by Inoue et al., 2019 and the GtP dataset. Of note, this comparison only considers the final reported couplings that in the Inoue’s study were based on the criteria of positive coupling if LogRAi ≥ –1 and negative coupling if LogRAi ≤ –1, and is influenced by the different cut-offs and normalization used in the two studies. A comparison of couplings using common Emax standard deviation cut-off, quantitative normalization and aggregation of G proteins into families is provided in the accompanying paper (Hauser et al., 2022). As can be seen in Supplementary file 4A, among the 70 receptors common to both studies, less couplings were detected in our study than reported in Inoue et al. for Gαs (21 vs. 28), Gαi1 (54 vs. 56), Gαq (31 vs. 34), and Gα14 (36 vs. 40). In contrast, more receptors activating Gα12 (29 vs. 23), Gαo (59 vs. 41), Gα13 (30 vs. 15), Gαz (52 vs. 37), and Gα15 (62 vs. 15) were detected in our study. When comparing with data collected in GtP, that reports couplings grouped for G protein families (i.e.: Gs, Gi/o, Gq/11, or G12/13) and not at the single G protein subtype level, we detected less couplings than what was reported in GtP for Gαs (32 vs. 37), but more for Gαi/o (89 vs. 69), Gαq/11 (81 vs. 48), and Gα12/13 (47 vs. 10), among the 99 receptors common to both datasets (Supplementary file 4B).
Altogether, the comparative analysis reveals 64% and 69% identity of couplings between the EMTA and Inoue’s or GtP datasets, respectively. Each dataset reporting unique couplings and missing couplings found in the other two datasets. The reasons for these differences are plausibly due to intrinsic differences in the assays used. For instance, for G12/13 and G15 specifically, the difference with the GtP dataset most likely results from the fact that in most cases G12/13 or G15 activation were determined indirectly since, until their recent description (G12/13: Quoyer et al., 2013; Schrage et al., 2015; G15:Inoue et al., 2019; Olsen et al., 2020), no robust readily available assay existed to monitor the activation of these G proteins.
Validation of newly identified G12/13 and G15 couplings
Given the overrepresentation of both G12/13 and G15 couplings, obtained with the EMTA assays vs. those reported by Inoue et al. and the GtP datasets, the validity of the EMTA assay to detect real productive couplings, was confirmed using orthogonal assays for selective examples not reported in the two other datasets. For G12/13, we used the PKN-based BRET biosensor detecting Rho activation downstream of either G12/13 or Gq/11 (Namkung et al., 2018) and the MyrPB-Ezrin-based BRET biosensor detecting the activation of Ezrin downstream of G12/13 (Leguay et al., 2021), both in the absence of heterologously expressed G proteins. Ligand stimulation of FP and CysLT2 receptors led to Rho and ezrin activation (Figure 3—figure supplement 3A), that were insensitive to the Gq/11 inhibitor YM-254890, confirming that these receptors activate Gα12/13.
For newly identified G15 couplings, we took advantage of the lack of Gα15 in HEK293 cells and assessed the impact of Gα15 heterologous expression on receptor-mediated calcium responses (Figure 3—figure supplement 3B). For prostaglandin E2 (EP2) and κ-opioid (κOR) receptors, which couple to G15 but no other Gq/11 members, expression of Gα15 significantly increased the PGE2- and Dynorphin A- promoted calcium responses. For α2A adrenergic (α2AAR) and vasopressin 2 (V2) receptors that couple other Gq/11 family members, treatment with YM-254890 completely abolished the agonist-promoted calcium response in the absence of Gα15. In contrast, the calcium response evoked by α2AAR and V2 agonists following Gα15 expression was completely insensitive to YM-254890 (Figure 3—figure supplement 3B), confirming that these receptors can activate this YM-254890-insensitive G protein subtype (Takasaki et al., 2004).
EMTA platform detects constitutive receptor activity and biased signaling
We went on to assess the ability of the EMTA platform to detect receptor constitutive activity. Transfection of increasing amounts of adenosine A1 receptor (A1) led to a receptor-dependent increase in basal ebBRET of the Gαi2-activation sensor (Figure 5A, left), reflecting A1 constitutive activity. The A1 inverse agonist DPCPX (Lu et al., 2014) dose-dependently decreased the constitutive A1-mediated activation of Gαi2 (Figure 5A, left), indicating that EMTA can detect inverse agonism. Although we can not exclude that the high basal activity resulted from activation by adenosine in the cell culture medium, the fact that high basal activity was observed for A1 but not A3, despite a similar potency of adenosine to activate these two receptors subtypes (see Figure 5—figure supplement 1A), supports the notion that the increased basal activity reflects A1 constitutive activity.
Figure 5 with 1 supplement see all
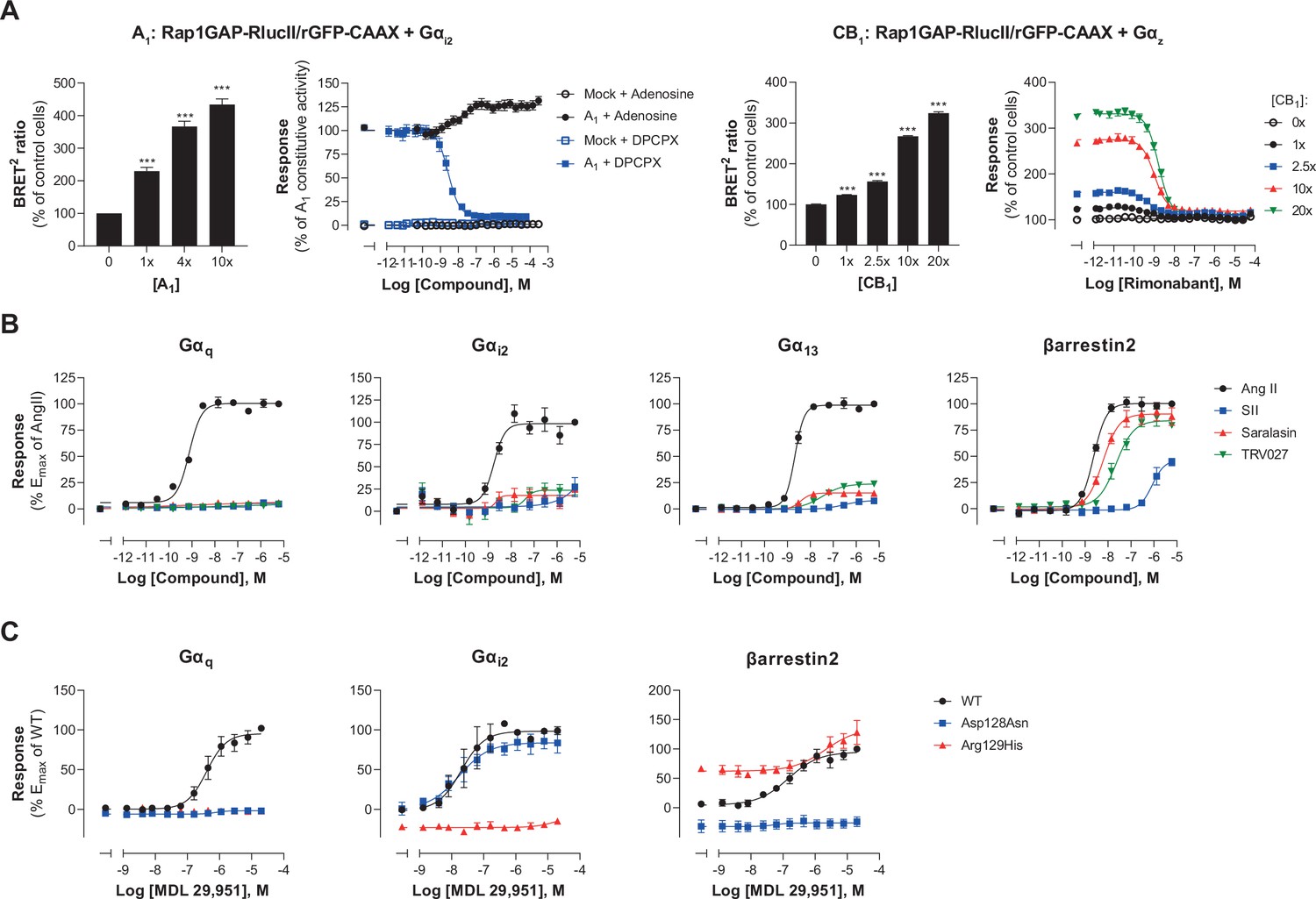
Multiple applications using the EMTA ebBRET platform.
(A) Inverse agonist activity detection. Left: Gαi2 activation in HEK293 cells transfected with the Rap1GAP-RlucII/rGFP-CAAX sensors with untagged Gαi2 and increasing amount of A1 receptor plasmid. Data are expressed in % of response obtained in control cells (0 ng of A1) and are the mean ± SEM of 4–6 independent experiments performed in two replicates. One Way ANOVA test: ***p < 0.001 compared to control cells. HEK293 cells expressing the Gαi2 activation sensor and control (Mock) or A1 receptor plasmid were stimulated (10 min) with increasing concentrations of the indicated compound. Data are expressed in % of constitutive response obtained in vehicle-treated A1 transfected cells and are the mean ± SEM of 4-6 independent experiments performed in one replicate. Right: Gαz activation in HEK293 cells transfected with the Rap1GAP-RlucII/rGFP-CAAX sensors with untagged Gαz and increasing amount of CB1 receptor plasmid. Data are expressed in % of response obtained in control cells (0 ng of CB1) and are the mean ± SEM of 4 independent experiments performed in one replicate. One Way ANOVA test: ***p < 0.001 compared to control cells. HEK293 cells expressing the Gαz activation sensor and increasing amount of CB1 receptor plasmid were directly stimulated (10 min) with increasing concentrations of the CB1 inverse agonist rimonabant. Data are expressed as % of the response obtained in control cells (0 ng of CB1) treated with vehicle and are the mean ± SEM of 4 independent experiments performed in one replicate. (B) Ligand-biased detection. Concentration-response curves of AT1 for the endogenous ligand (Angiotensin II, AngII) and biased agonists [Sar1-Ile4-Ile8] AngII (SII), saralasin or TRV027. G protein and βarrestin2 signaling activity were assessed by EMTA platform. Data are expressed in % of maximal response elicited by AngII and are the mean ± SEM of 3–6 independent experiments performed in one replicate. (C) Functional selectivity of naturally occurring receptor variants. Concentration-response curves for WT or E/DRY motif Asp128Asn and Arg129His variants of GPR17 upon agonist stimulation in HEK293 cells co-expressing the indicated EMTA biosensor. Data are expressed in % of maximal response elicited by WT receptor and are the mean ± SEM of 3 independent experiments performed in one replicate.
-
Figure 5—source data 1
Raw data of Figure 5.
- https://cdn.elifesciences.org/articles/74101/elife-74101-fig5-data1-v2.xlsx
To further confirm that the platform can adequately detect inverse agonism, a second receptor for which no endogenous ligand should be present in the media, the CB1 receptor, was used. As illustrated in Figure 5A (right), increase CB1 expression led to a ligand-independent constitutive activation of Gz, that could be completely blocked by the CB1 inverse agonist rimonabant.
EMTA also faithfully detected biased signaling. Indeed, as previously reported (Namkung et al., 2018; Wei et al., 2003), angiotensin analogs such as SII, saralasin or TRV027 displayed biased signaling by promoting efficient βarrestin2 recruitment but marginal or no Gαq, Gαi2, or Gα13 activation as compared to angiotensin II that activated all G proteins and βarrestin2 (Figure 5B). The platform was also used to identify biased signaling resulting from single nucleotide polymorphisms. As shown in Figure 5C, two naturally occurring variants of human GPR17 (isoform 2) localised in the TM3 E/DRY motif resulted in altered functional selectivity profiles. Whereas the Asp128Asn variant displayed WT-like activity on Gαi2, it lost the ability to activate Gαq and βarrestin2. In contrast, variant Arg129His at the neighboring position resulted in an increased constitutive βarrestin2 recruitment and a loss of Gαi2 and Gαq protein signaling.
Combining Gz and G15 biosensors for safety panels and systems pharmacology
The G protein coupling profiles obtained for the 100 GPCRs revealed that 95% of receptors activate either Gαz (73%) or Gα15 (81%). Measuring activation of both pathways simultaneously provides an almost universal sensor applicable to screening. Combining the two sensors (Rap1GAP-RlucII/p63-RhoGEF-RlucII/rGFP-CAAX) in the same cells allowed to detect ligand concentration-dependent activation of a safety panel of 24 GPCRs, that are well established as contributors to clinical adverse drug reactions (Bowes et al., 2012; Figure 6—figure supplement 1). Indeed, the Gz/G15 sensor captured the activation of receptors largely or uniquely coupled to either Gαz (e.g. CB2) or Gα15 (e.g. A2A and A2B), as well as receptors coupled (to varying degrees) to both pathways. The usefulness of the Gz/G15 combined sensor to detect off-target ligand activity is illustrated in Figure 6A. Most ligands tested were specific for their primary target(s). However, certain ligands displayed functional cross-reactivity with GPCRs other than their cognate targets. These included the activation of the α2AAR by dopamine and serotonin, the D2 by noradrenaline and serotonin, and of the CB1 and CB2 receptors by acetylcholine (Figure 6B–C). The activation of D2 by noradrenaline and serotonin was confirmed by the ability of the D2-family selective antagonist eticlopride to block the dopamine-, serotonin-, and noradrenaline-promoted responses detected using the combined Gz/G15 or the Gi2- and GoB-selective sensors and βarrestin2 sensor (Figure 6B, top). Similarly, use of the α2AR selective antagonist, WB4101, allowed to confirm that dopamine can activate Gαi2, GαoB and βarrestin2 through the α2AAR (Figure 6B, bottom). Such pleiotropic activation of different monoaminergic receptors by catecholamines and serotonin has been previously observed (Roth et al., 2004; Sánchez-Soto et al., 2016; Sunahara et al., 1991). Direct activation of the α2AAR by dopamine was confirmed by showing that treatment with the D2-family receptor selective antagonist eticlopride had negligible effect on dopamine-mediated activation of Gαi2 and GαoB in cells heterologously expressing α2AAR, confirming that the response did not result from the activation of endogenously expressed dopamine receptor. In contrast, eticlopride blocked the activation of Gαi2 and GαoB in cells heterologously expressing D2 (Figure 6—figure supplement 2).
Figure 6 with 2 supplements see all
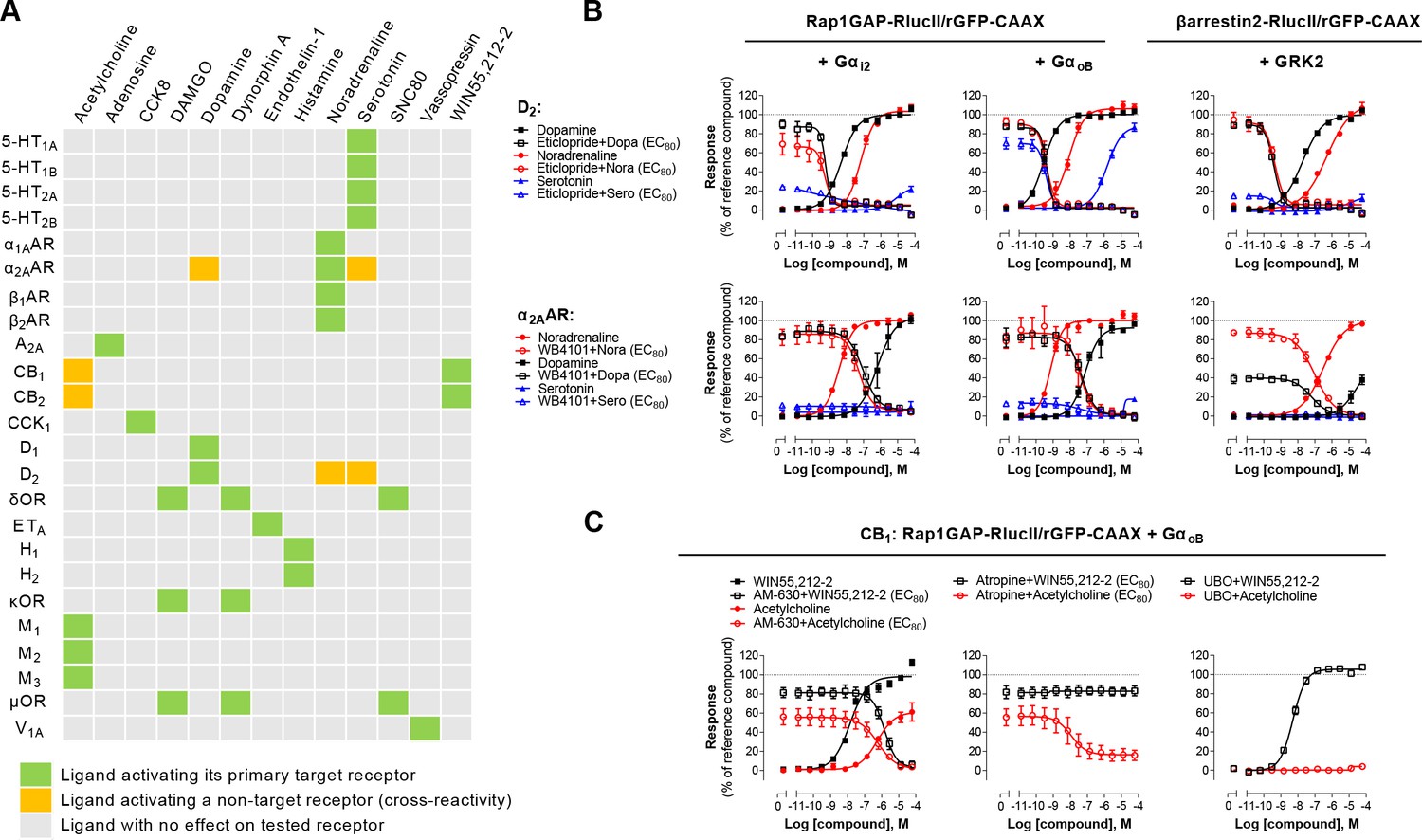
Detection of direct and indirect (trans) mechanisms of ligand polypharmacology using the Gz/G15 biosensor.
(A) Test of the Gz/G15 biosensor on a safety target panel. ebBRET signal was measured before and after stimulation with the indicated ligand in HEK293 cells transfected with the combined Gz/G15 biosensor and one of the 24 receptors listed. (B) Cross-activation of D2 and α2AAR by other natural ligands. For the agonist mode read, HEK293 cells expressing D2 or α2AAR and either the Gαi2, GαoB, or the βarrestin2 + GRK2 sensors were stimulated with increasing concentrations of the indicated ligand. For the antagonist mode read, cells were pretreated with increasing concentrations of the selective D2 antagonist eticlopride or the selective α2AAR antagonist WB4101 before stimulation with an EC80 of the indicated ligand. Data are the mean ± SEM from 3-4 independent experiments performed in one replicate and expressed in % of the response elicited by dopamine or noradrenaline for D2 and α2AAR expressing cells, respectively. (C) Indirect (trans) activation of CB1 by acetylcholine. For the agonist mode read, HEK293 cells expressing CB1 and the Rap1GAP-RlucII/rGFP-CAAX sensors with untagged GαoB were stimulated with increasing concentrations of the indicated ligand. For the antagonist mode read, same cells were pretreated or not with increasing concentrations of the CB inverse agonist AM-630 (left) or the cholinergic antagonist atropine (central) before stimulation with an EC80 of the indicated ligand. To evaluate the contribution of Gq/11-coupled receptor, cells were pretreated with the Gαq inhibitor UBO-QIC and then stimulated with increasing concentrations of the indicated ligand (right). Data are the mean ± SEM from 3-5 independent experiments performed in one replicate and expressed in % of the response elicited by WIN55,212–2.
-
Figure 6—source data 1
Raw data of Figure 6.
- https://cdn.elifesciences.org/articles/74101/elife-74101-fig6-data1-v2.xlsx
These cross-reactivity may be direct (i.e. via direct binding of a ligand to its non-cognate receptor) as suggested above, or indirect (e.g. ‘trans’, via ligand activation of its canonical receptor, leading to subsequent secretion of factors that activate the non-canonical target). One such example of trans-activation is provided by the activation of cannabinoid CB1 and CB2 receptors by acetylcholine (detected by the Gz/15 and confirmed with the GoB sensors; Figure 6A and C). Indeed, the activation was completely inhibited by both the CB inverse agonist AM-630 and by the cholinergic antagonist atropine (Figure 6C, left). Yet the response evoked by the CB selective agonist WIN55,212 2 was not blocked by atropine (Figure 6C, center). GαoB activation by acetylcholine did not result from direct activation of endogenous muscarinic receptors since no GαoB response was observed in parental cells (Figure 3—figure supplement 2). Given that the M3 muscarinic receptor, which is endogenously expressed at relatively high levels in HEK293 cells (Atwood et al., 2011), is strongly coupled to the Gq/11, CB1-expressing cells were pretreated with Gq/11/14 inhibitor UBO-QIC prior to stimulation with acetylcholine. UBO-QIC pre-treatment blocked acetylcholine- but not WIN55,212–2-mediated GαoB activation (Figure 6C, right). These results demonstrate that CB1 activation by acetylcholine is indirect and potentially involves the secretion of an endogenous CBR ligand following activation of Gq/11 by endogenous muscarinic acetylcholine receptors. The combined Gz/G15 sensor is therefore a useful tool to identify interplay between receptors and to explore systems pharmacology resulting from such cross-talks.
Discussion
This study describes the development and validation of a genetically encoded ebBRET-based biosensor platform allowing live-cell mapping of GPCR-G protein coupling preferences covering 12 heterotrimeric G proteins. The novel EMTA biosensors were combined with previously described ebBRET-based βarrestin trafficking sensors (Namkung et al., 2016), providing an unprecedented description of GPCR signaling partner couplings. In addition to providing a resource to study GPCR functional selectivity (Pándy-Szekeres et al., 2022) , the sensors provide versatile and readily usable tools to study, on a large-scale, pharmacological processes such as constitutive activity, inverse agonism, ligand-biased signaling, and signaling cross-talk.
Our EMTA-based biosensor platform offers several advantages relative to other available approaches. First, EMTA provides direct real-time measurement of proximal signaling events following GPCR activation (i.e. Gα protein activation and βarrestin recruitment) and resulting in lower level of amplification than those of assays relying on enzymatic activity of downstream effectors (i.e.: adenylyl cyclase or phospholipase C) or artificial detection systems (i.e.: gene-reporter or TGF-α shedding assays) that measure signal accumulation sometimes following extended incubation times. In addition, measuring proximal activity reduces the risk of cross-talks between pathways that may complicate data interpretation when considering downstream signaling as the readout (Mancini et al., 2015).
Second, EMTA uses native untagged GPCRs and G protein subunits (except for Gs), contrary to protein complementation (Laschet et al., 2019), FRET/BRET-based Gαβγ dissociation/receptor-G protein interaction (Bünemann et al., 2003; Galés et al., 2005; Galés et al., 2006; Hoffmann et al., 2005; Namkung et al., 2018; Olsen et al., 2020) or TGF-α shedding (Inoue et al., 2019) assays. Modifying these core-signaling components could alter responses, complicate interpretation and explain some of the discrepancies observed between the EMTA platform and other approaches used to study G protein activation. Moreover, the ability to work with unmodified receptors and G proteins (except for Gs) offers numerous advantages. First, it allows for the detection of endogenous GPCR signaling in either generic HEK293 cells (Figure 3—figure supplement 2) or more physiologically relevant cell lines such as induced pluripotent stem cell (iPSC)-derived cardiomyocytes (Figure 7A) and promyelocytic HL-60 cells (Figure 7B). Further it allows, in cells expressing sufficient endogenous level of the G proteins of interest, to detect activation of both native receptor and G proteins with no need of overexpression (Figure 7C–D). This is illustrated by the ability to detect the recruitment of Rap1GAP upon activation of the endogenous Gi/o family members by the formyl peptide receptor 2 (FPR2) in HL-60 cells (Figure 7C) or protease-activated receptor-2 (PAR2) in HEK293 cells (Figure 7D). The ability to detect the activation of endogenous G protein was also illustrated in Figure 2—figure supplement 1I, where the responses elicited by agonist stimulation were lost in cells genetically deleted of the G protein engaged by the studied receptor (i.e.: Gq/11 or Gi/o families). Recently, another BRET-based approach (Maziarz et al., 2020), taking advantage of a synthetic peptide recognizing the GTP-bound form of Gα subunits, also allows the detection of native G protein activation, offering alternative means to probe coupling selectivity profiles for both endogenously and heterologously expressed GPCRs.
Figure 7
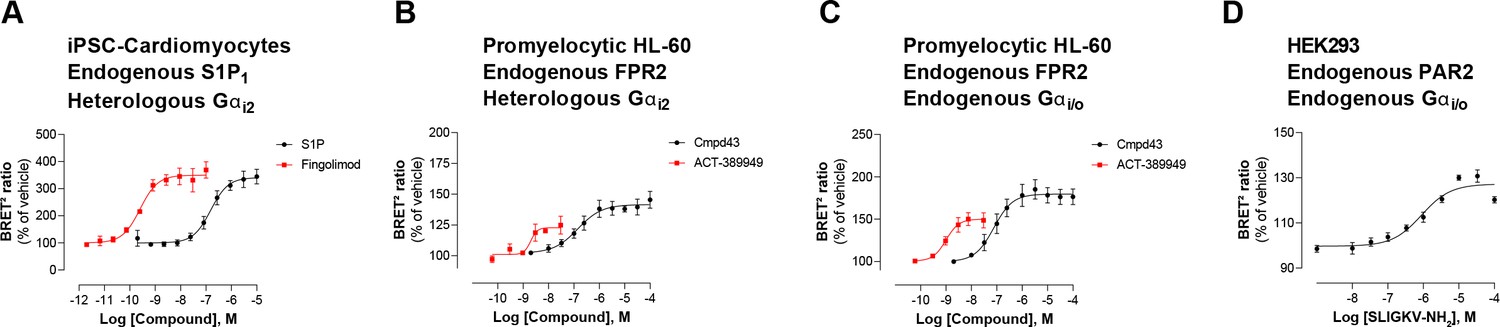
Detection of endogenous receptor- and/or G protein-mediated responses in cells with the EMTA ebBRET platform.
Concentration-dependent activation of Gαi2 protein by (A) endogenous S1P1 receptor in iPSC-derived cardiomyocytes transfected with heterologous Gαi2, (B) endogenous FPR2 in promyelocytic HL-60 cells transfected with heterologous Gαi2, (C) endogenous FPR2 in promyelocytic HL-60 cells with endogenous Gi/o proteins and (D) endogenous PAR2 receptor in HEK293 cells with endogenous Gi/o proteins. In all cases, cells were co-transfected with the Rap1GAP-RlucII/rGFP-CAAX biosensor. Data are the mean ± SEM of 3-4 independent experiments performed in one replicate and are expressed as BRET2 ratio in percentage of response induced by vehicle.
-
Figure 7—source data 1
Raw data of Figure 7.
- https://cdn.elifesciences.org/articles/74101/elife-74101-fig7-data1-v2.xlsx
Finally, similarly to BERKY, the EMTA assay platform detects the active form of the Gα subunits rather than the surrogate measurement of Gαβγ dissociation (Galés et al., 2005; Masuho et al., 2015; Maziarz et al., 2020; Mende et al., 2018), which can also detect non-productive binding as recently described for the V2 engagement of G12 (Okashah et al., 2020).
A potential caveat of EMTA is the use of common downstream effectors for all members of a given G protein family. Indeed, one cannot exclude that distinct members of a given family may display different relative affinities for their common effector. However, such differences are compensated by our data normalization that establishes the maximal response observed for a given subtype as the reference for this pathway (Figure 3A), as long as the number of the diversity of receptors included in the analysis is sufficient.
A second potential caveat of EMTA is that, when using heterologously expressed GPCRs and G proteins, some of the responses could result from favorable stoichiometries that may not exist under physiological conditions. It follows that such profiling represents the coupling possibilities of a given GPCR and not necessarily the coupling that will be observed in all cell types. Any couplings observed in such high-throughput studies requires further validation to conclude on their physiological relevance in cells or tissues of interest, and to form hypothesis for futures studies. Because we elected to use unmodified receptors (i.e.: not bearing any tags), the expression level of receptors could not be directly monitored. However, the double normalization method developed (see Materials and methods) allows quantitative comparison of coupling preferences across different receptors curtailing the influence of the assay response windows as well as receptor expression levels. Indeed, the double normalization allows ranking the coupling propensity of the receptors first as a function of the receptor which shows the strongest coupling to a specific G protein subtype, and then establishing the maximal response observed for a given G protein subtype as the reference for all G protein activated by a given receptor. In addition, as illustrated using the ETA receptor as example, titrating receptor levels did not influence the pEC50 for the activation of the different G protein coupled to this receptor (Figure 2—figure supplement 3B and Supplementary file 1C). Similarly, the pEC50 was not affected when titrating the amount of G protein subtype expressed (Figure 2—figure supplement 3A and Supplementary file 1B). As expected, only the amplitude of the response was affected.
It could be argued that overexpressing the G protein effectors (i.e.: p63-RhoGEF, Rap1GAP or PDZ-RhoGEF) used as sensors could influence the couplings observed. This potential caveat is mitigated by the fact that we used truncated part and/or modified versions of these effectors that limit the possibilities of interference with other components of the signaling machinery, and served essentially as a binding detector of the active forms of the G proteins (see Materials and methods). Supporting this notion, titrating the amount of the biosensor effector component did not affect the pEC50 of G protein activation (Figure 2—figure supplement 3C and Supplementary file 1D).
Another limitation of the EMTA platform is the lack of a soluble effector protein selective for activated Gαs thus requiring tagging of the Gαs subunit (Figure 1B, bottom) and monitoring its dissociation from the plasma membrane. Yet, our data show that this translocation reflects Gs activation state, justifying its use in a G protein activation detection platform.
Finally, because EMTA is able to detect constitutive activity, high receptor expression levels may lead to an elevated basal signal level that may obscure an agonist-promoted response. Such an example can be appreciated for the A1 receptor for which the agonist-promoted Gαi2 response did not reach the activation threshold criteria because of a very high constitutive activity level (Figure 5A). The impact of receptor expression on the constitutive activity and the narrowing on the agonist-promoted response is illustrated for Gαq activation by the 5-HT2C (Figure 5—figure supplement 1B).
A limitation of any large-scale signaling study and drug discovery program is that ligands may elicit responses downstream of receptors other than the one under study. The development of a Gz/G15 quasi-universal biosensor enables efficient screening and detection of such polypharmacology and cross-talk. Using a combination of EMTA and appropriate pharmacological tools, we also proposed a systematic approach to distinguish off-target action of ligands from cross-talk. Interestingly, the cross-talk between the M3 and CB receptors detected (Figure 6) may have physiological relevance since activation of muscarinic acetylcholine receptors has been shown to enhance the release of endocannabinoids in the hippocampus (Kim et al., 2002). The combined Gz/G15 biosensor should be particularly useful for early profiling of compound activity on safety panels and for the design of drugs displaying polypharmacology, an approach that is increasingly considered for the development of neuropsychiatric drugs (Roth et al., 2004).
The EMTA platform undoubtedly represents a novel tool-set that could be amenable for high throughput screening of small molecules and biologics across an array of signaling pathways, allowing for the discovery of functionally selective molecules or for GPCR deorphanization campaigns. The ability of the EMTA platform to quantitatively assess G protein coupling selectivity firmly expands the concept of functional selectivity and potential ligand bias beyond the dichotomic G protein vs. βarrestin view and provides plausible functional selectivity profiles that could be tested for their biological and pharmacological outcomes.
Materials and methods
Cells
HEK293 clonal cell line (HEK293SL cells), hereafter referred as HEK293 cells, were a gift from S. Laporte (McGill University, Montreal, Quebec, Canada) and previously described (Namkung et al., 2016). HEK293 cells devoid of functional Gαs (ΔGs), Gα12 and Gα13 (ΔG12/13), Gαq, Gα11, Gα14 and Gα15 (ΔGq/11) and, Gαi, and Gαo (ΔGi/o) proteins were a gift from Dr. A. Inoue (Tohoku University, Sendai, Miyagi, Japan) and previously described (Devost et al., 2017; Namkung et al., 2018; Schrage et al., 2015; Stallaert et al., 2017). Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Wisent, Saint-Jean-Baptiste, QC, Canada) supplemented with 10% fetal bovine serum (FBS, Wisent) and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin (PS); Wisent). HL-60 cells were obtained from ATCC and maintained in RPMI 1640 medium containing L-Glutamine and 25 mM HEPES (Gibco) supplemented with 20% FBS (Wisent) and 1/100 volume PS (Wisent). Differentiation of HL-60 cells into neutrophil-like cells was induced by maintaining the cells in growth medium containing 1.3% DMSO (Bioshop) during 5 days. Cardiomyocytes derived from induced pluripotent stem cells (iPSCs; iCell Cardiomyocytes) were obtained from FUJIFILM Cellular Dynamics (Madison, WI, USA) and maintained in maintenance medium provided with the cells (special formulation by FujiFilm). Cells were grown at 37 °C in 5% CO2 and 90% humidity and checked for mycoplasma contamination.
Plasmids and ebBRET biosensor constructs
Request a detailed protocolOnly human GPCRs and human Gα subunits were used in this study. An open reading frame of each full-length GPCR was cloned into pcDNA3.1(+) expression plasmid. Except when otherwise specified, GPCRs sequences were devoid of epitope tags.
Gαs-67-RlucII (Carr et al., 2014), Gαi1-loop-RlucII and GFP10-Gγ1 (Armando et al., 2014), Gαi2-loop-RlucII and βarrestin2-RlucII (Quoyer et al., 2013), GαoB-99-RlucII (Mende et al., 2018), Gαq-118-RlucII (Breton et al., 2010), Gα12-136-RlucII and PKN-RBD-RlucII (Namkung et al., 2018), Gα13-130-RlucII (Avet et al., 2020), GFP10-Gγ2 (Galés et al., 2006), βarrestin1-RlucII (Zimmerman et al., 2012), rGFP-CAAX (Namkung et al., 2016), EPAC (Leduc et al., 2009), MyrPB-Ezrin-RlucII (Leguay et al., 2021), HA-β2AR (Lavoie et al., 2002), signal peptide-Flag-AT1 (Goupil et al., 2015), and EAAC-1 (Brabet et al., 1998) were previously described. Full-length, untagged Gα subunits, Gβ1 and Gγ9 were purchased from cDNA Resource Center. GRK2 was generously provided by Dr. Antonio De Blasi (Istituto Neurologico Mediterraneo Neuromed, Pozzilli, Italy).
To selectively detect Gi/o activation, a construct coding for aa 1–442 of Rap1 GTPase-activating protein (comprising a Gi/o binding domain) fused to Rluc8, was sequence-optimized, synthetized and subcloned at TopGenetech (St-Laurent, QC, Canada). From this construct, a RlucII-tagged version of Rap1GAP (1-442) with a linker sequence (GSAGTGGRAIDIKLPAT) between Rap1GAP and RlucII was created by Gibson assembly in pCDNA3.1_Hygro (+) GFP10-RlucII, replacing GFP10. Three substitutions (i.e. S437A/S439A/S441A) were introduced into the Rap1GAP sequence by PCR-mediated mutagenesis. These putative (S437 and S439) and documented (S441) (McAvoy et al., 2009) protein kinase A phosphorylation sites were removed in order to eliminate any Gs-mediated Rap1GAP recruitment to the plasma-membrane.
To selectively detect Gq/11 activation, a construct encoding the Gq binding domain of the human p63 Rho guanine nucleotide exchange factor (p63RhoGEF; residues: 295–502) tagged with RlucII was done from IMAGE clones (OpenBiosystems; Burlington, ON, Canada) and subcloned by Gibson assembly in pCDNA3.1_Hygro (+) GFP10-RlucII, replacing GFP10. The Gq binding domain of p63RhoGEF and RlucII were separated by the peptidic linker ASGSAGTGGRAIDIKLPAT. N-term part containing palmitoylation sites maintaining p63 to plasma membrane and part of its DH domain involved in RhoA binding/activation (Aittaleb et al., 2010; Aittaleb et al., 2011) are absent of the sensor.
To selectively detect G12/13 activation, a construct encoding the G12/13 binding domain of the human PDZ-RhoGEF (residues: 281–483) tagged with RlucII was done by PCR amplification from IMAGE clones (OpenBiosystems) and subcloned by Gibson assembly in pCDNA3.1_Hygro (+) GFP10-RlucII, replacing GFP10. The peptidic linker GIRLREALKLPAT is present between RlucII and the G12/13 binding domain of PDZ-RhoGEF. The sensor is lacking the PDZ domain of PDZ-RhoGEF involved in protein-protein interaction, as well as actin-binding domain and DH/PH domains involved in GEF activity and RhoA activation (Aittaleb et al., 2010).
The sequence of each EMTA biosensors is provided in the Supplementary file 5.
Transfection
Request a detailed protocolFor BRET experiments, HEK293 cells (1.2 mL at 3.5 × 105 cells per mL) were transfected with a fixed final amount of pre-mixed biosensor-encoding DNA (0.57 μg, adjusted with salmon sperm DNA; Invitrogen) and human receptor DNA. Transfections were performed using a polyethylenimine solution (PEI, 1 mg/mL; Polysciences, Warrington, PA, USA) diluted in NaCl (150 mM, pH 7.0; 3:1 PEI/DNA ratio). Gelatin solution (1%; Sigma-Aldrich, Saint-Louis, Missouri) was used to stabilize DNA/PEI transfection mixes. Following addition of cells to the stabilized DNA/PEI transfection mix, cells were immediately seeded (3.5 × 104 cells/well) into 96-well white microplates (Greiner Bio-one; Monroe, NC, USA) and maintained in culture for the next 48 hr in DMEM containing 2% FBS and 1% PS. DMEM medium without L-glutamine (Wisent) was used for transfection of cells with mGluR to avoid receptor activation and desensitization. For Neutrophil-like differentiated HL-60 cells, cells were resuspended in electroporation medium (growth medium containing an extra 15 mM of HEPES pH 7.0) at 25 × 106 cells/mL. Electroporation reactions were prepared by adding 50 µL of DNA mastermix (20 µg total of DNA adjusted with salmon sperm DNA, supplemented with 210 mM NaCl) to 200 µL of cell suspension and transferring into 0.4 cm gap electroporation cuvettes (Bio-Rad). The cells were electroporated at 350 µF/400 V using a Bio-Rad Gene Pulser II electroporation system, washed in electroporation medium, and seeded in 96-well plates at 0.8 × 106 cells/well in 200 µL of growth medium. BRET assays were performed 6 hr post-electroporation. For iPSC Cardiomyocytes, cells were seeded in 96-well plates pretreated with fibronectin (10 µg/ml 60 min; Sigma-Aldrich) at 3.5 × 104 cells /well. After 48 hr, attached iPSCs cells were transfected with the indicated biosensor components, using TransIT-LT1 reagent (Mirus; Madison, WI, USA), according to manufacturer recommendation. BRET assays were performed 48 hr after transfection.
For Ca2+ experiments, cells (3.5 × 104 cells/well) were co-transfected with the indicated receptor, with or without Gα15 protein, using PEI and seeded in poly-ornithine-coated 96-well clear-bottom black microplates (Greiner Bio-one) and maintained in culture for the next 48 hr.
For BRET-based imagery, cells (4 × 105 cells/dish) were seeded into 35 mm poly-d-lysine-coated glass-bottom culture dishes (Mattek Corporation; Ashland, MA, USA) in 2 ml of fresh medium and incubated at 37 °C in 5% CO2, 3 day before imaging experiments. Twenty-four hours later, cells were transfected with EMTA ebBRET biosensors and the indicated receptor (i.e. p63-RhoGEF-RlucII/rGFP-CAAX + Gαq and AT1, Rap1GAP-RlucII/rGFP-CAAX + Gαi2 and D2 or PDZ-RhoGEF-RlucII/rGFP-CAAX + Gα13 and TPαR) using X-tremeGENE 9 DNA transfection reagent (3:1 reagent/DNA ratio; Roche) diluted in OptiMEM (Gibco) and maintained in culture for the next 48 hr in DMEM containing 10% FBS and 1% PS.
Bioluminescence resonance energy transfer measurement
Request a detailed protocolEnhanced bystander BRET (ebBRET) was used to monitor the activation of each Gα protein, as well as βarrestin 1 and 2 recruitment to the plasma membrane. Gαs protein activation was measured between the plasma membrane marker rGFP-CAAX and human Gαs-RlucII in the presence of human Gβ1, Gγ9 and the tested receptor. Gαs downstream cAMP production was determined using the EPAC biosensor and GPBA receptor. Gαi/o protein family activation was followed using the selective-Gi/o effector Rap1GAP-RlucII and rGFP-CAAX along with the human Gαi1, Gαi2, GαoA, GαoB, or Gαz subunits and the tested receptor. Gαq/11 protein family activation was determined using the selective-Gq/11 effector p63-RhoGEF-RlucII and rGFP-CAAX along with the human Gαq, Gα11, Gα14, or Gα15/16 subunits and the tested receptor. Gα12/13 protein family activation was monitored using the selective-G12/13 effector PDZ-RhoGEF-RlucII and rGFP-CAAX in the presence of either Gα12 or Gα13 and the tested receptor. The expression level of the Gα subunits was monitored by western blot in HEK293 cells that endogenously expressed Gαi1, Gαi2, Gα12, Gα13, Gαq, Gα11, Gα14, and Gαs but not GαoA, GαoB, Gαz, and Gα15 (Figure 2—figure supplement 6). Gα12/13-downstream activation of the Rho pathway was measured using PKN-RBD-RlucII or Ezrin-RlucII and rGFP-CAAX with the indicated receptor. βarrestin recruitment to the plasma membrane was determined using DNA mix containing rGFP-CAAX and βarrestin1-RlucII with GRK2 or βarrestin2-RlucII alone or with GRK2 and the tested receptor. Glutamate transporters EAAC-1 and EAAT-1 were systematically co-transfected with the mGluR to prevent receptor activation and desensitization by glutamate secreted in the medium by the cells (Brabet et al., 1998). All ligands were also tested for potential activation of endogenous receptors by transfecting the biosensors without receptor DNA. The Gz/G15 biosensor consists of a combination of the following plasmids: rGFP-CAAX, Rap1GAP-RlucII, Gαz, p63-RhoGEF-RlucII and Gα15. For G protein activation detection using the BRET-based Gαβγ dissociation sensors, cells were co-transfected with untagged Gβ1 and Gαq-118-RlucII, Gα12-136-RlucII or Gα13-130-RlucII with GFP10-Gγ1, or Gαi1-loop-RlucII, Gαi2-loop-RlucII or GαoB-99-RlucII with GFP10-Gγ2, along with the indicated receptor.
The day of the BRET experiment, cells were incubated in HBSS for 1 hr at room temperature (RT). Cells were then co-treated with increasing concentrations of ligand (see Appendix 1—key resources table and Supplementary file 2 for details) and the luciferase substrate coelenterazine prolume purple (1 µM, NanoLight Technologies; Pinetop, AZ, USA) for 10 min at RT. Plates were read on a Synergy Neo microplate reader (BioTek Instruments, Inc; Winooski, VT, USA) equipped with 410 ± 80 nm donor and 515 ± 30 nm acceptor filters or with a Spark microplate reader (Tecan; Männedorf, Switzerland) using the BRET2 manufacturer settings. The BRET signal (BRET²) was determined by calculating the ratio of the light intensity emitted by the acceptor over the light intensity emitted by the donor. To validate the specificity of the biosensor responses, cells were pretreated in the absence or presence of either the Gαq inhibitor UBO-QIC (100 nM, 30 min; Institute for Pharmaceutical Biology of the University of Bonn, Germany), the Gαi/o inhibitor PTX (100 ng/mL, 18 hr; List Biological Laboratories, Campbell, California, USA) or the Gαs activator CTX (0–200 ng/mL, 4 hr; Sigma-Aldrich) before stimulation with agonist. For inverse agonist activity detection of A1 or CB1 receptors, cells were stimulated during 10 min with increasing concentrations of DPCPX or rimonabant, respectively. For ligand-cross receptor activation experiments, cells were pretreated for 10 min with increasing concentrations of antagonists or inverse agonist (eticlopride for D2, WB4101 for α2AAR, atropine for muscarinic receptors and AM-630 for CB1) before a 10 min stimulation with an EC80 concentration of the indicated agonist. BRET was measured as described above. For the safety target panel ligand screen using the combined Gz/G15 sensor, basal ebBRET level was first measured 10 min following the addition of coelenterazine prolume purple (1 µM) and ebBRET level was measured again following a 10 min stimulation with a single dose of the indicated ligand (1 μM for endothelin-1 and 10 μM for all other ligands). Technical replicates for each receptor were included on the same 96-well plate. For kinetics experiment of ETA activation, basal BRET was measured during 150 s before cells stimulation with either vehicle (DMSO) or 1 µM of endothelin-1 (at time 0 sec) and BRET signal was recorded each 30 s during 3570 s. For the validation of G12/13-mediated signal by new identified G12/13-coupled receptor using PKN- or Ezrin-based BRET biosensors, cells were pretreated or not with the Gαq inhibitor YM-254890 (1 µM, 30 min; Wako Pure Chemical Industries (Fujifilm), Osaka, Japan) before agonist stimulation for 10 min. For G protein activation detection using the BRET-based Gαβγ dissociation sensors, and for titration experiments of either Gα proteins subunit with GEMTA sensors, GPCRs with GEMTA sensors or Effector-RlucII (p63-RhoGEF-RlucII for Gαq/11, Rap1GAP-RlucII for Gαi/o or PDZ-RhoGEF-RlucII for Gα12/13) from GEMTA sensors, cells were stimulated with increasing concentrations of the indicated agonist in the presence of prolume purple for 10 min before BRET measurement. For BRET in iPSC cardiomyocytes and HL-60 cells, cells were incubated in Tyrode Hepes buffer (137 mM NaCl, 0.9 mM KCl, 1 mM MgCl2, 11.9 mM NaHCO3, 3.6 mM NaH2PO4, 25 mM HEPES, 5.5 mM D-Glucose and 1 mM CaCl2, pH 7.4) 30 min at RT before being treated with increasing concentrations of agonist for 15 min, using prolume purple (2 µM) as luciferase substrate, and BRET measured.
BRET data analyses and coupling efficiency evaluation
Request a detailed protocolAll BRET ratios were standardized using the equation below and represented as universal BRET (uBRET) values: uBRET = ((BRET ratio – A)/(B-A)) * 10,000. Constants A and B correspond to the following values:
A = pre-established BRET ratio obtained from transfection of negative control (vector coding for RlucII alone).
B = pre-established BRET ratio obtained from transfection of positive control (vector coding for a GFP10-RlucII fusion protein).
For a given signaling pathway, uBRET values at each agonist concentration were normalized as the % of the response obtained in the absence of agonist (vehicle) and concentration-response curves were fitted in GraphPad Prism 8.3 software using a four-parameter logistic nonlinear regression model. Results are expressed as mean ± SEM of at least three independent experiments.
A ligand-promoted response was considered real when the Emax value was ≥to the mean + 2*SD of the response obtained in vehicle condition and that a pEC50 value could be determined in the agonist concentration range used to stimulate the receptor. Consequently, a score of 0 or 1 was assigned to each signaling pathway depending on an agonist’s ability to activate the tested pathway (0 = no activation; 1 = activation). In the case were responses associated to endogenous receptor were detectable, we considered as ‘distorted’ and exclude all the responses observed in the presence of transfected receptor for which Emax was ≤to 2*mean of the Emax value obtained with endogenous receptors or pEC50 was ≥to 2*mean of the pEC50 value obtained with endogenous receptors. Consequently, a score of 0 was assigned for these distorted responses in radial graph representation (Figure 3—figure supplement 1) and concentration-response curves were placed on a gray background in signaling signature profile panels (Supplementary file 3). Whenever transfected receptors produced an increase in Emax or a left-shift in pEC50 values compared to endogenous receptors, responses were considered ‘true’ and were assigned with a score of 1 for radial graph representation (Figure 3—figure supplement 1) and concentration-response curves were placed on a yellow background in signaling signature profile panels to indicate a partial contribution of endogenous receptors (Supplementary file 3).
We used a double normalization of Emax and pEC50 values to compare the signaling efficiency obtained for the 100 GPCRs across all receptors and pathways. Emax and pEC50 values deduced from concentration-response curves were first normalized between 0 and 1 across receptors by ranking the receptors as a function of the receptor that most efficiently activate a given pathway and then using the activation value for the pathway (including G protein and βarrestin subtypes) that a given receptor most efficiently activate as a reference for the other pathways that can be activated by this receptor. This double normalization can be translated in the following formalized equation:
STEP1: For each receptor and for each pathway:
Pathway A = Pathway specific normalized score for GPCRx on pathway A ([PSNS GPCRx]Pathway A)
where: GPCRx is receptor being analyzed, GPCRRef is the receptor giving greatest Emax on pathway A of all receptors studied (i.e. reference receptor for pathway A). A PSNS was determined for every receptor and every pathway coupled to that receptor.
STEP2: For any given receptor:
= Normalized pathway A coupling score for GPCRx
where: [PSNS GPCRx] Pathway A is the pathway specific normalized score for GPCRx on pathway A, and [PSNS GPCRx] Ref pathway is the pathway specific normalized score for the pathway giving the highest PSNS for GPCRx (i.e., reference pathway for GPCRx).
For the safety target panel ligand screen using the combined Gz/G15 sensor, the fold ligand-induced stimulation was calculated for each receptor by dividing the BRET ratio after ligand addition (measured at 10 min post stimulation) by the basal BRET ratio prior to receptor stimulation. Activation thresholds were defined as the mean + 2*SD of the ligand-stimulated response obtained with receptor-null cells expressing only the combined Gz/G15 sensor.
Ca2+ mobilization assay
Request a detailed protocolThe day of the experiment, cells were incubated with 100 μL of a Ca2+-sensitive dye-loading buffer (FLIPR calcium five assay kit, Molecular Devices; Sunnyvale, CA, USA) containing 2.5 mM probenecid (Sigma-Aldrich) for 1 hr at 37 °C in a 5% CO2 incubator. During a data run, cells in individual wells were exposed to an EC80 concentration of agonist, and fluorescent signals were recorded every 1.5 s for 3 min using the FlexStation II microplate reader (Molecular Devices). For receptors that also activate other Gq/11 family members, cells were pretreated with the Gq/11 inhibitor YM-254890 (1 µM, 30 min) before agonist stimulation. Gα15 is resistant to inhibition by YM-254890, thus allowing to measure Ca2+ responses generated specifically by Gα15.
BRET-based imaging
Request a detailed protocolBRET images were obtained as previously described (Kobayashi et al., 2019). Briefly, the day of imaging experiment, cells were carefully rinsed with HBSS, and images were acquired before and after agonists addition (100 nM for Angiotensin II and U46619, and 1 µM for dopamine) diluted in HBSS in the presence of the luciferase substrate coelenterazine prolume purple (20 µM).
Images were recorded using an inverted microscope (Nikon Eclipse Ti-U) equipped with x60 objective lens (Nikon CFI Apochromat TIRF) and EM-CCD camera (Nuvu HNu 512). Measurements were carried out in photon counting mode with EM gain 3000. Exposure time of each photon counting was 100ms. Successive 100 frames were acquired alternatively with 480 nm longpass filter (acceptor frames) or without filter (total luminescence frames), and integrated. Image integrations were repeated 10 times and 1000 frames (Video 1) or 5 times and 500 frames (Videos 2 and 3) of acceptor and total luminescence were used to generate each image.
BRET values were obtained by dividing acceptor counts by total luminescence counts pixelwise. BRET values from 0.0 to 0.8 (Video 1) or 0.0–0.5 (Videos 2 and 3) were allocated to ‘jet’ heatmap array using MATLAB 2019b. Brightness of each pixel was mapped from the signal level of total luminescence image. 0% and 99.9% signal strength were allocated to the lowest and highest brightness to exclude the influence of defective pixels with gamma correction factor of 2.0.
The movies were generated using ImageJ 1.52 a. Frame rate is 10 (Video 1) or 3 (Videos 2 and 3) frames/s, and frame interval is 20 or 100 s for Videos 1 and 2–3, respectively. The field of view of the movie is 137 µm x 137 µm.
Western blot analysis
Request a detailed protocolCells were transfected or not with the indicated biosensors mix as previously described and whole-cell extracts were prepared 48 hr later. Briefly, cells were washed with ice-cold PBS and lysed in a buffer containing 10 mM Tris buffer (pH 7.4), 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 1% Triton X-100, 10% Glycerol supplemented with protease inhibitors cocktails (Thermo Fisher Scientific). Cell lysates were centrifuged at 13,000 × g for 30 min at 4 °C. Equal amounts of proteins were separated by SDS-PAGE and transferred onto polyvinylidene fluoride membrane. The membranes were blocked (1 hr incubation at RT in TBS, 0.1% Tween-20, 5% BSA) and successively probed with primary antibody and appropriate goat secondary antibodies coupled to horseradish peroxidase (see Appendix 1-Key Resources Table). Western blots were visualized using enhanced chemiluminescence and detection was performed using a ChemiDoc MP Imaging System (BioRad). Relative densitometry analysis on protein bands was performed using MultiGauge software (Fujifilm). Results were normalized against control bands.
Statistical analyses
Request a detailed protocolCurve fitting and statistical analyses were performed using GraphPad Prism 8.3 software and methods are described in the legends of the figures. Significance was determined as p < 0.05.
Appendix 1
Key Resources Table
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (Homo-sapiens) | HEK293 | 10.1038/ncomms12178 (Namkung et al., 2016) | HEK293 clonal cell line (HEK293SL cells) | |
| Cell line (Homo-sapiens) | ΔGs HEK293 cells | Dr. A. Inoue (Tohoku University, Sendai, Miyagi, Japan) 10.1124 /mol.116.106419 (Stallaert et al., 2017) | HEK293 cells devoid of functional Gαs | |
| Cell line (Homo-sapiens) | ΔG12/13 HEK293 cells | Dr. A. Inoue (Tohoku University, Sendai, Miyagi, Japan) 10.1074/jbc.M116.763854 (Devost et al., 2017) | HEK293 cells devoid of functional Gα12 and Gα13 | |
| Cell line (Homo-sapiens) | ΔGq/11 HEK293 cells | Dr. A. Inoue (Tohoku University, Sendai, Miyagi, Japan) 10.1038/ncomms10156 (Schrage et al., 2015) | HEK293 cells devoid of functional Gαq, Gα11, Gα14 and Gα15 | |
| Cell line (Homo-sapiens) | ΔGi/o HEK293 cells | Dr. A. Inoue (Tohoku University, Sendai, Miyagi, Japan) | HEK293 cells devoid of functional Gαi and Gαo | |
| Cell line (Homo-sapiens) | HL-60 | ATCC | Cat. #: CCL-240 | |
| Cell line (Homo-sapiens) | iCell Cardiomyocytes, 01434 | FUJIFILM Cellular Dynamics | Cat. #: R1057 | |
| Transfected construct (Homo sapiens) | Human Gα subunits-encoding plasmid library | Missouri S&T cDNA Resource Center (https://www.cdna.org/) | Cat. #: GNAI100000; GNAI200000; GNA0OA0000; GNA0OB0000; GNA0Z00000; GNA1200000; GNA1300001; GNA0Q00000; GNA1100000; GNA1400000; GNA1500000; GNA0SL0000 | |
| Transfected construct (Homo sapiens) | Gβ1 | Missouri S&T cDNA Resource Center (https://www.cdna.org/) | Cat. #: GNB0100000 | |
| Transfected construct (Homo sapiens) | Gγ9 | Missouri S&T cDNA Resource Center (https://www.cdna.org/) | Cat. #: GNG0900000 | |
| Transfected construct (Homo sapiens) | Gαs-67-RlucII | 10.1074/jbc.M114.618819(Carr et al., 2014) | ||
| Transfected construct (Homo sapiens) | Gαi1-loop-RlucII | 10.1096/fj.13–242446 (Armando et al., 2014) | ||
| Transfected construct (Homo sapiens) | Gαi2-loop-RlucII | 10.1073/pnas.1312515110 (Quoyer et al., 2013) | ||
| Transfected construct (Homo sapiens) | GαoB-99-RlucII | 10.1073/pnas.1804003115(Mende et al., 2018) | ||
| Transfected construct (Homo sapiens) | Gαq-118-RlucII | 10.1016 /j.bpj.2010.10.025 (Breton et al., 2010) | ||
| Transfected construct (Homo sapiens) | Gα12–136-RlucII | 10.1126/scisignal.aat1631(Namkung et al., 2018) | ||
| Transfected construct (Homo sapiens) | Gα13–130-RlucII | 10.1038 /s42003-020-01453-8 (Avet et al., 2020) | ||
| Transfected construct (Homo sapiens) | GFP10-Gγ1 | 10.1096/fj.13–242446 (Armando et al., 2014) | ||
| Transfected construct (Homo sapiens) | GFP10-Gγ2 | 10.1038/nsmb1134(Galés et al., 2006) | ||
| Transfected construct (Homo sapiens) | EPAC | 10.1124/jpet.109.156398(Leduc et al., 2009) | ||
| Transfected construct (Homo sapiens) | rGFP-CAAX | 10.1038/ncomms12178(Namkung et al., 2016) | ||
| Transfected construct (Homo sapiens) | Rap1GAP-RlucII | This paper | See Materials and Methods | |
| Transfected construct (Homo sapiens) | p63-RhoGEF-RlucII | This paper | See Materials and Methods | |
| Transfected construct (Homo sapiens) | PDZ-RhoGEF-RlucII | This paper | See Materials and Methods | |
| Transfected construct (Homo sapiens) | PKN-RBD-RlucII | 10.1126/scisignal.aat1631(Namkung et al., 2018) | ||
| Transfected construct (Homo sapiens) | MyrPB-Ezrin-RlucII | 10.1242/jcs.255307(Leguay et al., 2021) | ||
| Transfected construct (Homo sapiens) | βarrestin1-RlucII | 10.1126/scisignal.2002522(Zimmerman et al., 2012) | ||
| Transfected construct (Homo sapiens) | βarrestin2-RlucII | 10.1073/pnas.1312515110 (Quoyer et al., 2013) | ||
| Transfected construct (Homo sapiens) | GRK2 | This paper | See Materials and Methods | |
| Transfected construct (Homo sapiens) | RAMP3 | Domain Therapeutics North America | N/A | |
| Transfected construct (Homo sapiens) | EAAC-1 | 10.1016/s0028-3908(98)00091-4 (Brabet et al., 1998) | ||
| Transfected construct (Homo sapiens) | EAAT-1 | Domain Therapeutics North America | N/A | |
| Transfected construct (Homo sapiens) | signal peptide-Flag-AT1 | 10.1074/jbc.M114.631119(Goupil et al., 2015) | ||
| Transfected construct (Homo sapiens) | FLAG-α2BAR | Domain Therapeutics North America | N/A | |
| Transfected construct (Homo sapiens) | HA-β2AR | 10.1074/jbc.M204163200(Lavoie et al., 2002) | ||
| Antibody | Gαi1 (I-20) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-391 RRID: AB_2247692 | WB (1:500) |
| Antibody | Gαi2 (T-19) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-7276 RRID:AB_2111472 | WB (1:500) |
| Antibody | Gαo (K-20) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-387 RRID:AB_2111641 | WB (1:500) |
| Antibody | Gαz (Rabbit monoclonal) | Abcam | Cat. #: ab154846 | WB (1:1000) |
| Antibody | Gαs (K-20) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-823 RRID:AB_631538 | WB (1:500) |
| Antibody | Gα12 (S-20) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-409 RRID:AB_2263416 | WB (1:500) |
| Antibody | Gα13 (A-20) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-410 RRID:AB_2279044 | WB (1:500) |
| Antibody | Gαq (E-17) (Rabbit polyclonal) | Santa Cruz | Cat. #: sc-393 RRID:AB_631536 | WB (1:500) |
| Antibody | Gα11 (C-terminal) (Rabbit polyclonal) | Sigma-Aldrich | Cat. #: SAB2109181 | WB (1:500) |
| Antibody | Gα14 (Rabbit polyclonal) | Sigma-Aldrich | Cat. #: SAB4300771 | WB (1:500) |
| Antibody | Gα15 (Rabbit polyclonal) | ThermoFisher scientific (Pierce) | Cat. #: PA1-29022 RRID:AB_1958024 | WB (1:5,000) |
| Antibody | βactin (Mouse monoclonal) | Sigma-Aldrich | Cat. #: A5441 RRID:AB_476744 | WB (1:5,000) |
| Antibody | Anti-rabbit HRP-coupled (Donkey polyclonal) | GE Healthcare | Cat. #: NA934 RRID:AB_772206 | WB (1:5,000) |
| Antibody | Anti-mouse HRP-coupled (Sheep polyclonal) | GE Healthcare | Cat. #: NA931 RRID:AB_772210 | WB (1:10,000) |
| Commercial assay or kit | FLIPR Calcium 5 Assay Kit | Molecular Devices | Cat. #: R8185 | |
| Chemical compound, drug | α-linolenic acid | Cayman Chemical | Cat. #: 21,910 | |
| Chemical compound, drug | α-MSH | Sigma-Aldrich | Cat. #: M4135 | |
| Chemical compound, drug | γ-MSH | Tocris | Cat. #: 4,272 | |
| Chemical compound, drug | [Pyr1]-Apelin 13 | Tocris | Cat. #: 2,420 | |
| Chemical compound, drug | [Sar1, Ile4,8]-Angiotensin II | Peptides International | Cat. #: PAN-4476-V-1EA | |
| Chemical compound, drug | 3-hydroxyoctanoic acid (3-HOA) | Sigma-Aldrich | Cat. #: H3898 | |
| Chemical compound, drug | 7α–25 dihydroxycholesterol | Sigma-Aldrich | Cat. #: SML0541 | |
| Chemical compound, drug | Acetylcholine chloride | Tocris | Cat. #: 2,809 | |
| Chemical compound, drug | ACT-389949 | Provided by Bristol-Myers Squibb | N/A | |
| Chemical compound, drug | Adenosine | Sigma-Aldrich | Cat. #: A9251 | |
| Chemical compound, drug | AM-630 | Tocris | Cat. #: 1,120 | |
| Chemical compound, drug | Amylin | Tocris | Cat. #: 3,418 | |
| Chemical compound, drug | Angiotensin II (Ang II) | Sigma-Aldrich | Cat. #: A9525 | |
| Chemical compound, drug | Arginine vasopressin (AVP) | Sigma-Aldrich | Cat. #: V9879 | |
| Chemical compound, drug | Atropine | Sigma-Aldrich | Cat. #: A0132 | |
| Chemical compound, drug | Bovine serum albumin | Sigma-Aldrich | Cat. #: A7030 | |
| Chemical compound, drug | C5a | Complement Technology | Cat. #: A144(300) | |
| Chemical compound, drug | Calcitonin | Bachem | Cat. #: H-2250 | |
| Chemical compound, drug | CCK Octapeptide, sulfated (CCK8) | Tocris | Cat. #: 1,166 | |
| Chemical compound, drug | CCL20 | R&D Systems | Cat. #: 360-MP/CF | |
| Chemical compound, drug | CCL3 (MIP-1a) | R&D Systems | Cat. #: 270-LD/CF | |
| Chemical compound, drug | Cholera Toxin (CTX) from Vibrio cholerae | Sigma-Aldrich | Cat. #: C8052 | |
| Chemical compound, drug | Cmpd43 | Provided by Bristol-Myers Squibb | N/A | |
| Chemical compound, drug | Corticotropin-Releasing Factor (CRF) | Bachem | Cat. #: H-2435 | |
| Chemical compound, drug | CXCL12 | R&D Systems | Cat. #: 350-NS | |
| Chemical compound, drug | CXCL13 | R&D Systems | Cat. #: 801 CX/CF | |
| Chemical compound, drug | CXCL8 | R&D Systems | Cat. #: 208-IL/CF | |
| Chemical compound, drug | DAMGO | Tocris | Cat. #: 1,171 | |
| Chemical compound, drug | Dopamine | Sigma-Aldrich | Cat. #: H8502 | |
| Chemical compound, drug | DPCPX | Tocris | Cat. #: 0439 | |
| Chemical compound, drug | Dynorphin A | Tocris | Cat. #: 3,195 | |
| Chemical compound, drug | Endothelin-1 | Tocris | Cat. #: 1,160 | |
| Chemical compound, drug | Eticlopride | Tocris | Cat. #: 1,847 | |
| Chemical compound, drug | Fingolimod | Provided by Bristol-Myers Squibb | N/A | |
| Chemical compound, drug | Gastric Inhibitory Peptide (GIP) | Bachem | Cat. #: H-5645 | |
| Chemical compound, drug | Ghrelin | Tocris | Cat. #: 1,463 | |
| Chemical compound, drug | Glucagon (Aittaleb et al., 2010; Aittaleb et al., 2011; Armando et al., 2014; Atwood et al., 2011; Avet et al., 2020; Azzi et al., 2003; Bowes et al., 2012; Brabet et al., 1998; Breton et al., 2010; Bünemann et al., 2003; Carr et al., 2014; Casey et al., 1990; Chandan et al., 2021; De Haan and Hirst, 2004; Devost et al., 2017; Fukuhara et al., 2001; Galandrin et al., 2007; Galés et al., 2005; Galés et al., 2006; Goupil et al., 2015; Hauser et al., 2017; Hauser et al., 2022; Hoffmann et al., 2005; Inoue et al., 2019; Jordan et al., 1999; Kawamata et al., 2003; Kenakin, 2019; Kim et al., 2002) | Bachem | Cat. #: H-6790 | |
| Chemical compound, drug | Glucagon-like peptide-1 GLP-1 (Bowes et al., 2012; Brabet et al., 1998; Breton et al., 2010; Bünemann et al., 2003; Carr et al., 2014; Casey et al., 1990; Chandan et al., 2021; De Haan and Hirst, 2004; Devost et al., 2017; Fukuhara et al., 2001; Galandrin et al., 2007; Galés et al., 2005; Galés et al., 2006; Goupil et al., 2015; Hauser et al., 2017; Hauser et al., 2022; Hoffmann et al., 2005; Inoue et al., 2019; Jordan et al., 1999; Kawamata et al., 2003; Kenakin, 2019; Kim et al., 2002; Kobayashi et al., 2019; Laschet et al., 2019; Lavoie et al., 2002; Leduc et al., 2009; Leguay et al., 2021; Lu et al., 2014; Lutz et al., 2007) | Bachem | Cat. #: H-6795 | |
| Chemical compound, drug | Glucagon-like peptide-2 GLP-2 (Aittaleb et al., 2010; Aittaleb et al., 2011; Armando et al., 2014; Atwood et al., 2011; Avet et al., 2020; Azzi et al., 2003; Bowes et al., 2012; Brabet et al., 1998; Breton et al., 2010; Bünemann et al., 2003; Carr et al., 2014; Casey et al., 1990; Chandan et al., 2021; De Haan and Hirst, 2004; Devost et al., 2017; Fukuhara et al., 2001; Galandrin et al., 2007; Galés et al., 2005; Galés et al., 2006; Goupil et al., 2015; Hauser et al., 2017; Hauser et al., 2022; Hoffmann et al., 2005; Inoue et al., 2019; Jordan et al., 1999; Kawamata et al., 2003; Kenakin, 2019; Kim et al., 2002; Kobayashi et al., 2019; Laschet et al., 2019; Lavoie et al., 2002; Leduc et al., 2009) | Bachem | Cat. #: H-7742 | |
| Chemical compound, drug | Glutamate | Sigma-Aldrich | Cat. #: 49,621 | |
| Chemical compound, drug | GnRH (LH-RH) | Peptides International | Cat. #: PLR-4013 | |
| Chemical compound, drug | Histamine | Tocris | Cat. #: 3,545 | |
| Chemical compound, drug | Kallidin | Anaspec | Cat. #: 22,853(AN) | |
| Chemical compound, drug | Leukotriene B4 (LTB4) | Cayman Chemical | Cat. #: 20,110 | |
| Chemical compound, drug | Leukotriene D4 (LTD4) | Cayman Chemical | Cat. #: 20,310 | |
| Chemical compound, drug | Litocholic acid | Sigma-Aldrich | Cat. #: L6250 | |
| Chemical compound, drug | Melatonin | Bachem | Cat. #: Q-1300 | |
| Chemical compound, drug | MDL 29,951 | Cayman Chemical | Cat. #: 16,266 | |
| Chemical compound, drug | Neuropeptide FF (NPFF) | Tocris | Cat. #: 3,137 | |
| Chemical compound, drug | Neuropeptide Y (NPY) | Bachem | Cat. #: H-6375 | |
| Chemical compound, drug | Nicotinic acid | Abcam | Cat. #: ab120145 | |
| Chemical compound, drug | Nociceptin | Tocris | Cat. #: 910 | |
| Chemical compound, drug | Noradrenaline | Tocris | Cat. #: 5,169 | |
| Chemical compound, drug | Oleoyl-Lysophosphatidic acid (O-LPA) | Sigma-Aldrich | Cat. #: L7260 | |
| Chemical compound, drug | Orexin-A | Bachem | Cat. #: H-4172 | |
| Chemical compound, drug | Oxytocin | Tocris | Cat. #: 1910 | |
| Chemical compound, drug | Parathyroid Hormone (Aittaleb et al., 2010; Aittaleb et al., 2011; Armando et al., 2014; Atwood et al., 2011; Avet et al., 2020; Azzi et al., 2003; Bowes et al., 2012; Brabet et al., 1998; Breton et al., 2010; Bünemann et al., 2003; Carr et al., 2014; Casey et al., 1990; Chandan et al., 2021; De Haan and Hirst, 2004; Devost et al., 2017; Fukuhara et al., 2001; Galandrin et al., 2007; Galés et al., 2005; Galés et al., 2006; Goupil et al., 2015; Hauser et al., 2017; Hauser et al., 2022; Hoffmann et al., 2005; Inoue et al., 2019; Jordan et al., 1999; Kawamata et al., 2003; Kenakin, 2019; Kim et al., 2002; Kobayashi et al., 2019; Laschet et al., 2019; Lavoie et al., 2002; Leduc et al., 2009; Leguay et al., 2021) | Sigma-Aldrich | Cat. #: P3796 | |
| Chemical compound, drug | Pertussis toxin (PTX) from Bordetella pertussis | List Biological Laboratories | Cat. #: 179 A(LB) | |
| Chemical compound, drug | pH (proton) (Hydrochloric acid) | Sigma-Aldrich | Cat. #: 320,331 | |
| Chemical compound, drug | Probenecid | Sigma-Aldrich | Cat. #: P8761 | |
| Chemical compound, drug | Prolume Purple (methoxy e-Coelenterazine; Me-O-e-CTZ) | Nanolight | Cat. #: 369 | |
| Chemical compound, drug | Propionate (sodium salt) | Sigma-Aldrich | Cat. #: P1880 | |
| Chemical compound, drug | Prostaglandin D2 (PGD2) | Cayman Chemical | Cat. #: 12,010 | |
| Chemical compound, drug | Prostaglandin E2 (PGE2) | Sigma-Aldrich | Cat. #: P0409 | |
| Chemical compound, drug | RFamide-related peptide 3 (RFRP3) | Tocris | Cat. #: 4,683 | |
| Chemical compound, drug | Rimonabant | Cayman Chemical | Cat. #: 9000484 | |
| Chemical compound, drug | Saralasin | ApexBio | Cat. #: B5063 | |
| Chemical compound, drug | Serotonin | Cayman Chemical | Cat. #: 14,332 | |
| Chemical compound, drug | SLIGKV-NH2 (PAR2 AP) | Tocris | Cat. #: 3,010 | |
| Chemical compound, drug | SNC80 | Sigma-Aldrich | Cat. #: S2812 | |
| Chemical compound, drug | Somatostatin-14 | Bachem | Cat. #: H-6276 | |
| Chemical compound, drug | Sphingosine 1-phosphate | Cayman | Cat. #: 62,570 | |
| Chemical compound, drug | TFLLR-NH2 (PAR1 AP) | Tocris | Cat. #: 1,464 | |
| Chemical compound, drug | TRV027 | Provided by Bristol-Myers Squibb | N/A | |
| Chemical compound, drug | UBO-QIC (FR900359) | Institute for Pharmaceutical Biology of the University of Bonn | N/A | |
| Chemical compound, drug | Undecanoic acid | Sigma-Aldrich | Cat. #: 171,476 | |
| Chemical compound, drug | Urocortin II | Phoenix Pharmaceutical | Cat. #: 019–30 | |
| Chemical compound, drug | UTP | Sigma-Aldrich | Cat. #: U1006 | |
| Chemical compound, drug | Vasoactive Intestinal Peptide (VIP) | Tocris | Cat. #: 1911 | |
| Chemical compound, drug | WB4101 | Tocris | Cat. #: 946 | |
| Chemical compound, drug | WIN55,212–2 | Enzo Life Sciences | Cat. #: BMLCR105 | |
| Chemical compound, drug | YM-254890 | Wako Pure Chemical Industries (Fujifilm) | Cat. #: 257–00631 | |
| Chemical compound, drug | Zinc chloride (Zn2+) | Sigma-Aldrich | Cat. #: 229,997 | |
| Software, algorithm | Prism, Version 8.3 | GraphPad | ||
| Software, algorithm | MATLAB, Version R2019b | MathWorks | ||
| Software, algorithm | ImageJ, Version 1.52 a | NIH https://imagej.nih.gov/ij/ | ||
| Software, algorithm | Scipy, Version 1.4.1 | https://www.scipy.org | ||
| Other | 96 W white plate | Greiner Bio-one | Cat. #: 655,083 | |
| Other | 96 W black plate, clear-bottom | Greiner Bio-one | Cat. #: 655,090 | |
| Other | OptiPlate-384, White Opaque 384-well Microplate | Perkin Elmer | Cat. #: 6007290 | |
| Other | 35 mm poly-d-lysine-coated glass-bottom culture dishes | Mattek | Cat. #: P35GC-1.5–14 C | |
| Other | Microplate washer | BioTek Instruments | Cat. #: 405TSUS | |
| Other | D300e Digital Dispenser | Tecan | ||
| Other | T8 + Dispensehead Cassettes | Hp (Tecan) | Cat. #: 30097370 | |
| Other | Synergy NEO Luminescence microplate reader | BioTek Instruments | ||
| Other | FlexStation 2 Multi-mode, auto-pipetting microplate reader | Molecular Devices | ||
| Other | Inverted microscope | Nikon Eclipse Ti-U | ||
| Other | x60 objective lens | Nikon CFI Apochromat TIRF | ||
| Other | EMCCD camera | Nuvu HNu 512 |
Data availability
All data generated or analysed during this study are included in the manuscript and supporting file; Source Data files have been provided for Figures 2, 4, 5, 6 and 7 and associated figure supplements. Supplementary file 1 contains the numerical data used to generate Figure 2-figure supplement 1 and Figure 2-figure supplement 3. Supplementary File 2 contains the numerical data used to generate figure 3. Further data are available in the companion paper co-published in eLife: https://doi.org/10.7554/eLife.74107.
References
-
Plasma membrane association of p63 Rho guanine nucleotide exchange factor (p63RhoGEF) is mediated by palmitoylation and is required for basal activity in cellsThe Journal of Biological Chemistry 286:34448–34456.https://doi.org/10.1074/jbc.M111.273342
-
Reducing safety-related drug attrition: the use of in vitro pharmacological profilingNature Reviews. Drug Discovery 11:909–922.https://doi.org/10.1038/nrd3845
-
Multiplexing of multicolor bioluminescence resonance energy transferBiophysical Journal 99:4037–4046.https://doi.org/10.1016/j.bpj.2010.10.025
-
Development and characterization of pepducins as Gs-biased allosteric agonistsThe Journal of Biological Chemistry 289:35668–35684.https://doi.org/10.1074/jbc.M114.618819
-
Gz, a guanine nucleotide-binding protein with unique biochemical propertiesThe Journal of Biological Chemistry 265:2383–2390.
-
Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review)Molecular Membrane Biology 21:77–92.https://doi.org/10.1080/09687680410001663267
-
Conformational Profiling of the AT1 Angiotensin II Receptor Reflects Biased AgonismG Protein Coupling, and Cellular Context. J Biol Chem 292:5443–5456.https://doi.org/10.1074/jbc.M116.763854
-
The evasive nature of drug efficacy: implications for drug discoveryTrends in Pharmacological Sciences 28:423–430.https://doi.org/10.1016/j.tips.2007.06.005
-
Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexesNature Structural & Molecular Biology 13:778–786.https://doi.org/10.1038/nsmb1134
-
Angiotensin II type I and prostaglandin F2alpha receptors cooperatively modulate signaling in vascular smooth muscle cellsThe Journal of Biological Chemistry 290:3137–3148.https://doi.org/10.1074/jbc.M114.631119
-
Trends in GPCR drug discovery: new agents, targets and indicationsNature Reviews. Drug Discovery 16:829–842.https://doi.org/10.1038/nrd.2017.178
-
Modulation of rap activity by direct interaction of Galpha(o) with Rap1 GTPase-activating proteinThe Journal of Biological Chemistry 274:21507–21510.https://doi.org/10.1074/jbc.274.31.21507
-
A G protein-coupled receptor responsive to bile acidsThe Journal of Biological Chemistry 278:9435–9440.https://doi.org/10.1074/jbc.M209706200
-
Biased Receptor Signaling in Drug DiscoveryPharmacological Reviews 71:267–315.https://doi.org/10.1124/pr.118.016790
-
Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampusThe Journal of Neuroscience 22:10182–10191.https://doi.org/10.1523/JNEUROSCI.22-23-10182.2002
-
A dynamic and screening-compatible nanoluciferase-based complementation assay enables profiling of individual GPCR-G protein interactionsThe Journal of Biological Chemistry 294:4079–4090.https://doi.org/10.1074/jbc.RA118.006231
-
Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacyThe Journal of Biological Chemistry 277:35402–35410.https://doi.org/10.1074/jbc.M204163200
-
Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligandsThe Journal of Pharmacology and Experimental Therapeutics 331:297–307.https://doi.org/10.1124/jpet.109.156398
-
Development of conformational BRET biosensors that monitor ezrin, radixin and moesin activation in real timeJournal of Cell Science 134:jcs255307.https://doi.org/10.1242/jcs.255307
-
Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRsScience (New York, N.Y.) 318:1923–1927.https://doi.org/10.1126/science.1147554
-
Exploring the Technology Landscape of 7TMR Drug Signaling ProfilingCurrent Topics in Medicinal Chemistry 15:2528–2542.https://doi.org/10.2174/1568026615666150701113344
-
Activated G Protein Galphas Samples Multiple Endomembrane CompartmentsThe Journal of Biological Chemistry 291:20295–20302.https://doi.org/10.1074/jbc.M116.729731
-
Functional interaction between Galpha(z) and Rap1GAP suggests a novel form of cellular cross-talkThe Journal of Biological Chemistry 274:36663–36669.https://doi.org/10.1074/jbc.274.51.36663
-
Heterotrimeric G protein activation by G-protein-coupled receptorsNature Reviews. Molecular Cell Biology 9:60–71.https://doi.org/10.1038/nrm2299
-
TRUPATH, an open-source biosensor platform for interrogating the GPCR transduceromeNature Chemical Biology 16:841–849.https://doi.org/10.1038/s41589-020-0535-8
-
The G protein database, GproteinDbNucleic Acids Research 50:D518–D525.https://doi.org/10.1093/nar/gkab852
-
Galphaq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domainThe Journal of Biological Chemistry 282:29201–29210.https://doi.org/10.1074/jbc.M703458200
-
Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophreniaNature Reviews. Drug Discovery 3:353–359.https://doi.org/10.1038/nrd1346
-
The experimental power of FR900359 to study Gq-regulated biological processesNature Communications 6:10156.https://doi.org/10.1038/ncomms10156
-
A novel Galphaq/11-selective inhibitorThe Journal of Biological Chemistry 279:47438–47445.https://doi.org/10.1074/jbc.M408846200
-
Activation-induced subcellular redistribution of Gs alphaMolecular Biology of the Cell 7:1225–1233.https://doi.org/10.1091/mbc.7.8.1225
Article and author information
Author details
Alexander S Hauser

Hiroyuki Kobayashi
Funding
Canada Research Chairs
- Michel Bouvier
Canadian Institutes of Health Research (FDN-148431)
- Michel Bouvier
Lundbeckfonden (R218-2016-1266)
- David E Gloriam
Lundbeckfonden (R313-2019-526)
- David E Gloriam
Novo Nordisk Fonden (NNF18OC0031226)
- David E Gloriam
Lundbeckfonden (R278-2018-180)
- Alexander S Hauser
Bristol-Myers Squibb
- Michel Bouvier
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Shane C Wright for scientific discussion and Monique Lagacé for critical reading of the manuscript. The authors are grateful to the funding from Bristol-Myers Squibb that supported the detection of Gα proteins by endogenous receptors in iPSC cardiomyocytes and promyelocytic HL-60 cells.
Copyright
© 2022, Avet et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 9,165
- views
-
- 1,489
- downloads
-
- 177
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs
eLife 11:e74101.
https://doi.org/10.7554/eLife.74101
Further reading
-
- Biochemistry and Chemical Biology
In healthy cells, cyclin D1 is expressed during the G1 phase of the cell cycle, where it activates CDK4 and CDK6. Its dysregulation is a well-established oncogenic driver in numerous human cancers. The cancer-related function of cyclin D1 has been primarily studied by focusing on the phosphorylation of the retinoblastoma (RB) gene product. Here, using an integrative approach combining bioinformatic analyses and biochemical experiments, we show that GTSE1 (G-Two and S phases expressed protein 1), a protein positively regulating cell cycle progression, is a previously unrecognized substrate of cyclin D1–CDK4/6 in tumor cells overexpressing cyclin D1 during G1 and subsequent phases. The phosphorylation of GTSE1 mediated by cyclin D1–CDK4/6 inhibits GTSE1 degradation, leading to high levels of GTSE1 across all cell cycle phases. Functionally, the phosphorylation of GTSE1 promotes cellular proliferation and is associated with poor prognosis within a pan-cancer cohort. Our findings provide insights into cyclin D1’s role in cell cycle control and oncogenesis beyond RB phosphorylation.
-
- Biochemistry and Chemical Biology
- Microbiology and Infectious Disease
Teichoic acids (TA) are linear phospho-saccharidic polymers and important constituents of the cell envelope of Gram-positive bacteria, either bound to the peptidoglycan as wall teichoic acids (WTA) or to the membrane as lipoteichoic acids (LTA). The composition of TA varies greatly but the presence of both WTA and LTA is highly conserved, hinting at an underlying fundamental function that is distinct from their specific roles in diverse organisms. We report the observation of a periplasmic space in Streptococcus pneumoniae by cryo-electron microscopy of vitreous sections. The thickness and appearance of this region change upon deletion of genes involved in the attachment of TA, supporting their role in the maintenance of a periplasmic space in Gram-positive bacteria as a possible universal function. Consequences of these mutations were further examined by super-resolved microscopy, following metabolic labeling and fluorophore coupling by click chemistry. This novel labeling method also enabled in-gel analysis of cell fractions. With this approach, we were able to titrate the actual amount of TA per cell and to determine the ratio of WTA to LTA. In addition, we followed the change of TA length during growth phases, and discovered that a mutant devoid of LTA accumulates the membrane-bound polymerized TA precursor.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}