Inhibition of mutant RAS-RAF interaction by mimicking structural and dynamic properties of phosphorylated RAS
- Graduate School of Engineering and Natural Sciences, Istanbul Medipol University, Turkey
- Medical Biology and Genetics Program, Graduate School for Health Sciences, Istanbul Medipol University, Turkey
- Cancer Research Center, Institute for Health Sciences and Technologies (SABITA), Istanbul Medipol University, Turkey
- Faculty of Engineering and Natural Sciences, Sabanci University, Turkey
- Department of Medical Biology, International School of Medicine, Istanbul Medipol University, Turkey
- Department of Computer Engineering, School of Engineering and Natural Sciences, Istanbul Medipol University, Turkey
- Regenerative and Restorative Medicine Research Center (REMER), Institute for Health Sciences and Technologies (SABITA), Istanbul Medipol University, Turkey
Figures
Figure 1
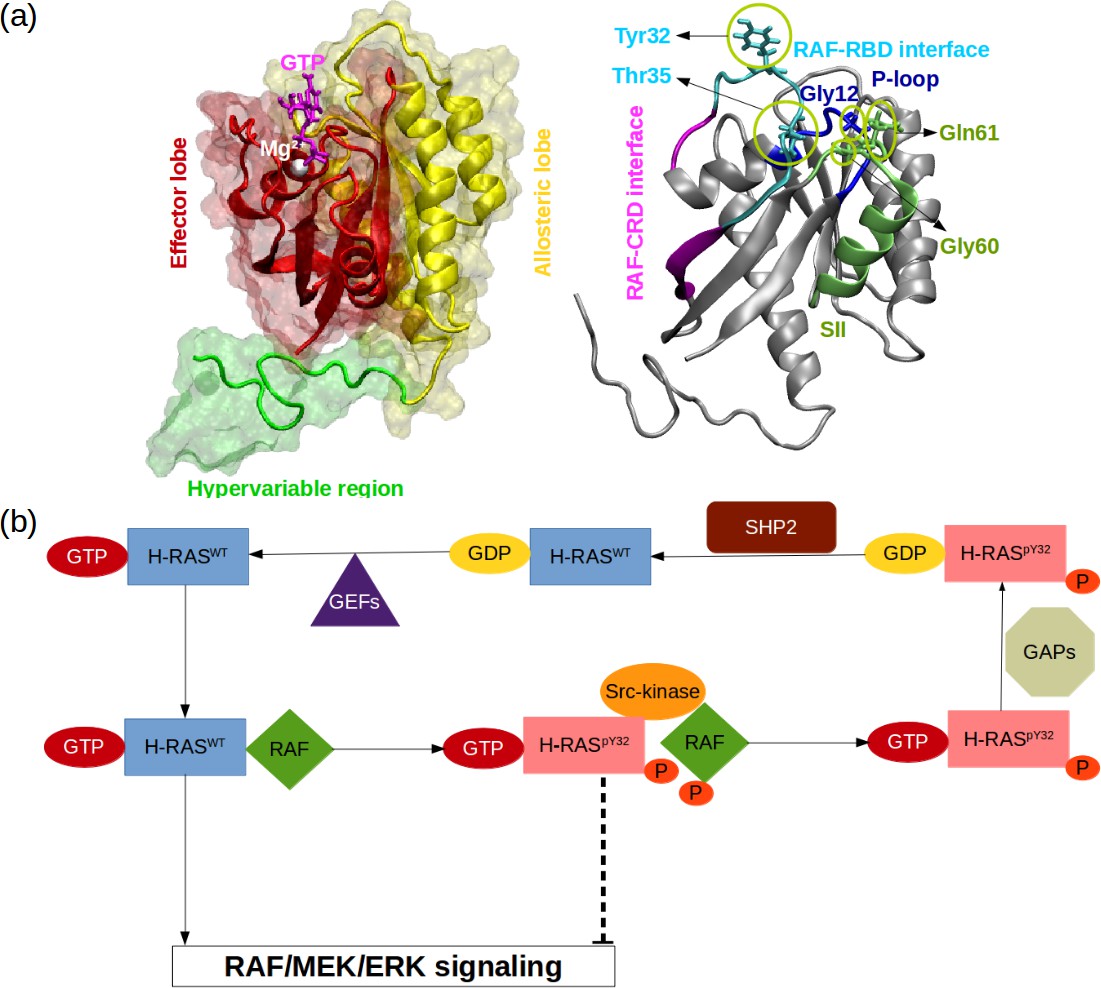
RAS phosphorylation/dephosphorylation cycle.
(a) Important residues/regions that play pivotal role in RAS function are shown. (b) A schematic that illustrates the impact of tyrosyl phosphorylation on the GTPase cycle of HRAS. The tyrosyl phosphorylation at the 32nd position, which is mediated by Src kinase, causes impairment of RAF binding, thus terminating RAF/MEK/ERK signaling pathway as long as the phosphoryl group of Y32 is not detached by SHP2 Bunda et al., 2014.
Figure 2 with 1 supplement
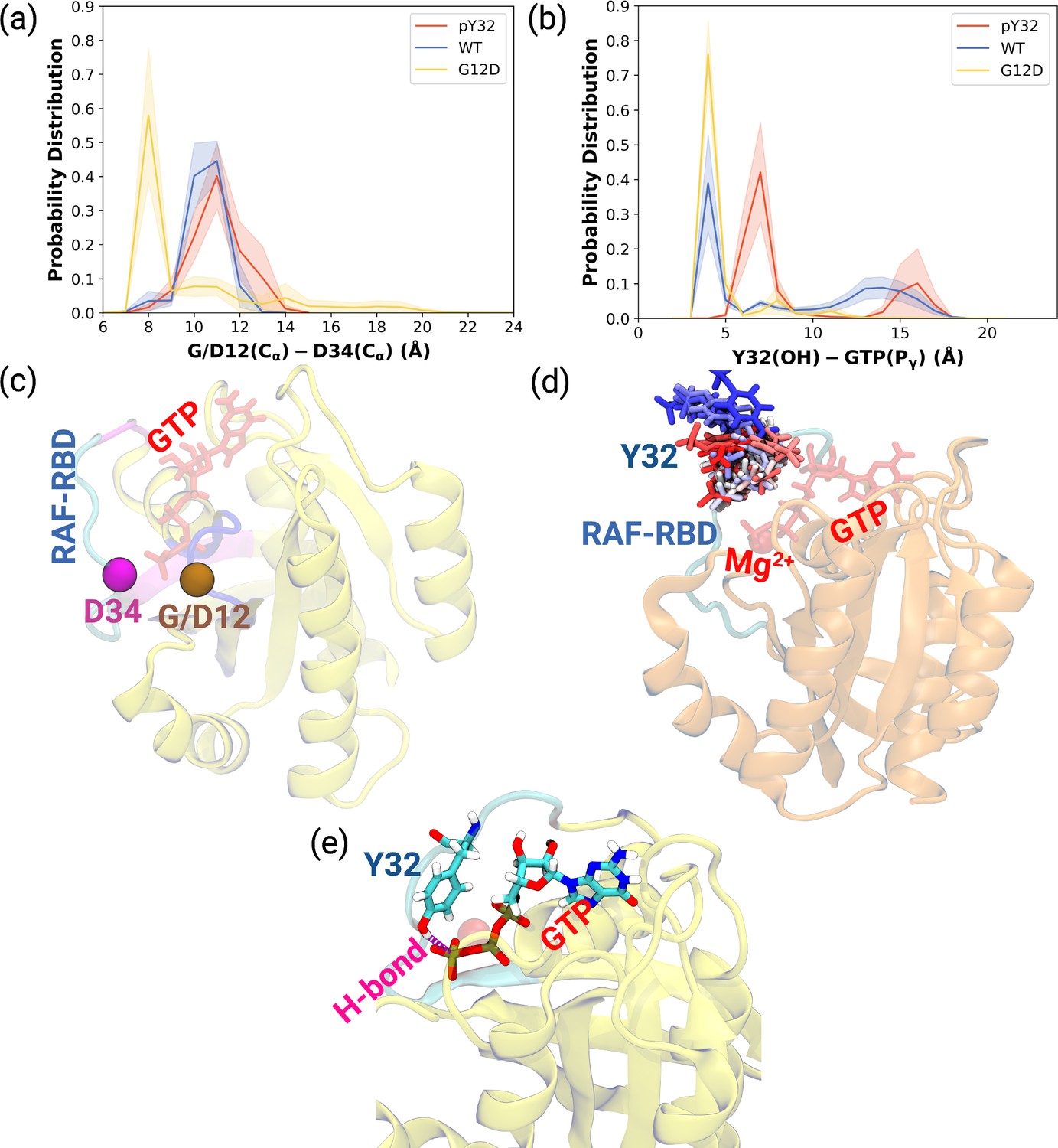
Probability distance distributions pertaining to nucleotide binding pocket and respective schematic representation of the residues used for the calculations.
(a) The normalized probability density distribution of the distance measured between the Cα atom of 12th 34th residues. The standard error (SE) of the measurements for HRASpY32, HRASWT, and HRASG12D are measured as 0.01, 0.004, and 0.01 Å, respectively. (b) The normalized probability distribution of the distance between the side-chain oxygen atom of Y32 and Pγ of GTP. The SE of the measurements for HRASpY32, HRASWT, and HRASG12D are calculated as 0.02, 0.02, and 0.003 Å, respectively. (c) Cα atoms of 12th and 34th residues are shown on the crystal structure of HRASWT (PDB ID: 5P21) in vdW representation and colored with ocher and purple, respectively, whereas GTP is shown in the licorice and colored with red. (d) The orientational dynamics of Y32 in the HRASpY32 trajectory. (e) The H-bond formed between side-chain of Y32 and Pγ of GTP in HRASG12D is shown in purple.
Figure 2—figure supplement 1
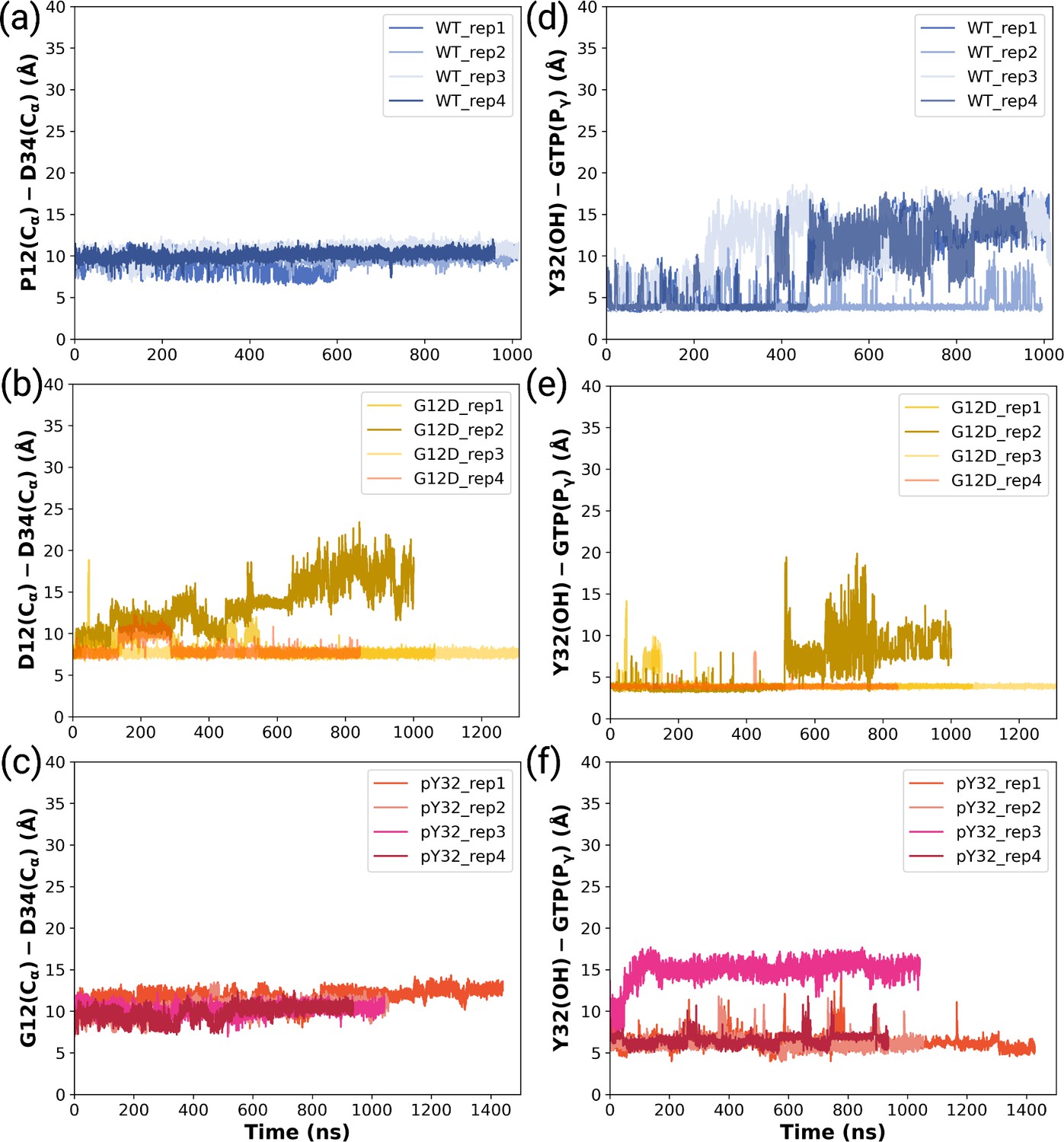
The timeline interatomic distances between Cα atoms of G/D12 and D34 throughout (a) HRASWT, (b) HRASG12D, and (c) HRASpY32 trajectories.
The timeline interatomic distances between OH atom of Y32 and Pγ atom of GTP throughout (d) HRASWT, (e) HRASG12D, and (f) HRASpY32 trajectories.
Figure 3 with 1 supplement
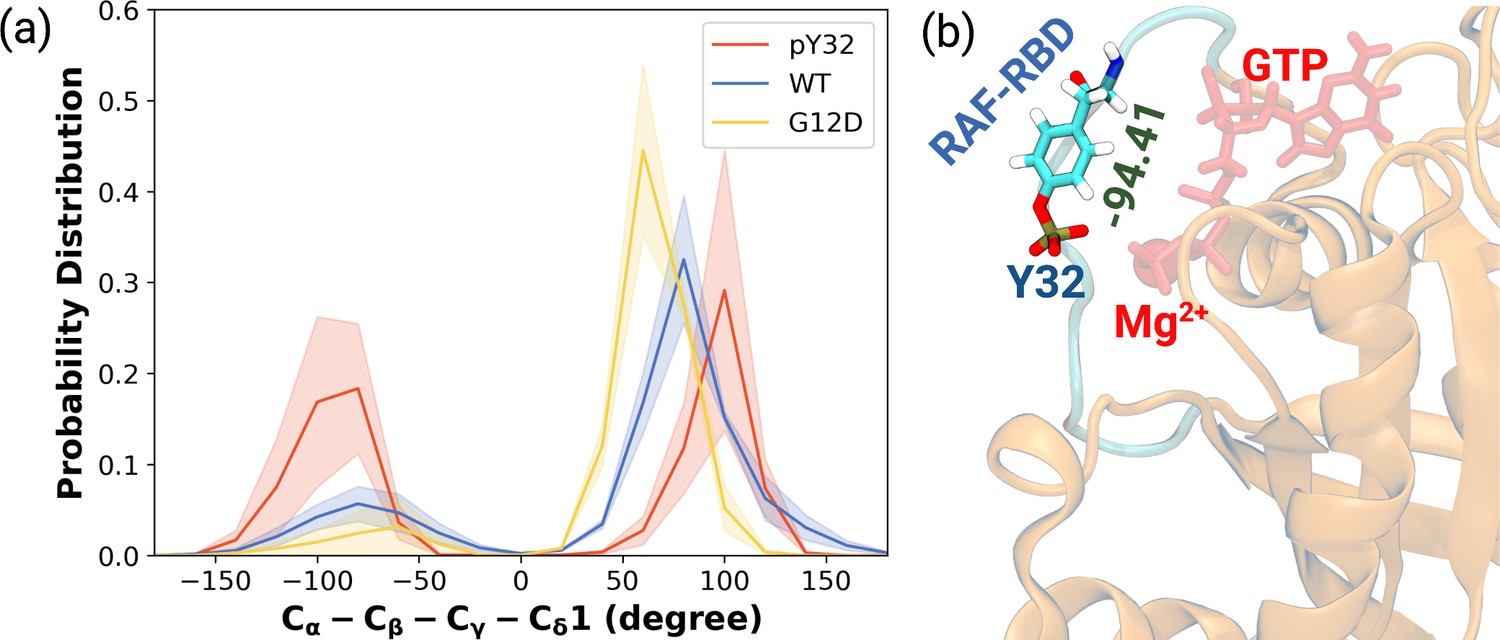
The normalized probability density distribution plots pertaining to chi2 angle of residue Y32 along with respective snapshot.
(a) The normalized probability density distribution of the measured χ2 angles of HRASWT, HRASG12D, and HRASpY32. The SE of χ2 pertaining to the phosphorylated, wild-type, and mutant systems are 0.44, 0.35, and 0.09, respectively. (b) A representative exposed state of Y32 obtained from the trajectory of the phosphorylated system.
Figure 3—figure supplement 1
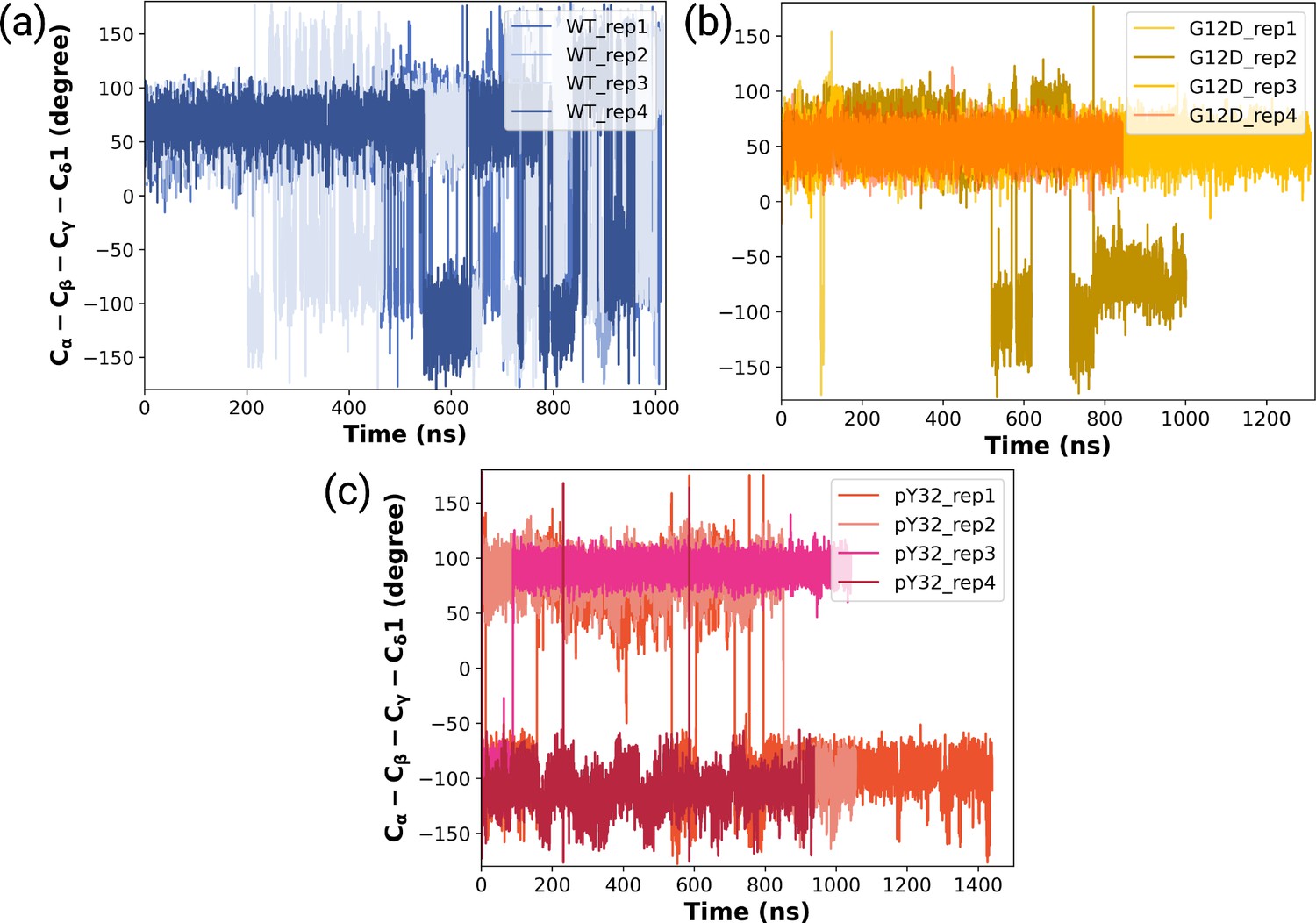
The timeline measured χ2 angles of Y32 throughout (a) HRASWT, (b) HRASG12D, and (c) HRASpY32 trajectories.
Figure 4 with 2 supplements
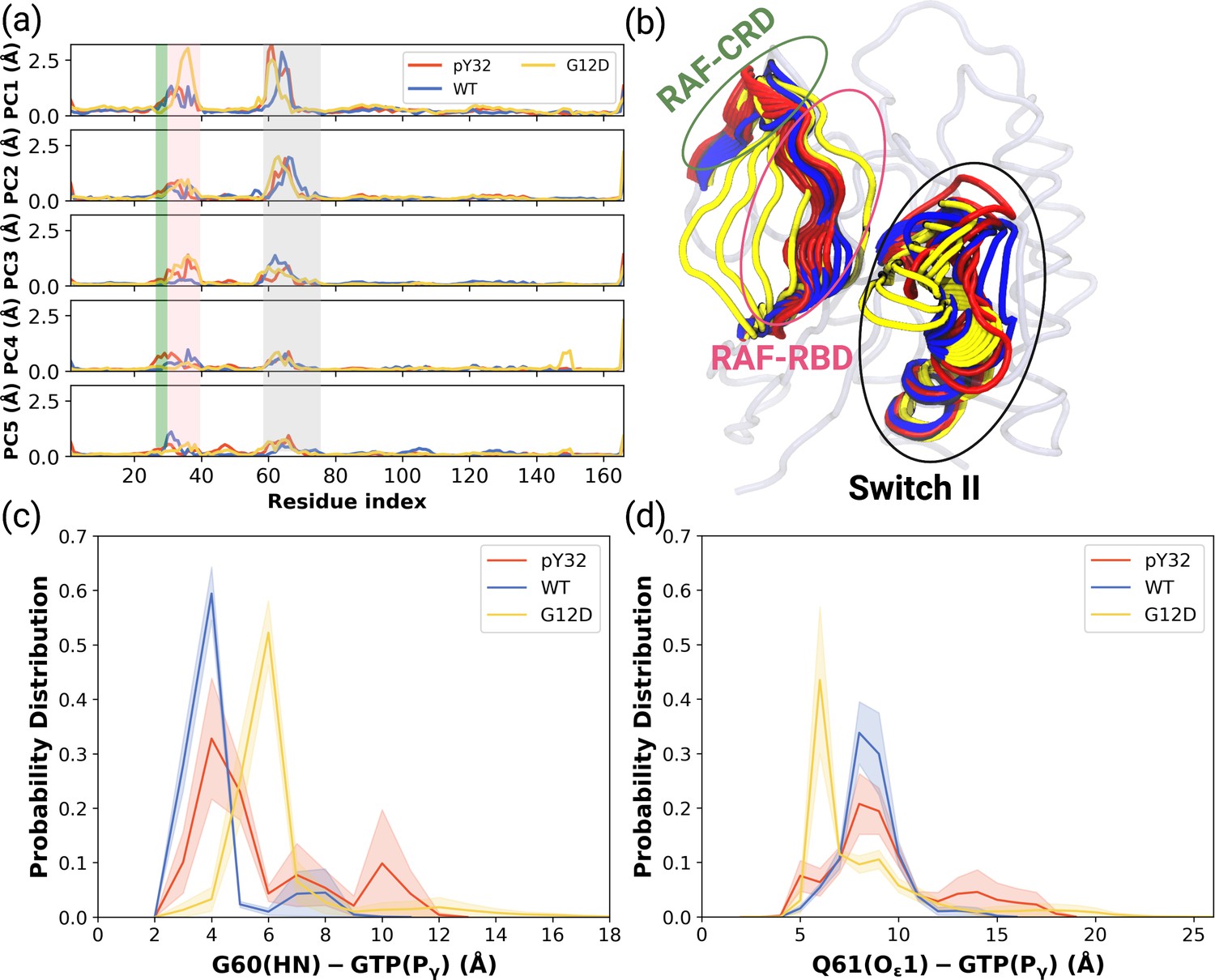
PCA analysis and Distance Probability Distributions pertaining to G60/Q61 and the nucleotide.
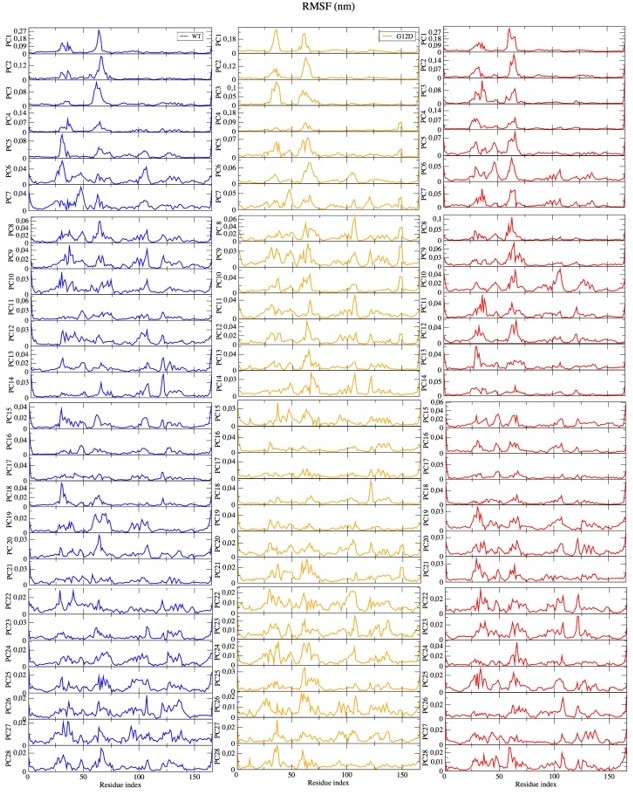
(a) Fluctuation of Cα atoms pertaining to HRASWT, HRASG12D, and HRASpY32 along the first five eigenvectors. The RAF-CRD & -RBD interaction interfaces, as well as Switch II, are shaded in the green, pink, and black rectangles, respectively. The eigen RMSF of Y32 pertaining to the phosphorylation system is pointed out by a dark violate bead. (b) The projected trajectories of the systems studied along with the first principal component, where the thickness of the ribbons are correlated the contribution of domain to the collective dynamics. The probability density distribution of the distance between (c) the backbone amide of G60 and Pγ of GTP is shown, and (d) the side-chain oxygen of Q61 and Pγ of GTP is shown. The SE of the distance between G60 and GTP pertaining to the phosphorylated, wild-type, and mutant systems are 0.01, 0.01, and 0.004 Å, respectively. The SE of the distance between Q61 and GTP pertaining to the phosphorylated, wild-type, and mutat systems are 0.02, 0.01, and 0.01, respectively.
Figure 4—figure supplement 1
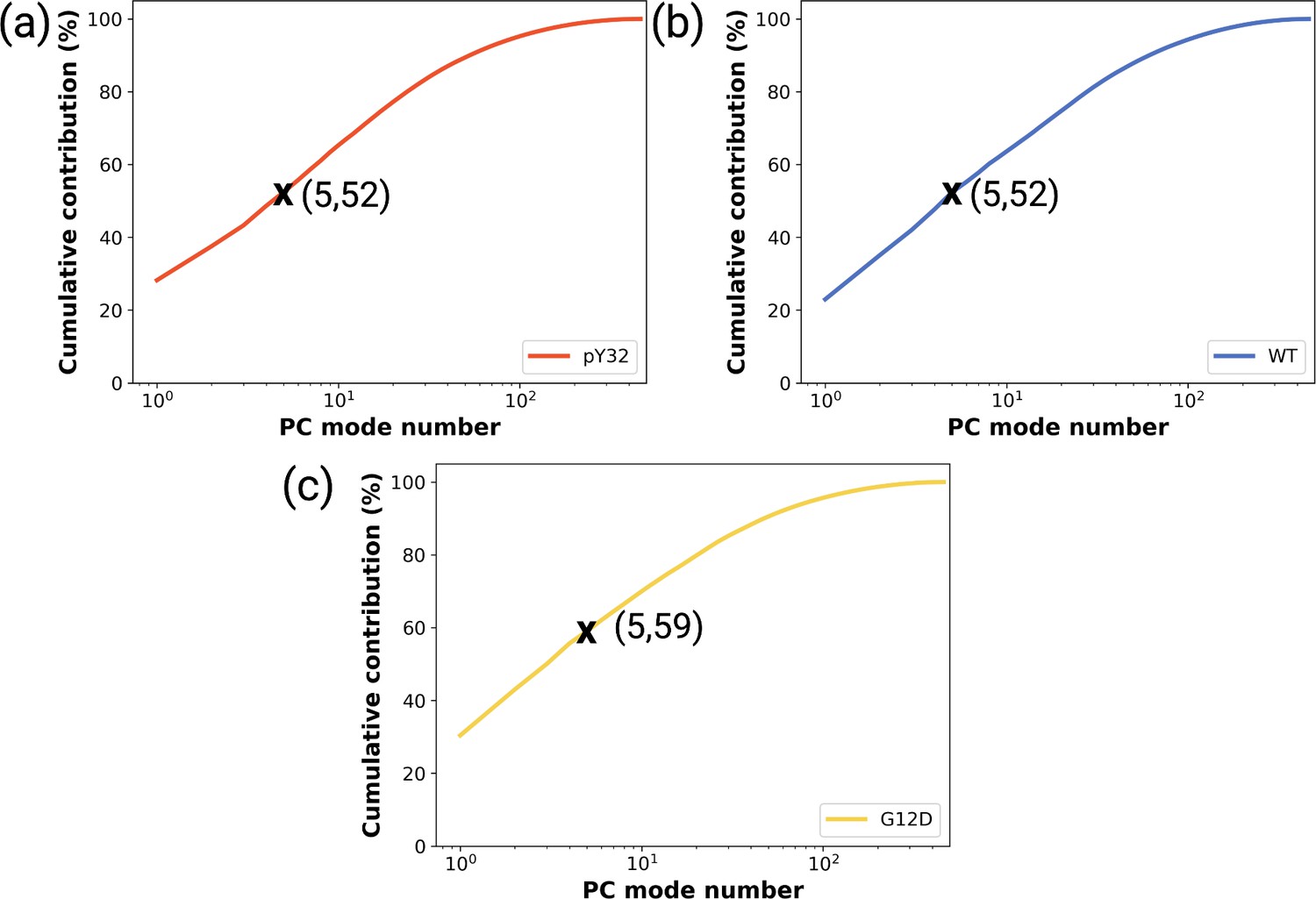
The cumulative contribution of the principal components pertaining to the (a) phosphorylated, (b) wild-type, and (c) G12D mutant systems to the overall dynamics.
The contribution of the first five principal components are denoted by a cross symbol on the plots.
Figure 4—figure supplement 2
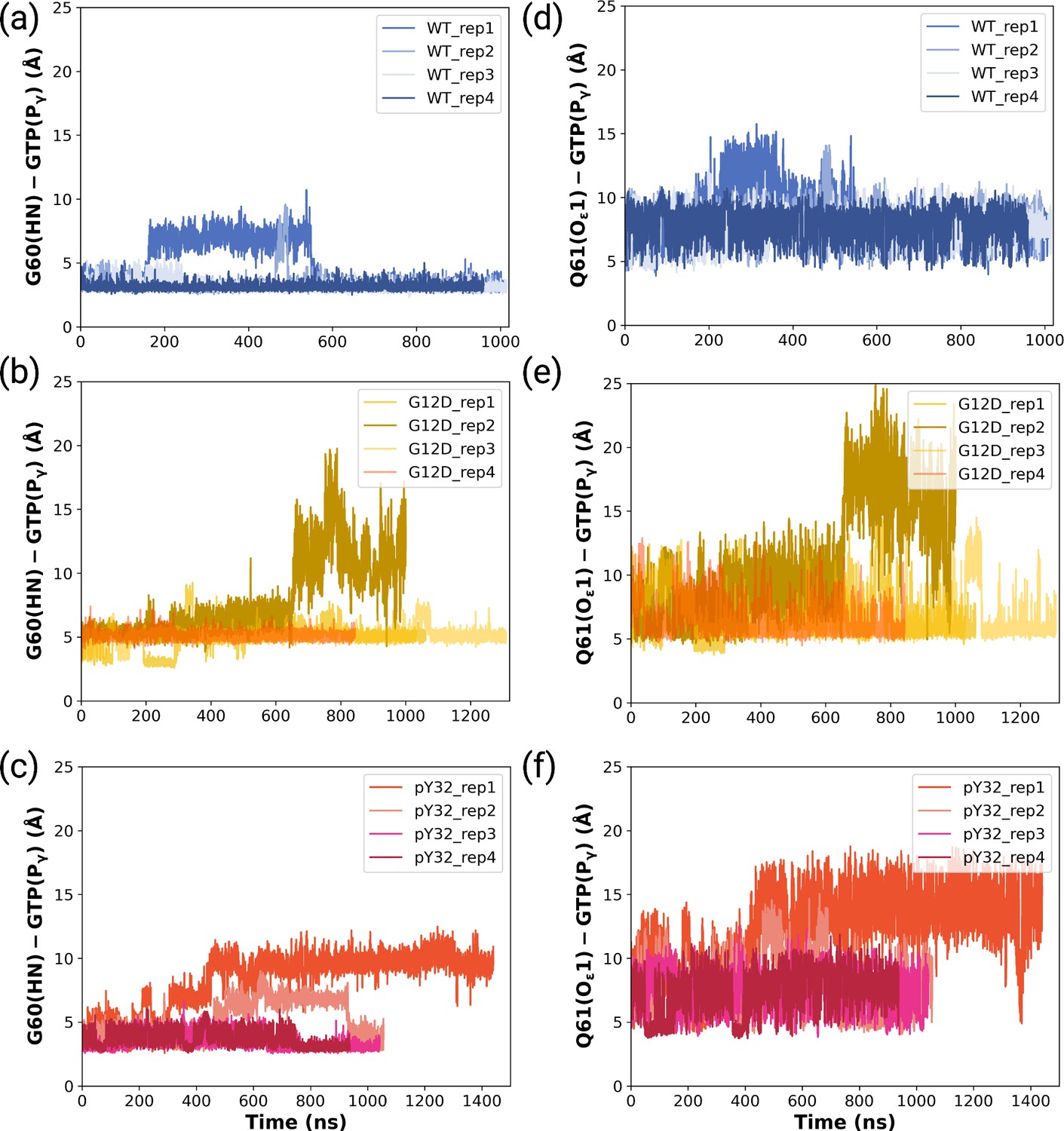
The timeline of the measured distances between HN of G60 and Pγ of GTP for (a) wild-type, (b) mutant, and (c) phosphorylated systems.
The timeline of the measured distances between Oε1 of Q61 and Pγ of GTP for (d) wild-type, (e) mutant, and (f) phosphorylated systems.
Figure 5 with 1 supplement
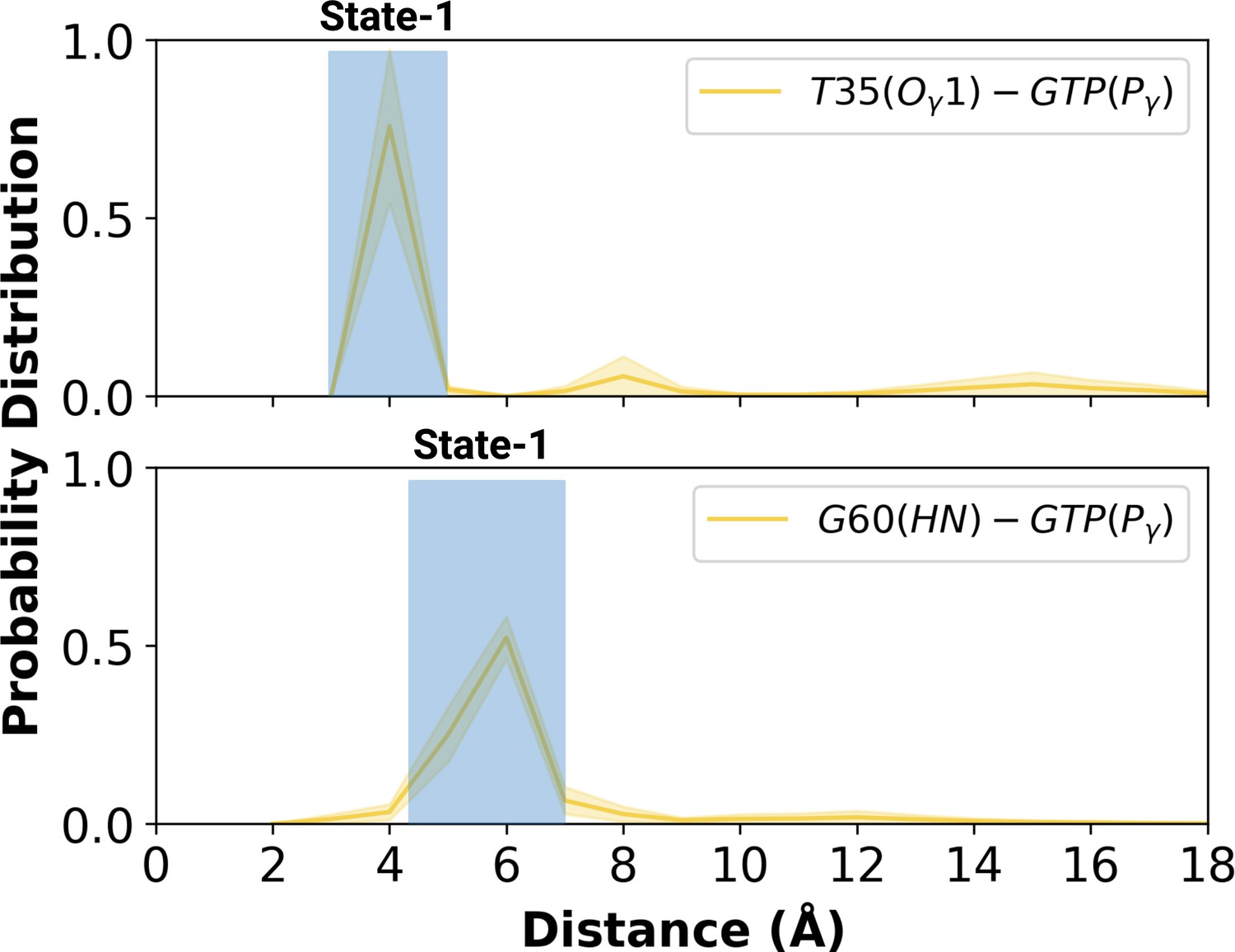
Probability distribution plot pertaining to distance measured between T35 (Ogamma)-GTP(Pgamma) and G60 (HN)-GTP (Pgamma) for the most populated conformational state of mutant H-RAS.
The normalized probability density distribution of the measured distance between the side-chain oxygen atom of T35 and Pγ of GTP, and the backbone amide of G60 and Pγ of GTP in the mutant system, where the sampling range for calculating the frequency of each interval was adjusted as 1 Å. The SE of the distance between T35 and GTP is calculated as 0.001 Å, whereas that of the distance between G60 and GTP is 0.004 Å.
Figure 5—figure supplement 1
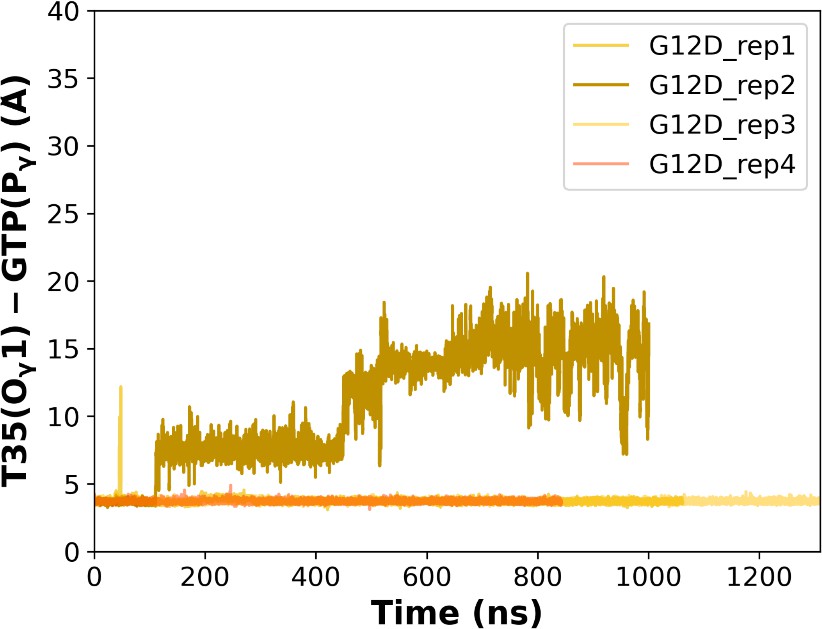
The (a) timeline and (b) histogram of the measured distance between O1 of T35 and Pγ of GTP for the mutant system.
Figure 6
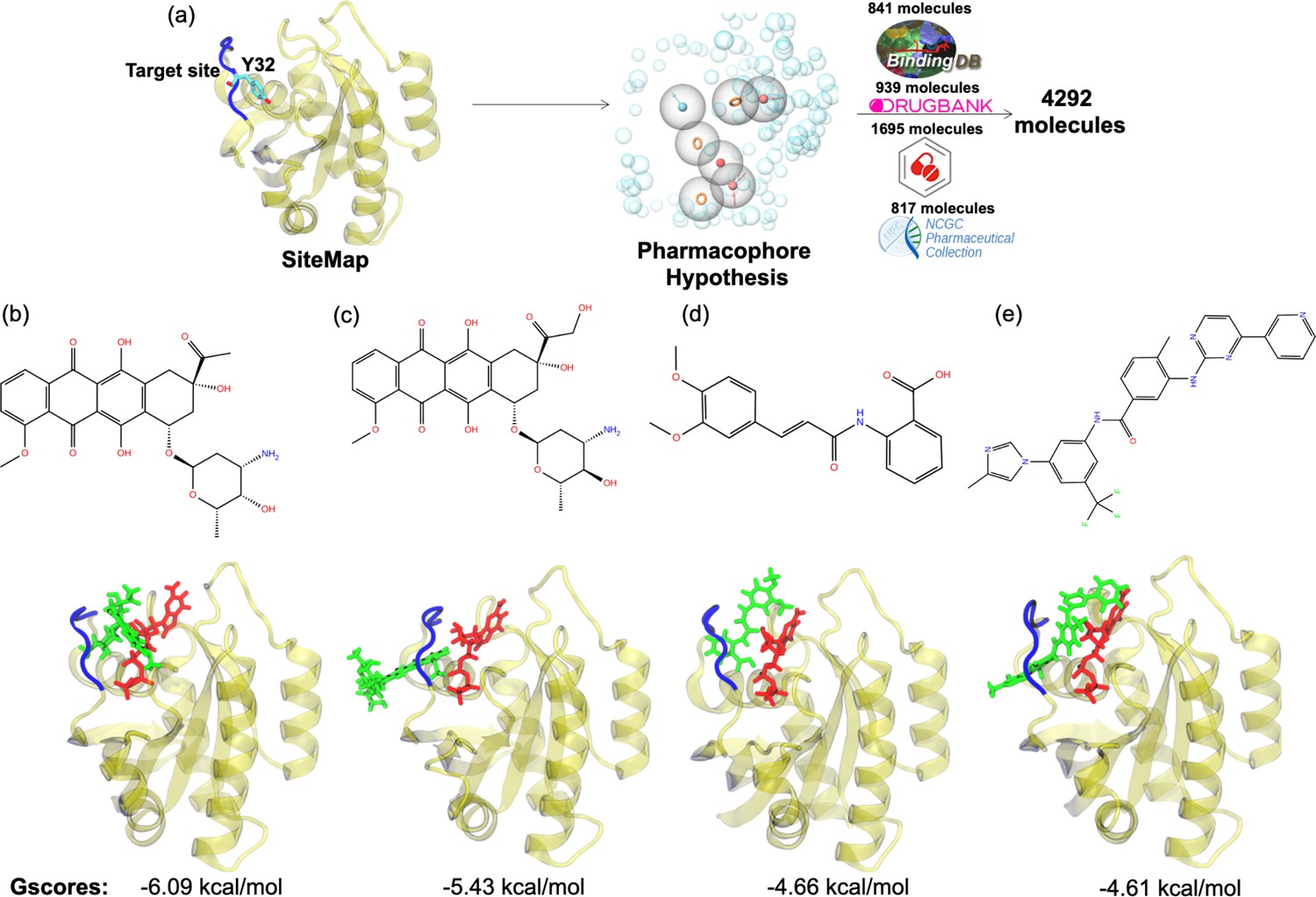
Virtual screening scheme used, and candidate molecules achieved along with their chemical structures and docking energy values.
(a) A schematic that summarizes the virtual screening workflow done for the identified binding pocket on the most frequently sampled conformation of HRASG12D. The 3D structures and corresponding GScores of (b) cerubidine, (c) epirubicin, (d) tranilast, and (e) nilotinib are shown. GTP is shown in licorice and red.
Figure 7 with 2 supplements
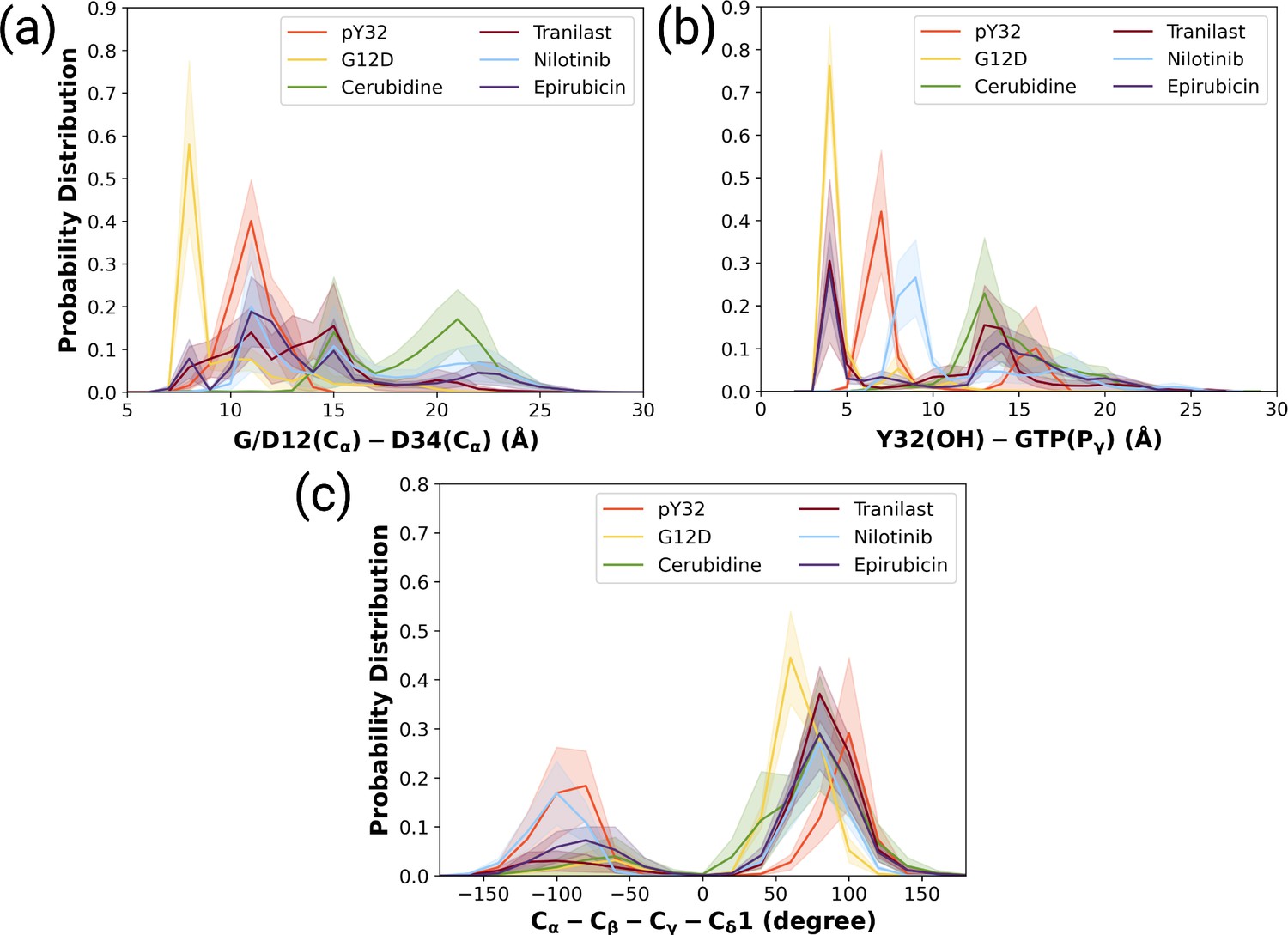
The normalized probability density distribution plots pertaining to distances of G/D12 (Calpha)- and D34 (Calpha), Y32 (OH)-GTP (Pgamma) and chi2 angle of Y32 measured in ligand-bound mutant H-RAS trajectories.
The normalized probability density distribution of (a) the distance between Cα atoms of G/D12 and P34, (b) the distance between the side-chain oxygen atom of Y32 and Pγ of GTP, (c) χ2 in HRASG12D, HRASpY32, and cerubidine-, tranilast-, nilotinib-, and epirubicin-bound HRASG12D. The SE of the G/D12-D34 distance pertaining to the above-mentioned systems, in seriatim, 0.01, 0.01, 0.02, 0.02, 0.03, and 0.03 Å. The SE of the same systems for Y32-GTP distance is 0.003, 0.02, 0.02, 0.03, 0.02, 0.04 Å, respectively. The SE of χ2 is 0.09, 0.44, 0.37, 0.39, 0.51, and 0.39 degree for the mutant, phosphorylated, cerubidine-, tranilast-, nilotinib, and epirubicin-bound HRASG12D systems, respectively.
Figure 7—figure supplement 1
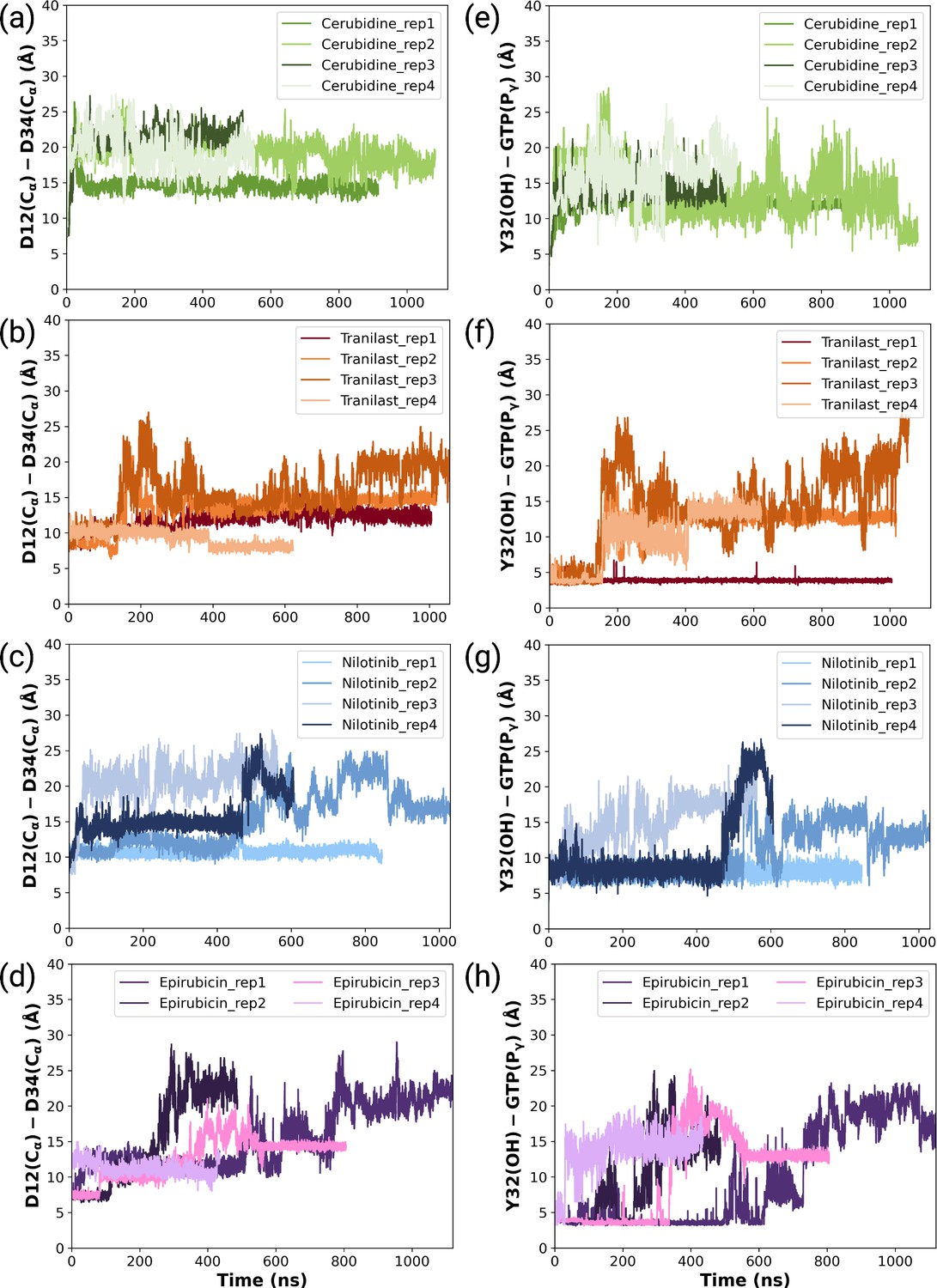
The timeline and histogram of the measured distances between Cα atoms of D12 and D34 for (a,e) cerubidine-bound, (b,f) tranilast-bound, (c,g) nilotinib-bound, and (d,h) epirubicin-bound HRASG12D systems, respectively.
Figure 7—figure supplement 2

The timeline and histogram of the measured χ2 angles pertaining to Y32 for (a) cerubidine-bound, (b) tranilast-bound, (c) nilotinib-bound, and (d) epirubicin-bound HRASG12D systems.
Figure 8
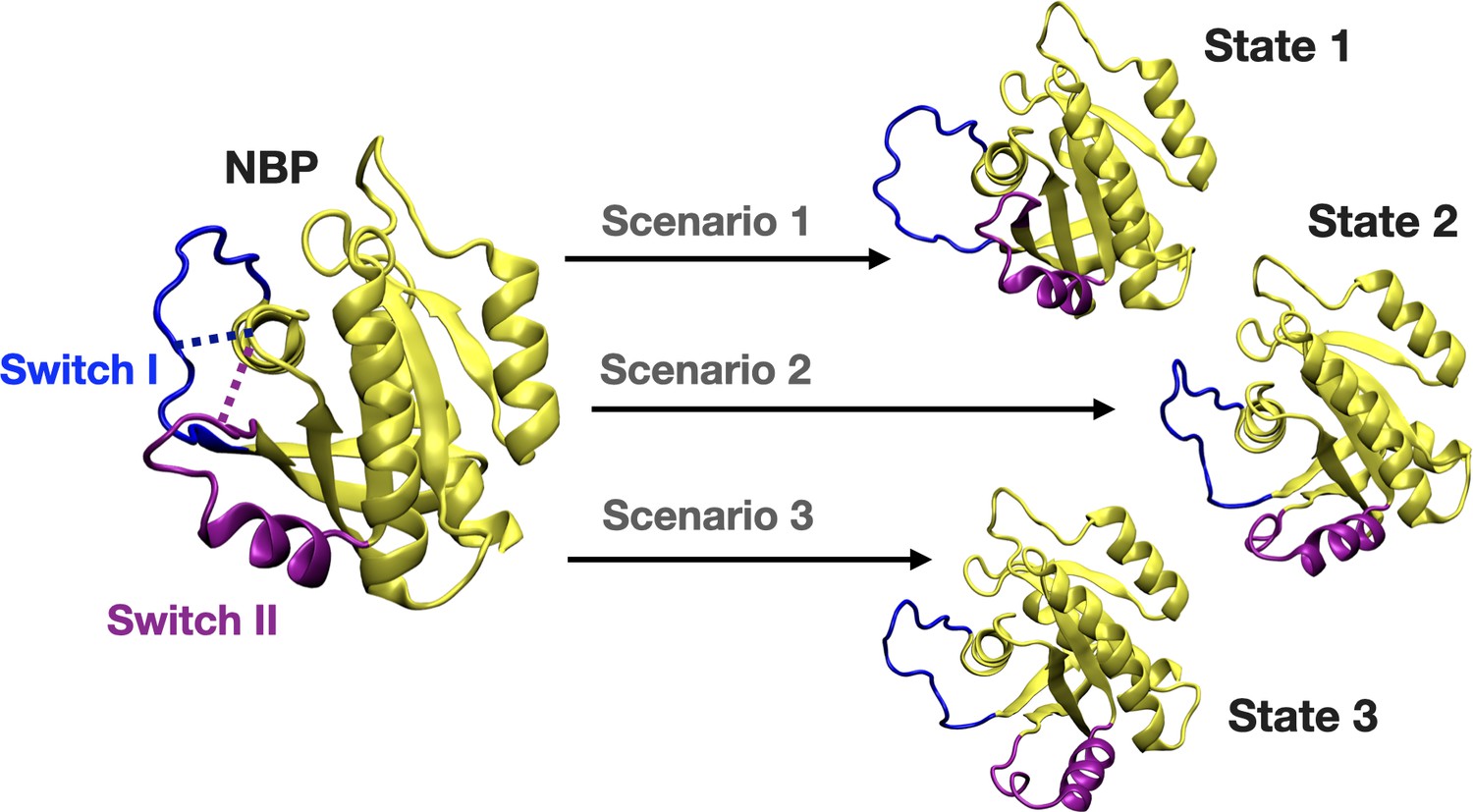
A schematic that illustrates the PRS calculations made for examining the transition between the initial and target states.
The initial state represents the conformation of the closed-state of Switch I and II. The target state-1 is described as the open state of Switch I (blue) and close state of Switch II (purple). The target state-2 represents the partially open state of Switch I and open state of Switch II. The target state-3 corresponds to open state of Switch I and II.
Figure 9 with 1 supplement
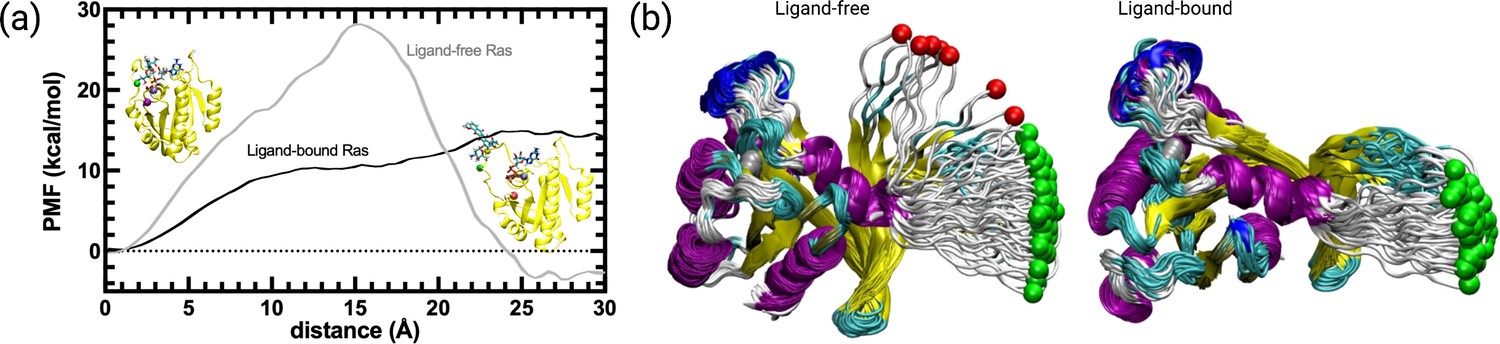
PMF along the PSP predicted coordinate with the highest overlap for the transition scenario 1 (Switch I opening motion) as a function of distance.
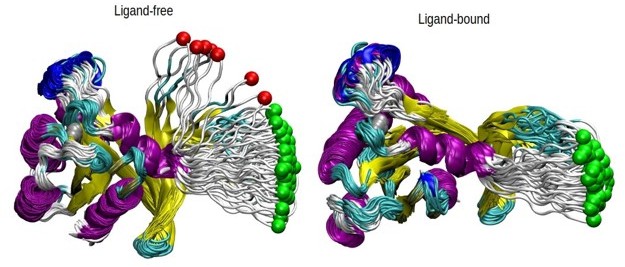
PMF is calculated for the HRASG12D system in the presence and absence of cerubidine, and each simulation was repeated 70 times. The reaction coordinate refers to the distance covered by the SMD atom (shown as a yellow bead) during the course of the pulling experiments. The initial and final structures of an SMD simulation are illustrated on the left and right sides of the figure, respectively. Yellow bead: Y32; Green bead: P34; Iceblue bead: G60; GTP and Cerubidine: Licorice representation. Errors are indicated by the thickness of the curves and are less than 0.3 kcal/mol and 0.1 kcal/mol in ligand-free and ligand-bound simulations, respectively.
Figure 9—figure supplement 1
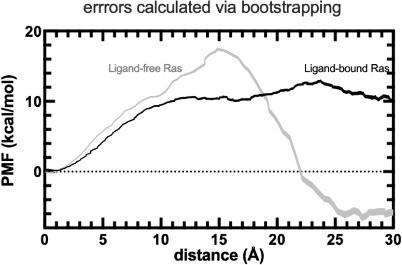
The free energy profile, where the error estimate was calculated by usingbootstrapping.
Figure 10

Engineering HEK-293T cells expressing mutant HRASG12D.
(a) Schematic representation of the cloning HRASG12D gene region into the eukaryotic expression plasmid (with PuroR gene to select transgene positive population) using the Gibson Assembly method and engineering HEK-293T cell line to overexpress HRASG12D protein upon transfection followed by puromycin selection. (b) Fluorescent images 293T cells transfected with GFP-encoding plasmid. (c) RT-PCR analysis showing expression levels of HRASG12D in 293T cells transfected with HRASG12D plasmid. (d) ImageJ analysis of band densities from ‘C’. (e) Western blot analysis showing expression levels of HRASG12D in 293T-HRASG12D cells. (f) ImageJ analysis of western-blot band densities. Data represent the means of three independent assays. Unpaired t-test analysis was used to test the difference between each experimental group and the control group. BF: bright field, FL: Fluorescence, ***: p<0,0001.
Figure 11
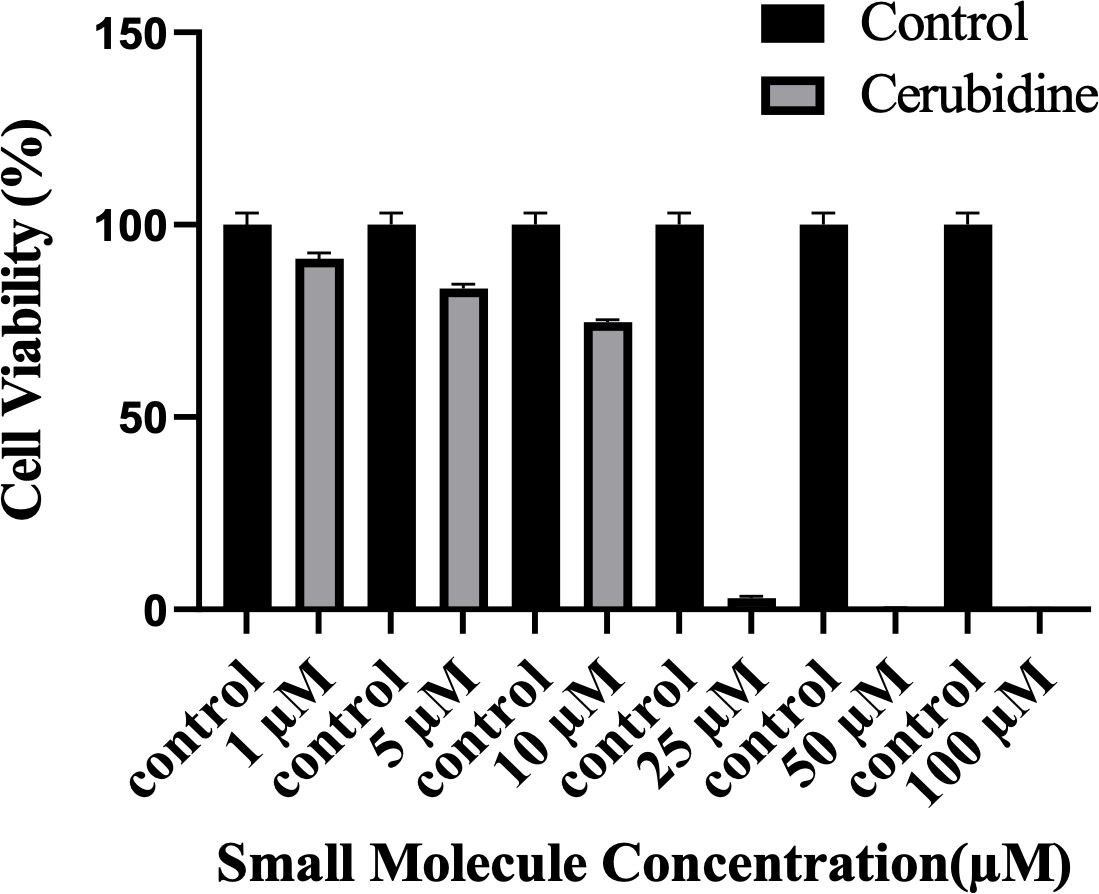
Cell viability assay in control and 24 hr treated 293T cells.
The plot indicates the cell viability of 293T cells upon treatment with cerubidine in a dose-dependent manner (1, 5, 10, 25, 50, 100 μM).
Figure 12
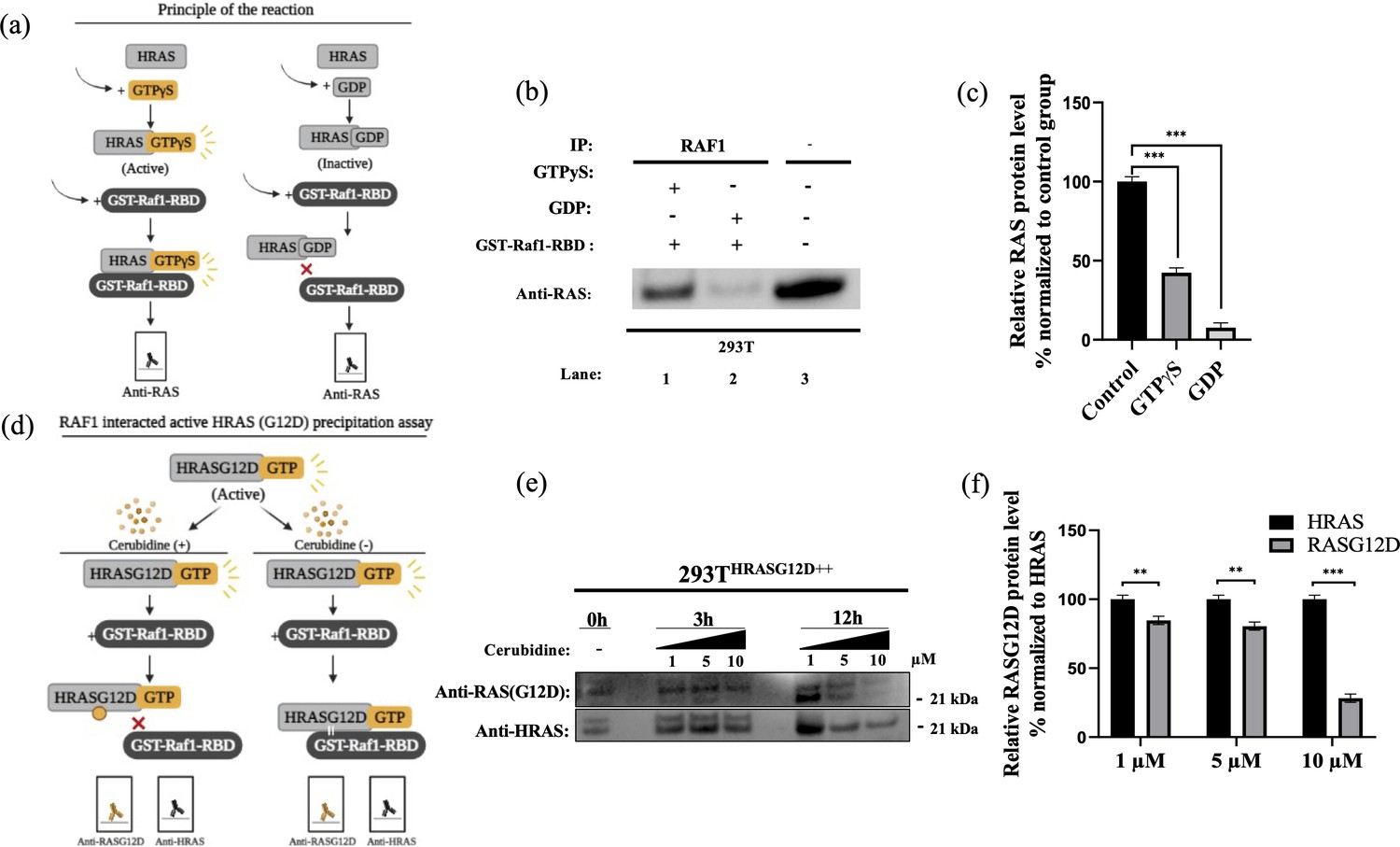
Cell viability assay in control and 24 hr treated 293T cells.
(a) Scheme to outline the principle of active Ras pull-down reaction. (b) Immunoprecipitation (IP) assays show interactions of RAS with RAF proteins in the presence (Lane 1–2) and absence (Lane 3) of GTPγS-GDP. Protein extracts were immunoprecipitated with Raf1-RBD probe and resolved by SDS PAGE. Protein-protein interactions were immunodetected using anti-RAS antibodies. (c) ImageJ analysis of Western-blot band densities. Unpaired t-test analysis was used to test the difference between each experimental group and the control group. (d) Scheme outlining the RAF1 interacted active HRASG12D precipitation assay. (e) Immunoprecipitation (IP) assays showing interactions of RAS with RAF proteins in 293THRASG12D++ cells treated with increasing doses (1,5 and 10 μM) of Cerubudine. Protein extracts obtained at different time points (0 hr, 3 hr, and 12 hr) were immunoprecipitated with the RAF1-RBD probe and resolved by SDS-PAGE. Protein-protein interactions were immunodetected using anti-RASG12D and anti-HRAS antibodies (f) ImageJ analysis of Western-blot band densities. Unpaired t-test analysis was used to test the difference between each RASG12D group and the HRAS group. **: p<0.001, ***: p<0.0001.
Figure 13
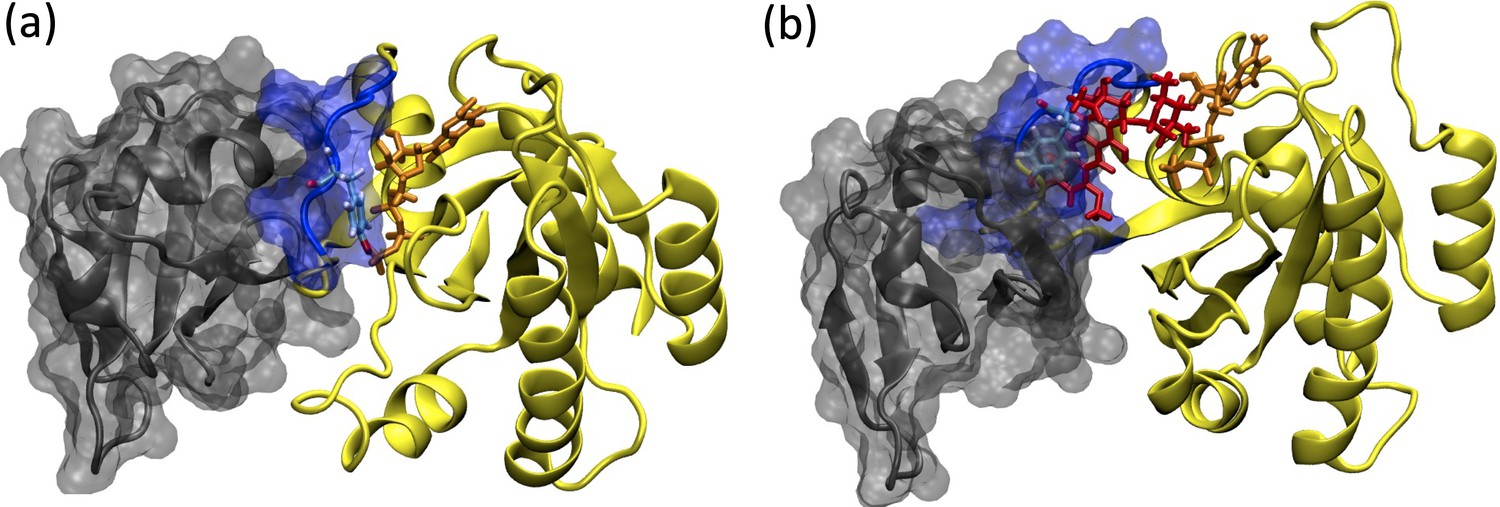
Depiction of the RAF/RAS interaction interface in the absence/presence of the ligand, cerubidine.
(a) RAF-RBD in complex with HRAS. Y32 and GTP are shown in licorice representation, whereas protein and RAF-RBD interaction interface is shown in New Cartoon, and surface representation, respectively. (b) The displacement of Y32 from the nucleotide-binding pocket by cerubidine, which is colored with red, causes steric clash at the RAS/RAF interface.
Author response image 1
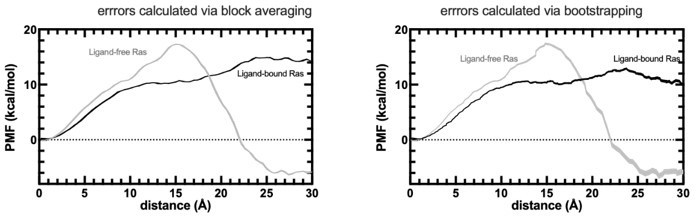
PMF profiles obtained by SMD experiments.
The errors are calculated by both block averaging (left) and bootstrapping (right).
Author response image 2
The pathway analysis of SMD simulations.
The displacement of the pulled atom, which is shown in green color, is shown throughout the 70 SMD trajectories in both ligand and ligand-free states. The protein is shown in new cartoon representation and colored according to the respective secondary structures. The trajectories excluded are shown in red while those included in green. The protein is aligned using all the regions except the loop.
Author response image 3
Tables
Table 1
The backbone RMSF values of key regions/residues pertaining to HRASWT, HRASpY32, and HRASG12D.
| Residue/Region-RMSF (Å) | HRASWT | HRASpY32 | HRASG12D |
|---|---|---|---|
| Y32 | 1.6 ± 0.2 | 1.5± 0.2 | 0.9± 0.1 |
| RAF-RBD interface residues | 1.2± 0.2 | 1.3± 0.2 | 0.8± 0.1 |
| RAF-CRD interface residues | 0.8± 0.1 | 0.9± 0.2 | 0.7± 0.03 |
| G60 | 1.0± 0.3 | 1.4± 0.5 | 1.1± 0.2 |
| Q61 | 1.2± 0.4 | 1.6± 0.4 | 1.5± 0.3 |
Table 2
The SiteMap scores of possible pockets found on the surface of the most probable conformation of HRASG12D.
| SiteScore | Size | DScore | Volume | Exposure | Enclosure | Contact | Phobic | Philic | Balance | Don/acc |
|---|---|---|---|---|---|---|---|---|---|---|
| 1.028 | 194 | 0.919 | 466.140 | 0.478 | 0.740 | 1.010 | 0.259 | 1.425 | 0.182 | 0.932 |
| 0.701 | 25 | 0.668 | 81.290 | 0.632 | 0.691 | 1.059 | 1.474 | 0.664 | 2.219 | 0.915 |
| 0.656 | 22 | 0.629 | 101.870 | 0.776 | 0.638 | 0.842 | 0.959 | 0.596 | 1.609 | 12.216 |
Table 3
The results of PRS calculations for the transition between initial and target states.
| Ligand | State | D12-P34 (Å) | G60-GTP (Å) | PRS selected residues | PRS overlap(Oi) |
|---|---|---|---|---|---|
| Cerubidinea | Initial stateb | 7.7 (closed) | 7.0 (closed) | - | - |
| Target state-1 | 26.3 (open) | 11.00 (closed) | 34, 35, 33, 32, 37, 36 | 0.74–0.70 | |
| Target state-2 | 13.9 (partially open) | 15.00 (open) | 35, 34, 33, 66, 16, 65 | 0.58–0.50 | |
| Target state-3 | 19.5 (open) | 16.9 (open) | 34, 66, 35, 64, 16, 33 | 0.59–0.50 |
Additional files
-
MDAR checklist
- https://cdn.elifesciences.org/articles/79747/elife-79747-mdarchecklist1-v3.pdf
-
Supplementary file 1
Result of the analyses for the number of water molecules, PRS calculations and total simulation time for ligand-mutant HRAS trajectories.
a The average number of water molecules within 5 Å of GTP were calculated over the course of the ligand-bound HRASG12D systems. b Total simulation time performed for ligand- HRASG12D complexes and changes in the backbone RMSF profiles of cerubidine-, tranilast-, nilotinib-, and epirubicin-bound HRASG12D systems with respect to those of HRASG12D. c The results of PRS calculations for the transition between initial and target states.
- https://cdn.elifesciences.org/articles/79747/elife-79747-supp1-v3.zip
-
Source data 1
Raw unedited gel.
- https://cdn.elifesciences.org/articles/79747/elife-79747-data1-v3.zip
-
Source data 2
Uncropped gels with relevant bands that are clearly labelled.
- https://cdn.elifesciences.org/articles/79747/elife-79747-data2-v3.zip
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Inhibition of mutant RAS-RAF interaction by mimicking structural and dynamic properties of phosphorylated RAS
eLife 11:e79747.
https://doi.org/10.7554/eLife.79747




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}