A lncRNA identifies Irf8 enhancer element in negative feedback control of dendritic cell differentiation
- Department of Cell Biology, Institute of Biomedical Engineering, RWTH Aachen University Medical School, Germany
- Helmholtz Institute for Biomedical Engineering, RWTH Aachen University, Germany
- Institute for Computational Genomics, RWTH Aachen University Medical School, Germany
- Department of Hematology, Oncology, Hemostaseology, and Stem Cell Transplantation, Faculty of Medicine, RWTH Aachen University, Germany
Abstract
Transcription factors play a determining role in lineage commitment and cell differentiation. Interferon regulatory factor 8 (IRF8) is a lineage determining transcription factor in hematopoiesis and master regulator of dendritic cells (DC), an important immune cell for immunity and tolerance. IRF8 is prominently upregulated in DC development by autoactivation and controls both DC differentiation and function. However, it is unclear how Irf8 autoactivation is controlled and eventually limited. Here, we identified a novel long non-coding RNA transcribed from the +32 kb enhancer downstream of Irf8 transcription start site and expressed specifically in mouse plasmacytoid DC (pDC), referred to as lncIrf8. The lncIrf8 locus interacts with the lrf8 promoter and shows differential epigenetic signatures in pDC versus classical DC type 1 (cDC1). Interestingly, a sequence element of the lncIrf8 promoter, but not lncIrf8 itself, is crucial for mouse pDC and cDC1 differentiation, and this sequence element confers feedback inhibition of Irf8 expression. Taken together, in DC development Irf8 autoactivation is first initiated by flanking enhancers and then second controlled by feedback inhibition through the lncIrf8 promoter element in the +32 kb enhancer. Our work reveals a previously unrecognized negative feedback loop of Irf8 that orchestrates its own expression and thereby controls DC differentiation.
Editor's evaluation
Authors provide valuable evidence identifying a lncRNA transcribed specifically in the pDC subtype from the +32Kb promoter region which is also the region for the enhancer for Irf8 specifically in the cDC1 subtype. With convincing methodology, they provide in-depth analysis about the possible role of lncIrf8, and its promoter region and cross-talk with Irf8 promoter to identify that it is not the lncIRF8 itself but its promoter region that is crucial for pDC and cDC1 differentiation conferring feedback inhibition of Irf8 transcription. The work will be of interest to immunologists working on immune cell development.
https://doi.org/10.7554/eLife.83342.sa0Introduction
Lineage-determining transcription factors (TF) are master regulators of gene programs that frequently initiate self-reinforcing loops by autoactivation. TF autoactivation is important for cells to pass restriction points during development (referred to as points of no return) and to enforce cellular identity. Molecular circuitries of autoactivation have been studied for several TF, such as GATA-binding factor 1 (GATA1), PU.1 (encoded by Spi1), CCAAT enhancer-binding protein α and ε (C/EBPα and ε; Graf and Enver, 2009; Loughran et al., 2020; Nishimura et al., 2000; Okuno et al., 2005; Theilgaard-Mönch et al., 2022). A further example is interferon regulatory factor 8 (IRF8), which shows autoactivation in cooperation with basic leucine zipper ATF-like transcription factor 3 (BATF3; Anderson et al., 2021; Grajales-Reyes et al., 2015). An important principle in nature is negative feedback control to avoid signal overshooting and toxicity. Negative feedback control applies also to lineage-determining TF; however, there is a paucity on our knowledge of the molecular mechanisms involved.
IRF8 is a hematopoietic TF positioned at the center of the regulatory gene network for dendritic cell (DC) development (Anderson et al., 2021; Belz and Nutt, 2012; Chauvistré and Seré, 2020; Kim et al., 2020; Lin et al., 2015; Nutt and Chopin, 2020; Tamura et al., 2015; Verlander et al., 2022). IRF8 is a member of the interferon regulatory factor (IRF) family of TF. Initially members of this TF family were found to mediate the induction of interferon induced genes, but are now known to serve diverse functions in regulating the immune system (Honda and Taniguchi, 2006; Tamura et al., 2008). Irf8 knockout mice show abnormal development of classical DC type 1 (cDC1) and plasmacytoid DC (pDC) (Durai et al., 2019; Schiavoni et al., 2002; Sichien et al., 2016; Tsujimura et al., 2003). Irf8 is prominently upregulated during DC development by autoactivation (Grajales-Reyes et al., 2015; Lin et al., 2015), yet how Irf8 autoactivation is controlled and eventually limited, and the epigenetic mechanisms involved is largely unknown.
Irf8 expression in hematopoietic cells is induced and maintained by enhancers located at –50 kb,+32 kb,+41 kb and +56 kb relative to Irf8 transcription start site (TSS) (Anderson et al., 2021; Bagadia et al., 2019; Durai et al., 2019; Grajales-Reyes et al., 2015; Murakami et al., 2021; Schönheit et al., 2013). Enhancers are cis-regulatory sequences with multiple TF binding sites that cooperatively bind TF and thereby activate transcription, as demonstrated by many studies including our work (Davidson et al., 1986; Long et al., 2016; Wildeman et al., 1986; Zenke et al., 1986). Enhancers regulate complex gene networks and can also produce non-coding RNA, referred to as enhancer RNA (eRNA). eRNA serve as an indicator for enhancer activity and some eRNA have an activity on their own and act in cis or trans to regulate cell fate decisions (Sartorelli and Lauberth, 2020; Statello et al., 2021). Enhancer-associated long non-coding RNA (lncRNA) represent a class of lncRNA transcribed from active enhancers. Thus, eRNA and enhancer-associated lncRNA provide opportunities to detect enhancer activity and to investigate enhancer function.
DC are highly specialized immune cells that play a critical role in regulating innate and adaptive immune responses (Cabeza-Cabrerizo et al., 2021). DC develop from hematopoietic stem cells (HSC) via successive steps of lineage commitment and differentiation. More specifically, HSC develop into multipotent progenitors (MPP) that are committed to DC restricted common DC progenitors (CDP) and differentiate into classic DC (cDC) type 1 and type 2 (cDC1 and cDC2, respectively) and pDC (Anderson et al., 2021; Cabeza-Cabrerizo et al., 2021; Ginhoux et al., 2022; Nutt and Chopin, 2020; Rodrigues and Tussiwand, 2020). pDC were recently also shown to develop from lymphoid progenitors (Dress et al., 2019; Rodrigues et al., 2018; Rodrigues and Tussiwand, 2020). Differential expression of Irf8 regulates DC and monocyte specification in a dose-dependent manner (Cytlak et al., 2020; Murakami et al., 2021). Irf8 expression starts at the CDP stage, and is high in pDC and cDC1, which is attributed to the autoactivation of Irf8 during DC subsets specification (Grajales-Reyes et al., 2015; Lin et al., 2015). Interestingly, IRF8 can act as a transcriptional activator or repressor in hematopoiesis by interacting with different partner TF and binding to specific DNA sequences (Tamura et al., 2015).
As an activator, IRF8 binds to its own promoter in DC differentiation, which is considered as the autoactivation capacity of Irf8 (Grajales-Reyes et al., 2015; Lin et al., 2015). For instance, IRF8 interacts with partner TF, such as PU.1, to initiate Irf8 autoactivation at the CDP stage (Grajales-Reyes et al., 2015). Inversely, IRF8 inhibits C/EBPα activity in neutrophil differentiation (Kurotaki et al., 2014). IRF8 also represses C/EBPβ to generate and maintain DC lineage-specific enhancer landscapes (Bornstein et al., 2014). In addition, IRF8 is important for the Myc-Mycl transition in DC differentiation (Anderson lll et al., 2021). IRF8 represses Myc expression in progenitors, while IRF8 at high levels interacts with PU.1 and drives Mycl expression (Anderson lll et al., 2021). All this emphasizes the central position of IRF8 in coordinating the gene network that regulates DC differentiation and function.
During DC differentiation, the Irf8 gene locus shows high epigenetic dynamics, including histone modifications and TF binding identified by ChIP-seq (Chauvistré and Seré, 2020; Durai et al., 2019; Grajales-Reyes et al., 2015; Lin et al., 2015), chromatin accessibility measured by ATAC-seq (Kurotaki et al., 2019; Li et al., 2019), and three-dimensional chromatin structure remodeling determined by chromosome conformation capture (3 C) (Kurotaki et al., 2022; Schönheit et al., 2013). All this emphasizes the impact of epigenetic regulators on Irf8 gene activity in DC differentiation. Notably, Irf8 is flanked by multiple enhancers at –50 kb,+32 kb,+41 kb, and +56 kb that regulate Irf8 expression in hematopoietic cells (Anderson et al., 2021; Murakami et al., 2021). These four enhancers were found to be driven by PU.1, BATF3, E proteins and Runt-related transcription factor (RUNX)-core binding factor beta (CBFβ) (RUNX-CBFβ), respectively (Bagadia et al., 2019; Durai et al., 2019; Grajales-Reyes et al., 2015; Murakami et al., 2021; Schönheit et al., 2013).
Chromatin conformation, particularly enhancer promoter interactions, provides a platform for TF-driven gene regulation and serves as a driving force for cell-fate determinations (Misteli and Finn, 2021; Oudelaar and Higgs, 2021; Stadhouders et al., 2019). Schönheit et al. demonstrated Irf8 promoter interactions with its upstream enhancers by quantitative 3 C (Schönheit et al., 2013). In this study PU.1 was found to regulate chromatin remodeling between the –50 kb enhancer and the Irf8 promoter in myeloid differentiation. In a recent study Kurotaki et al., 2022 determined the higher-order chromatin structure in DC progenitors, cDC1 and cDC2 on a genome-wide scale by Hi-C. In this study, reorganization of chromatin conformation at DC-specific gene loci was observed during cDC differentiation, and IRF8 was found to promote chromatin activation in DC progenitors leading to cDC lineage-specific gene expression. However, high resolution maps of the physical chromatin interactions of the Irf8 promoter with upstream and downstream enhancers in the full complement of DC subsets, including pDC, are required for understanding Irf8 regulation during DC differentiation.
Frequently, chromatin data, including ATAC-seq and/or ChIP-seq data, are used to identify regulatory elements of gene transcription. Here we embarked on a different approach and searched for lncRNA, which by themselves might have regulatory functions or are indicative of enhancer activity. We identified a novel lncRNA transcribed from the Irf8 +32 kb enhancer, which is specifically expressed in pDC, referred to as lncIrf8. We found that the lncIrf8 promoter element but not lncIrf8 itself impacts pDC and cDC1 development. Thus, lncIrf8 acts as an indicator for the Irf8 +32 kb enhancer activity. Importantly, our study revealed a previously unrecognized negative feedback loop of Irf8 in DC differentiation. Irf8 first activates its expression by autoactivation via the +32 kb enhancer and second limits its own expression through the lncIrf8 promoter element in the +32 kb enhancer.
Results
lncIrf8 marks a pDC-specific Irf8 enhancer element
Irf8 expression in DC development is subject to complex epigenetic regulation. Here, we used an integrated approach with RNA-seq, ATAC-seq, ChIP-seq and Capture-C to track the dynamics of gene expression, histone modification and chromatin conformation in the sequel MPP, CDP, pDC, cDC1, and cDC2 (Figure 1, Figure 1—figure supplements 1 and 2).
Figure 1 with 3 supplements see all
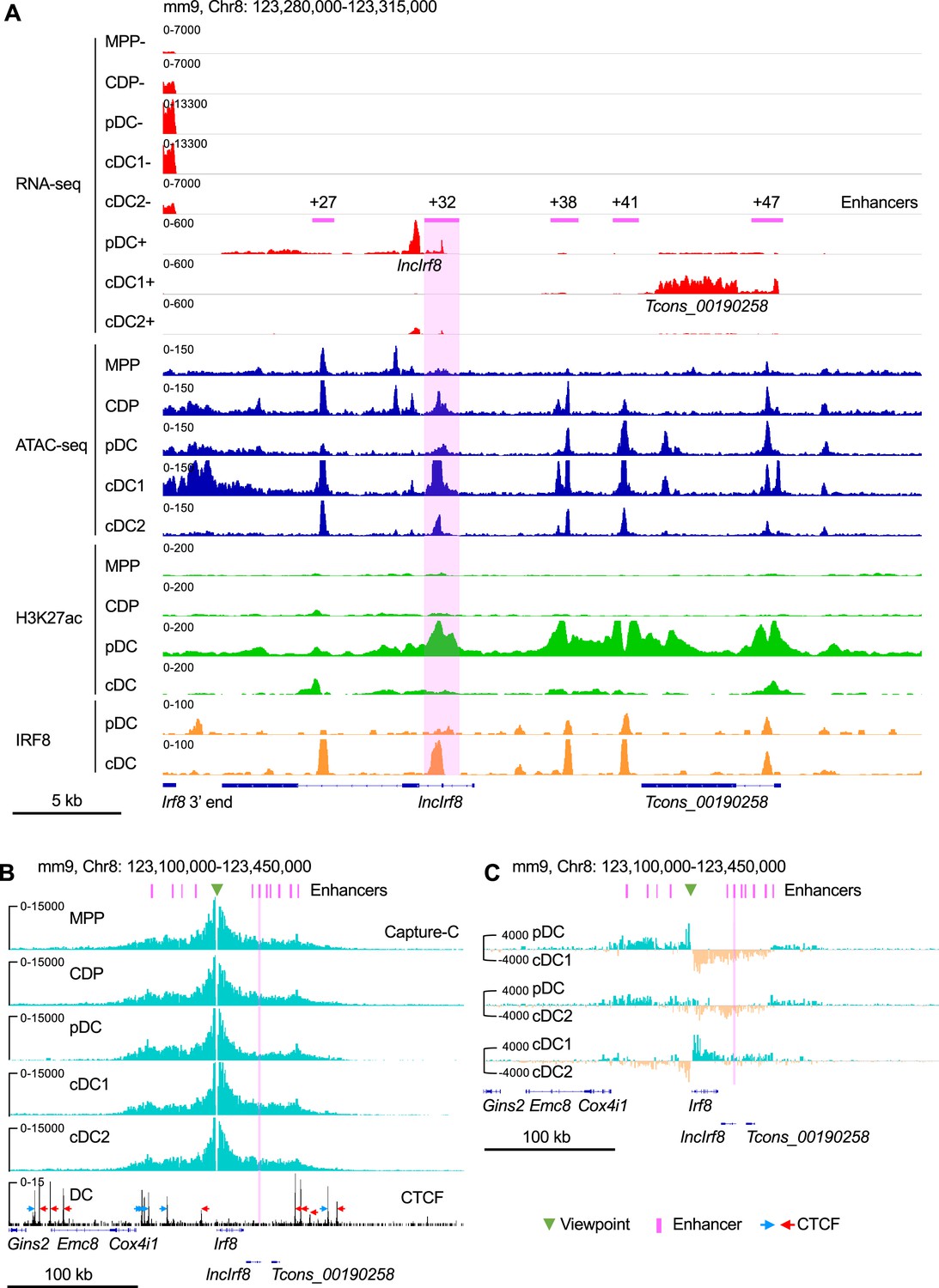
Irf8 epigenetic signatures and promoter-enhancer interaction maps during DC differentiation.
(A) Gene expression and epigenetic signatures of Irf8 downstream region in MPP, CDP, pDC, all cDC, cDC1, and cDC2 are visualized by IGV browser. Gene expression was measured by RNA-seq, chromatin accessibility by ATAC-seq, H3K27ac and IRF8 binding by ChIP-seq. Positions of Irf8 3’ end, Irf8 enhancers, pDC specific lncIrf8 and cDC1 specific Tcons_00190258 lncRNA are indicated. For RNA-seq - and +strands are shown. Scale bar: 5 kb. (B) Physical interactions of Irf8 promoter with flanking sequences in MPP, CDP, pDC, cDC1, and cDC2 by nuclear-titrated (NuTi) Capture-C (turquoise), and CTCF binding by ChIP-seq in DC (Garber et al., 2012). Mean numbers of unique interactions normalized to a 300 kb region around the Irf8 promoter viewpoint (green triangle) and scaled by a factor of 1,000,000 are shown (n=2). The orientations of CTCF binding are indicated with blue and red arrows. Tcons_00190258 refers to the cDC1-specific lncRNA shown in (A). Scale bar: 100 kb. (C) Comparations of the chromatin interactions with Irf8 promoter in pDC, cDC1 and cDC2. Differential tracks were created by subtraction of the mean normalized tracks of (B). Pairwise comparisons are shown and color coded. Turquoise and orange tracks represent specific interactions with the Irf8 promoter in the indicated cell types. Scale bar: 100 kb. Purple bars and lines indicate the position of flanking enhancers relative to Irf8 TSS. The purple bars from left to right represent –50 kb, –34 kb, –26 kb, –16 kb,+27 kb,+32 kb,+38 kb,+41 kb,+47 kb,+56 kb and +62 kb enhancer, respectively (panels B and C). Irf8 +32 kb enhancer is highlighted by purple box.
We performed de novo transcript assembly of the RNA-seq data and detected two previously unknown transcripts without coding potential downstream of Irf8: a pDC specific lncRNA (Tcons_00190250) in the following referred to as lncIrf8 and a cDC1 specific lncRNA (Tcons_00190258; Figure 1A and Figure 1—figure supplement 1). lncIrf8 and Tcons_00190258 show the same expression pattern in pDC and cDC1, respectively, in BM and spleen (Figure 1—figure supplement 3), as revealed by reanalyzing scRNA-seq and bulk RNA-seq data (Pang et al., 2022; Rodrigues et al., 2018). lncIrf8 is transcribed within an enhancer region located 32 kb downstream of the Irf8 TSS labeled by H3K27ac and H3K4me1 and occupied by DC differentiation-associated TF, such as IRF8 and PU.1 (Figure 1A and Figure 1—figure supplement 1). This region is largely devoid of H3K9me3, a chromatin modification frequently associated with heterochromatin, indicating an open chromatin configureuration in DC (Figure 1—figure supplement 1). In addition, sequences of this region have been implicated in DC development and referred to as +32 kb enhancer (Durai et al., 2019). Thus, we proceeded to study lncIrf8 in detail.
ATAC-seq analysis revealed further details of the lncIrf8 region in CDP, pDC, cDC1 and cDC2 (Figure 1A, Figure 2A and Figure 1—figure supplement 1). In cDC1 the prominent ATAC-seq and IRF8 peaks mark the cDC1 specific +32 kb enhancer (Durai et al., 2019). In pDC the ATAC-seq peak is smaller and shifted further towards downstream but aligns well with the valley in the prominent H3K27ac peak. This ATAC-seq peak marks the lncIrf8 promoter and aligns with p300 (Durai et al., 2019) and H3K4me3 (Figure 2A and Figure 1—figure supplement 1). All this indicates that this chromatin region is open and transcriptionally active in pDC, enabling lncIrf8 transcription.
Figure 2 with 4 supplements see all

IncIrf8 promoter KO compromises pDC and cDC1 development in vitro.
(A) Genomic anatomy of lncIrf8 locus determined by 3’ and 5’ RACE PCR. Blue box, exon 2 and 3 (48 bp and 468 bp, respectively). The 1010 bp intron and polyA tail are shown. Data of RNA-seq, ATAC-seq, ChIP-seq of H3K27ac (enhancer mark) and H3K4me3 (active promoter mark, near TSS) are visualized by IGV browser for the indicated cell populations (pDC, all cDC, cDC1 and cDC2). Grey box, lncIrf8 promoter KO region; open box, cDC1 specific +32 kb enhancer by Durai et al., 2019. Irf8 +32 kb enhancer based on the H3K27ac enhancer mark is indicated with a purple line. Scale bar: 1 kb. (B) Gene expression of lncIrf8 and Irf8 in lncIrf8 promoter KO and control at day 0, 5, and 7 of Flt3L directed DC differentiation. Gene expression was determined by RT-qPCR and normalized to GAPDH. n=4. (C) Representative flow cytometry analysis of Flt3L directed DC differentiation of lncIrf8 promoter KO HoxB8 MPP and control (Lutz et al., 2022; Xu et al., 2022). pDC, all cDC, cDC1, and cDC2 were gated as in Figure 2—figure supplement 1E and are shown. Bar diagrams depict quantification of pDC, cDC1 and cDC2 normalized to living single cells on DC differentiation day 0, 3, 5, 7, and 9. n=6–7. (D) Representative phase-contrast microscopy images of lncIrf8 promoter KO HoxB8 MPP and control on day 7 of Flt3L directed DC differentiation. Scale bar: 200 μm. (E) Representative flow cytometry analysis of spontaneous DC differentiation of lncIrf8 promoter KO HoxB8 MPP and control with growth factors but without E2 (Lutz et al., 2022; Xu et al., 2022) at day 8. Gr1+ monocytes and CD11c+ DC are shown. Quantification of Gr1+ monocytes of living single cells on day 3, 6, 8, and 10 of spontaneous DC differentiation. n=6, lncIrf8 promoter KO; n=4, control. Empty gRNA vector or non-targeting gRNA vector HoxB8 MPP were used as controls. Data represent mean ± SD of at least three independent experiments with different HoxB8 MPP clones of lncIrf8 promoter KO and control without deletion. *p<0.05, **p<0.01, ***p<0.001, multiple t-tests. Data that have no difference (p>0.05) are not labeled.
Next, we determined the chromatin conformation of the Irf8 locus and the lncIrf8 region. We generated interaction profiles by nuclear-titrated (NuTi) Capture-C in MPP, CDP, pDC, cDC1, and cDC2 (Figure 1—figure supplement 2A and B) using Irf8 promoter as viewpoint. The Irf8 promoter shows multiple interactions with regions spanning up to ~100 kb upstream and downstream of Irf8 (Figure 1B and Figure 1—figure supplement 1). In pDC, the Irf8 promoter interactions are stronger with the upstream sequences than with downstream sequences (Figure 1C and Figure 1—figure supplement 2C). In cDC1 Irf8 promoter interactions are more confined to the regions downstream of Irf8 compared to MPP, CDP and pDC (Figure 1C and Figure 1—figure supplement 2C). This suggests that upstream and downstream sequences of Irf8 gene are involved in differentially regulating Irf8 expression and controlling the development of pDC and cDC1, respectively.
The CCCTC-binding factor (CTCF) is important for regulation of chromatin conformation through loop extrusion (Sanborn et al., 2015) and we therefore visualized CTCF binding sites in the Irf8 locus in DC (Garber et al., 2012). Interestingly, most of the Irf8 flanking enhancers (Durai et al., 2019; Grajales-Reyes et al., 2015; Murakami et al., 2021; Schönheit et al., 2013) are located within convergent CTCF binding sites upstream and downstream of the Irf8 gene (Figure 1B and Figure 1—figure supplement 1). There are also multiple interactions within this region without convergent CTCF binding sites, suggesting interactions with Irf8 promoter in a CTCF independent manner, such as by TF binding, active histone modifications and gene transcription (Figure 1B and Figure 1—figure supplement 1; Owens et al., 2022).
Surprisingly, in pDC H3K27ac at the lncIrf8 promoter is high, but this locus shows less interactions with the Irf8 promoter in pDC compared to CDP, cDC1 and cDC2 (Figure 1C and Figure 1—figure supplement 2C). In addition, in pDC the IRF8 protein occupancy at the lncIrf8 promoter is low and much higher in cDC (Figure 1A and Figure 1—figure supplement 1; Durai et al., 2019; Grajales-Reyes et al., 2015).
These observations warrant further studies and we thus proceeded to investigate the lncIrf8 locus in detail.
lncIrf8 promoter KO compromises pDC and cDC1 development
First, we annotated lncIrf8. Our de-novo transcript assembly of RNA-seq data revealed different isoforms of lncIrf8, with the most prominent isoform comprising exon 2 and 3 (Figure 1A, Figure 2A and Figure 1—figure supplement 1). Additionally, 3' end and 5' end RACE PCR confirmed the anatomy of this lncIrf8 isoform: two exons, one intron, and a polyA tail (Figure 2A). As expected lncIrf8 is not conserved across species (data not shown), which is in line with the general characteristics of lncRNA. Then second, we deleted 300 bp in the lncIrf8 promoter by CRISPR/Cas9 editing in conditionally immortalized HoxB8 MPP (Figure 2A and Figure 2—figure supplement 1A–D). The lncIrf8 promoter is located in the Irf8 +32 kb enhancer region and is in close proximity to the cDC1 specific +32 kb enhancer (Durai et al., 2019; Figure 2A and Figure 2—figure supplement 2). The 300 bp deletion comprises the H3K4me3 promoter mark and is confined to open chromatin identified by ATAC-seq and positioned in the valley of the H3K27ac mark (Figure 2A). Additionally, it contains binding sites for IRF8, PU.1, and BATF3 TF, which are important for DC development (Figure 2A and Figure 2—figure supplement 2B).
Generation of a precise deletion requires clonal cell populations, which is hardly achieved in somatic cells due to their limited lifespan. Therefore, we developed a Mx-Cas9-GFP system of conditionally immortalized HoxB8 MPP, which upon differentiation faithfully recapitulate DC development (Figure 2—figure supplement 1A, B; Xu et al., 2022). HoxB8 MPP were obtained from bone marrow of Mx-Cas9-GFP mice by infection with the estrogen (E2) inducible HoxB8-ER. These HoxB9 MPP exhibited an extended lifespan and robust clonogenic potential and differentiated into all DC subsets in vitro and in vivo (Xu et al., 2022). Infection of gRNA targeting lncIrf8 promoter in Mx-Cas9-GFP HoxB8 MPP and induction of Cas9 by interferon generated single-cell lncIrf8 promoter KO clones. Five out of 71 single-cell colonies with homozygous deletions were further studied and subjected to DC differentiation (Figure 2C–E, Figure 2—figure supplement 1C–G and Figure 2—figure supplement 3).
lncIrf8 promoter KO abolished lncIrf8 expression during DC differentiation compared to control without deletion (Figure 2B). Surprisingly, Irf8 expression was also severely compromised, which points to a cross-talk of the lncIrf8 promoter element with the Irf8 promoter. To determine whether lncIrf8 promoter KO also impacts DC subsets, CD11c+ DC, pDC and cDC subsets cDC1 and cDC2 were analyzed (Figure 2C and Figure 2—figure supplement 1E–G). Frequencies of pDC and cDC1 were severely reduced, while cDC2 were unaffected (Figure 2C). Accordingly, lncIrf8 promoter KO cultures contained mainly cDC2 and some undifferentiated cells and were more homogenous compared to control without deletion, which contain multiple DC subsets (Figure 2C and D, Figure 2—figure supplement 1F, G).
lncIrf8 promoter KO affected also the differentiation propensity of progenitors upon E2 withdrawal from MPP/CDP culture (Figure 1—figure supplement 2A and Figure 2—figure supplement 3). lncIrf8 promoter KO showed a marked increase in strongly adhesive cells compared to control (Figure 2—figure supplement 3B). lncIrf8 promoter KO cultures had higher frequencies of Gr1+ monocytes (Figure 2E, Figure 2—figure supplement 3I, J) and lower frequencies of all DC subsets CD11c+ DC, pDC, cDC1, and cDC2 (Figure 2—figure supplement 3C–G) compared to control without deletion.
The lncIrf8 promoter element is in close proximity to the cDC1 specific +32 kb enhancer previously described in mice by Durai et al., 2019 and thus we generated the cDC1 specific +32 kb enhancer KO following the same procedure as for the lncIrf8 promoter KO. Five out of 165 single-cell clones with homozygous deletions of cDC1 +32 kb enhancer were subjected to DC differentiation and further analyzed (Figure 2—figure supplement 4). Similar to lncIrf8 promoter KO, cDC1 specific +32 kb enhancer KO abolished lncIrf8 expression and also decreased Irf8 expression during DC differentiation (Figure 2—figure supplement 4D). The cDC1 +32 kb enhancer KO also compromised the frequency of cDC1 in Flt3L directed DC differentiation, while cDC2 were unaffected (Figure 2—figure supplement 4E). Frequencies of pDC were also compromised at day 7; however, this was not statistically significant. These observations are in line with previous studies in mice that cDC1 +32 kb enhancer KO compromised cDC1 differentiation and left pDC and cDC2 largely unaffected (Durai et al., 2019; Murakami et al., 2021). In addition, cDC1 +32 kb enhancer KO cultures had higher frequencies of Gr1+ monocytes upon spontaneous DC differentiation by withdrawal of E2 (Figure 2—figure supplement 4F) and thus showed a similar phenotype as the lncIrf8 promoter KO upon spontaneous DC differentiation (Figure 2E).
Given the novel phenotype of the lncIrf8 promoter KO, we proceeded to investigate the impact of the lncIrf8 promoter KO on DC differentiation in vivo in mice. We transplanted CD45.2 lncIrf8 promoter KO and CD45.2 control HoxB8 MPP into irradiated CD45.1 recipient mice (Figure 3—figure supplement 1A). DC in bone marrow and spleen were analyzed by flow cytometry on day 7 and 14 after cell transplantation (Figure 3, Figure 3—figure supplement 1A, B). In bone marrow, lncIrf8 promoter KO cells mostly differentiated into Gr1+ monocytes, and lower frequencies of all DC subsets were observed on day 7 for lncIrf8 promoter KO cells compared to control (Figure 3A–F). In spleen, frequencies of cell populations from lncIrf8 promoter KO and control were similar to bone marrow, including lower frequencies of all DC subsets for lncIrf8 promoter KO (Figure 3G–L). CD45.2 donor HoxB8 cells were largely lost at day 14 after cell transplantation (Figure 3B–F and Figure 3H–L).
Figure 3 with 1 supplement see all
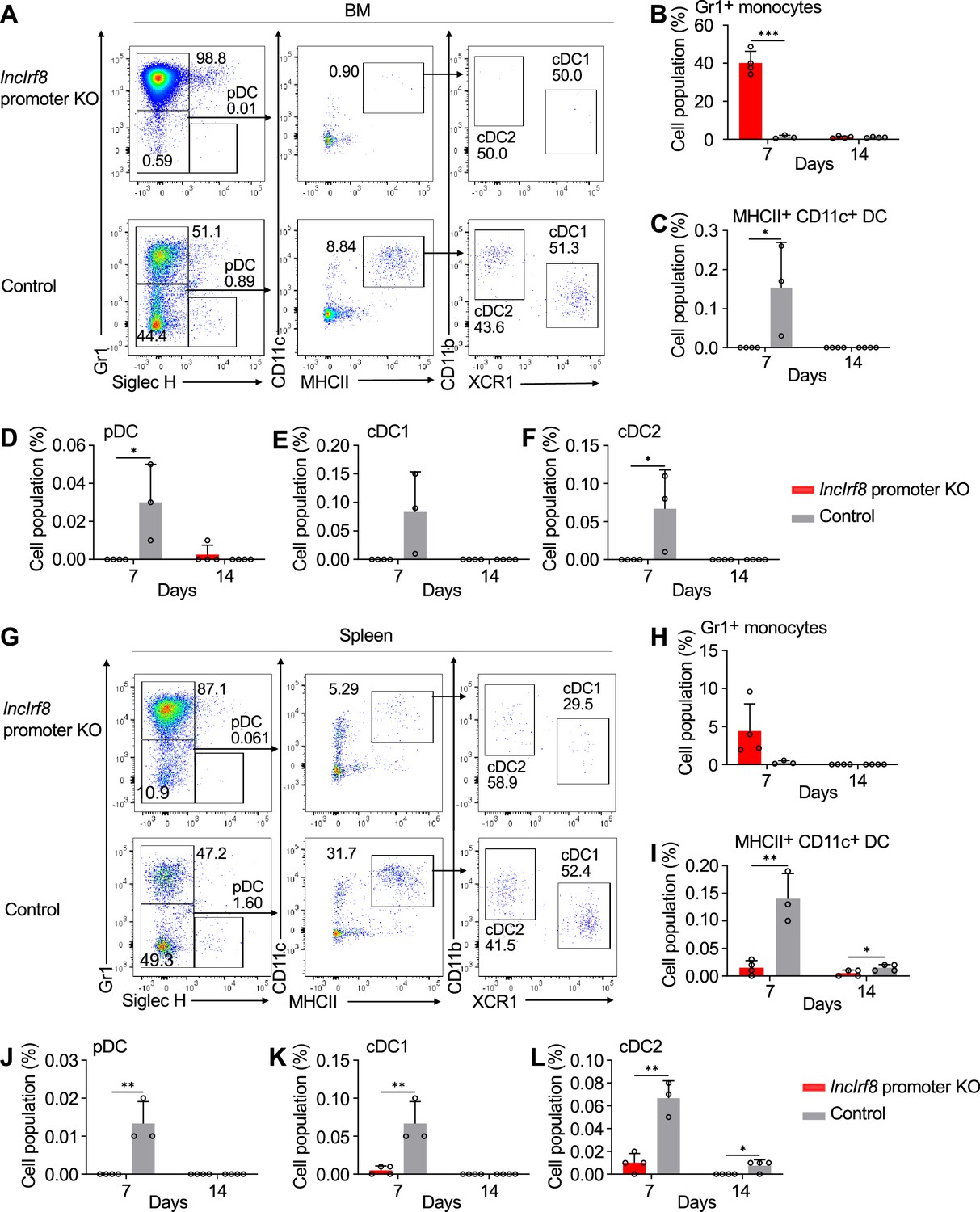
lncIrf8 promoter KO comprises pDC and cDC1 development in vivo upon cell transplantation.
(A) Representative flow cytometry analysis of CD45.2 lncIrf8 promoter KO and control HoxB8 MPP in BM at day 7 after cell transplantation (for details see Figure 3—figure supplement 1A, B). Donor cell populations were gated from 7-AAD- CD45.2+ Lin- cells and Gr1+ monocytes, pDC, cDC1 and cDC2 are shown. (B–F) Quantification of Gr1+ monocytes, MHCII+ CD11c+ DC, pDC, cDC1, and cDC2 of living single cells in BM on day 7 and 14 after cell transplantation (n=3–4). (G) Representative flow cytometry analysis of lncIrf8 promoter KO and control HoxB8 MPP in spleen at day 7 after cell transplantation. Gating was as in panel (A). (H–L) Quantification of Gr1+ monocytes, MHCII+ CD11c+ DC, pDC, cDC1 and cDC2 on day 7 and 14 after cell transplantation (n=3–4). Data represent mean ± SD from 3 to 4 mice. *p<0.05, **p<0.01, ***p<0.001, multiple t-tests. Data that have no difference (p>0.05) are not labeled.
Thus, lncIrf8 promoter KO compromised pDC and cDC1 development both in vitro and in vivo.
lncIrf8 acts as an indicator of Irf8 +32 kb enhancer activity in pDC
Knockout of lncIrf8 promoter and thus abolishment of lncIrf8 expression severely diminished pDC and cDC1 development in vitro and in vivo. The lncIrf8 promoter is located within Irf8 +32 kb enhancer (Durai et al., 2019) and thus it was important to determine whether lncIrf8 itself plays a role in regulating pDC and cDC1 development. To address this question, we (i) overexpressed lncIrf8 in wild-type MPP and (ii) re-expressed lncIrf8 in lncIrf8 promoter KO MPP and monitored its impact on DC development (Figure 4 and Figure 4—figure supplement 1).
Figure 4 with 1 supplement see all
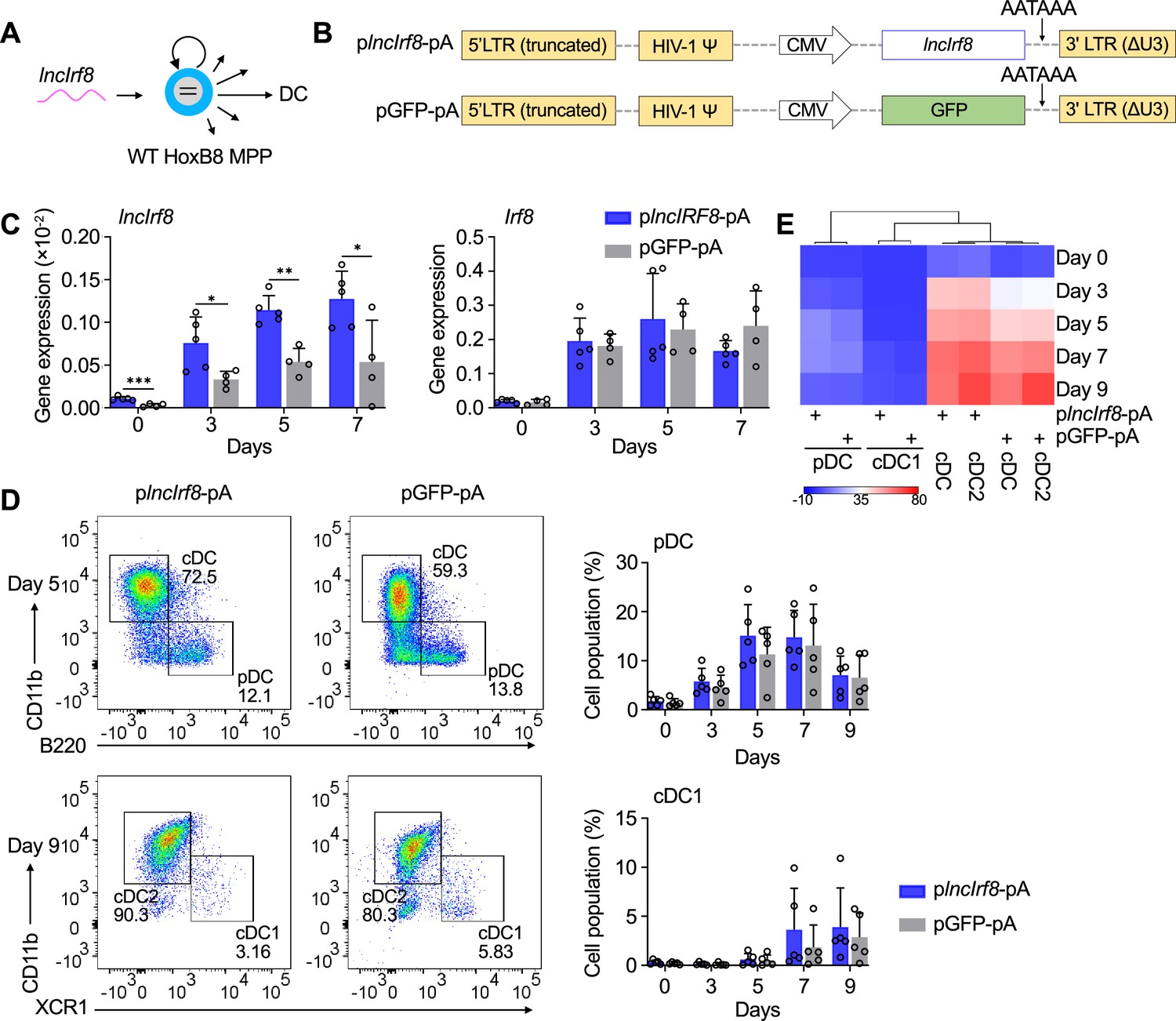
lncIrf8 overexpression leaves pDC and cDC1 development unaffected.
(A and B) Schematic representation of lncIrf8 overexpression in WT HoxB8 MPP and of plncIrf8-pA (lncIrf8 overexpression) and pGFP-pA (control) plasmids. A polyA signal AATAAA for transcription termination was inserted at the 3’ end of lncIrf8 and GFP. (C) Gene expression of lncIrf8 and Irf8 in plncIrf8-pA and pGFP-pA HoxB8 MPP on day 0, 3, 5, and 7 of Flt3L directed DC differentiation (n=4–5). Gene expression was by RT-qPCR and normalized to GAPDH. (D) Representative flow cytometry of DC subsets at day 5 and 9 of Flt3L directed DC differentiation of plncIrf8-pA and pGFP-pA HoxB8 MPP. Quantification of pDC and cDC1 of living single cells on Flt3L directed DC differentiation day 0, 3, 5, 7, and 9 (n=5) is shown. Gating for pDC and cDC1 was as in Figure 2—figure supplement 1E. (E) Heatmap representation of DC subsets of panel (D) at day 0, 3, 5, 7, and 9 of DC differentiation. Red, high frequency; white, intermediate frequency and blue, low frequency. Data represent mean ± SD of at least three independent experiments with different HoxB8 MPP clones of plncIrf8-pA and pGFP-pA. *p<0.05, **p<0.01, ***p<0.001, multiple t-tests. Data that have no difference (p>0.05) are not labeled.
lncIrf8 cDNA was cloned into a polyA lentivirus vector. An ‘AATAAA’ stop signal (Alvarez-Dominguez et al., 2015) was inserted at the 3’ end of lncIrf8 to avoid longer transcripts than lncIrf8 (plncIrf8-pA, Figure 4B). The respective pGFP-pA vector was used as control. lncIrf8 overexpressing single-cell clones were generated by limiting dilution (Figure 4—figure supplement 1A), expanded and subjected to DC differentiation. As expected lncIrf8 expression was markedly increased in plncIrf8-pA infected cells compared to control, while there were no significant differences in Irf8 expression between the two groups during DC differentiation (Figure 4C). Further, there were no differences in the frequencies of pDC, cDC1, and cDC2 between plncIrf8-pA infected cells and controls (Figure 4D and E, Figure 4—figure supplement 1B–D), indicating that lncIrf8 overexpression has no effect on Irf8 expression and DC differentiation.
To further extend this observation we performed a lncIrf8 rescue in lncIrf8 promoter KO MPP. lncIrf8 was re-expressed in lncIrf8 promoter KO MPP by lentiviral vector and cells were subjected to DC differentiation (Figure 4—figure supplement 1E). lncIrf8 RNA was effectively expressed and cells differentiated in response to Flt3L (Figure 4—figure supplement 1F). Yet frequencies of pDC and cDC1 were very low to absent and not rescued by lncIrf8 expression (Figure 4—figure supplement 1G, H).
In a nutshell, lncIrf8 overexpression and rescue had no effects on pDC and cDC1 development. This strongly suggests that lncIrf8 has no activity on its own in DC differentiation but rather acts as an indicator for the activation state of sequences within the Irf8 +32 kb enhancer. In addition, the lncIrf8 promoter comprises a sequence element with impact on pDC and cDC1 development.
Activation of lncIrf8 promoter promotes cDC1 development
Next, we proceeded to study the impact of the sequence element within lncIrf8 promoter on Irf8 expression and DC differentiation using CRISPR activation by dCas9-VP64 (Figure 5A and Figure 5—figure supplement 1). dCas9-VP64 is a mutated Cas9 deficient in nuclease activity, which is fused to the VP64 effector domain and confers gene activation. Targeting of dCas9-VP64 to the lncIrf8 promoter was achieved with specific gRNAs (Figure 5—figure supplement 1I). We also included targeting dCas9-VP64 to the Irf8 promoter to study the interplay with the lncIrf8 promoter. FACS sorted HoxB8 MPP expressing dCas9-VP64 and gRNA were subjected to DC differentiation and analyzed for lncIrf8 and Irf8 expression and DC subset composition (Figure 5B–E and Figure 5—figure supplement 1A–E).
Figure 5 with 1 supplement see all

Activation of lncIrf8 promoter promotes cDC1 development.
(A) Schematic representation of lncIrf8 and Irf8 promoter activation (top and middle, respectively) by CRISPR activation with dCas9-VP64. gRNAs were positioned upstream of lncIrf8 and Irf8 TSS for gene activation. dCas9-VP64 cells with non-targeting gRNA were used as control (bottom). Green and purple wavy lines represent Irf8 and lncIrf8 RNA, respectively. The number of wavy lines indicates levels of gene transcription determined by RT-qPCR in (C). Different length of blue arrows represents the frequencies of pDC and cDC1 according to panel B, D and E. Nc, No change; Ctrl, Control. (B) Representative flow cytometry analysis of CRISPR activation targeting the lncIrf8 and Irf8 promoters at day 7 and 9 of Flt3L directed DC differentiation. Two non-targeting gRNAs were used as controls and one representative non-targeting gRNA is shown (Non-targeting-VP64). Top row, CD11b+ B220- cDC and CD11b- B220+ pDC at day 7 of Flt3L directed DC differentiation; bottom row, CD11blow/- XCR1+ cDC1 and CD11b+ XCR1- cDC2 at day 9 of DC differentiation. For gating strategy see Figure 5—figure supplement 1B. (C) Gene expression of lncIrf8 and Irf8 in lncIrf8-VP64, Irf8-VP64 and non-targeting-VP64 HoxB8 MPP on day 0, 5, 7, and 9 of Flt3L directed DC differentiation (n=3). Gene expression analysis was by RT-qPCR and data are normalized to GAPDH. Data represent mean ± SD of three independent experiments. *p<0.05, **p<0.01, ****p<0.0001, two-way ANOVA, Tukey’s multiple comparisons test. Data that have no difference (p>0.05) are not labeled. (D and E) Quantification of pDC and cDC1 in percent of living single cells as in panel (B) on various days of Flt3L directed DC differentiation (n=3). Non-targeting-VP64 refers to both non-targeting-VP64 controls (n=6). Data represent mean ± SD of 3 independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA, Tukey’s multiple comparisons test. Data that have no difference (p>0.05) are not labeled.
Activation of the lncIrf8 promoter by dCas9-VP64 caused a massive increase of lncIrf8 expression at DC differentiation day 0 (Figure 5C). The activation of the lncIrf8 promoter led also to Irf8 upregulations at DC differentiation day 5, 7, and 9 compared to non-targeting control (Figure 5A and C). This demonstrates the positive impact of the lncIrf8 promoter element on Irf8 expression during DC differentiation and is in accord with the physical interaction of lncIrf8 and Irf8 promoters by Capture-C (Figure 1B and Figure 1—figure supplement 1).
Intriguingly, in lncIrf8-VP64 cells lncIrf8 expression was downregulated at DC differentiation day 5, 7 and 9, while Irf8 expression was upregulated (Figure 5A and C). This indicates a repressive effect of IRF8 on lncIrf8 promoter and is in accord with IRF8 binding to the lncIrf8 region by ChIP-seq (Figure 1 and Figure 1—figure supplement 1). This observation suggests a negative feedback loop of IRF8 on the lncIrf8 promoter during DC differentiation (Figure 5A and C).
Activation of Irf8 promoter by dCas9-VP64 increased Irf8 expression at DC differentiation day 0, 5, 7, and 9, while expression of lncIrf8 was unaffected compared to non-targeting control (Figure 5A and C). As expected Irf8 promoter activation led to higher pDC frequencies (Figure 5B and D), and also increased cDC1 frequencies compared to the non-targeting controls (Figure 5B and E). Importantly, lncIrf8 promoter activation by dCas9-VP64 also increased cDC1 frequencies and this was particular prominent at day 9 of DC differentiation (Figure 5B and E). Frequencies of cDC2 were decreased at day 9 and other populations, including pDC, remained unchanged (Figure 5B and D, Figure 5—figure supplement 1C–E).
Taken together our CRISPR activation of the lncIrf8 and Irf8 promoters by dCas9-VP64 suggest a negative feedback loop of Irf8 for pDC and cDC1 development.
Negative feedback regulation of lncIrf8 and Irf8 promoters controls DC differentiation
To directly investigate the negative feedback loop of Irf8 regulation, we repressed lncIrf8 and Irf8 promoters by targeted repression with dCas9-KRAB and analyzed the DC subsets during DC differentiation (Figure 6A–F and Figure 5—figure supplement 1A, B, F-H). dCas9-KRAB is a nuclease deficient Cas9 fused to the KRAB effector domain, which confers gene repression when positioned with specific gRNA (Figure 5—figure supplement 1I).
Figure 6
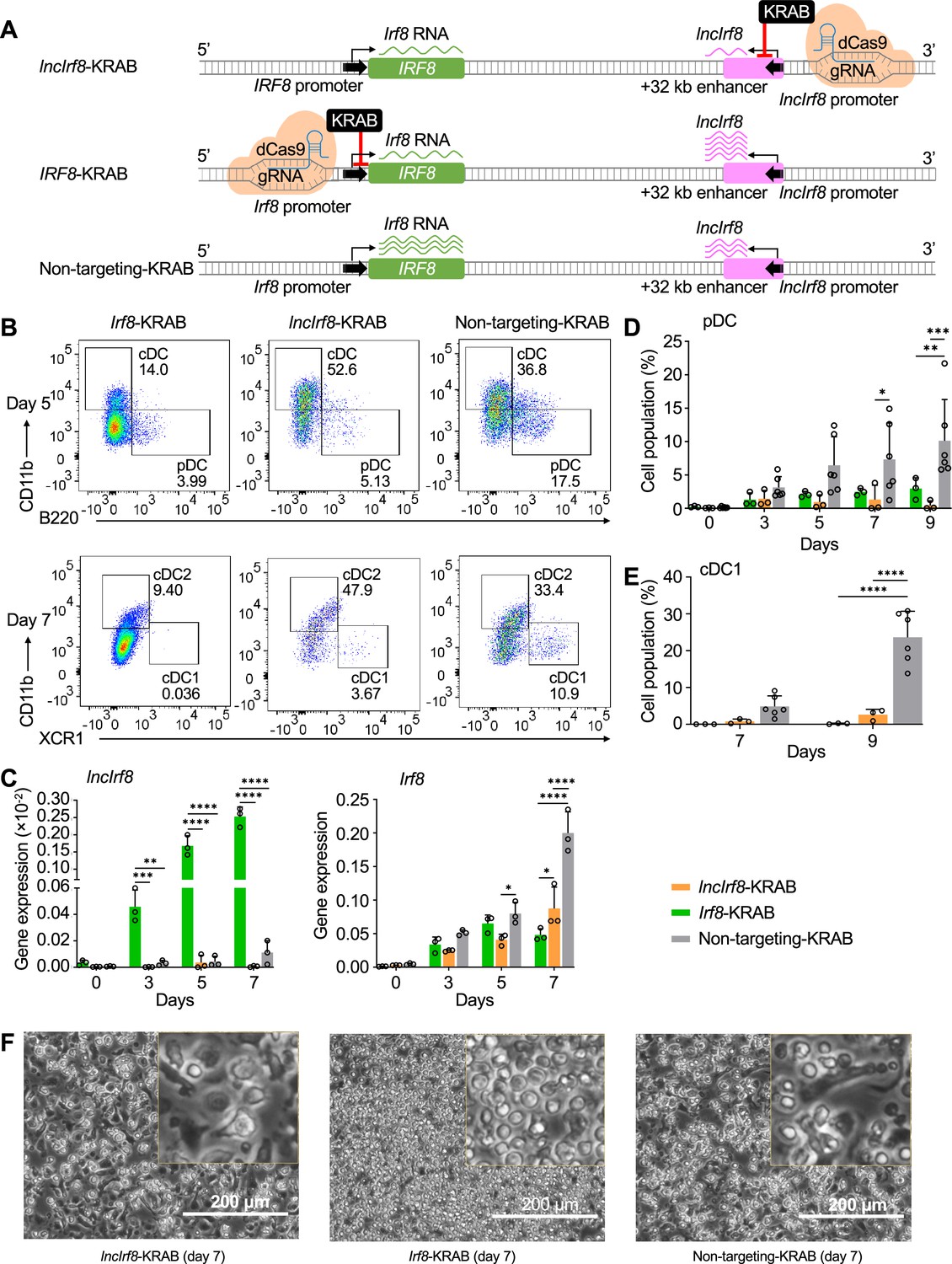
Repression of lncIrf8 promoter compromises pDC and cDC1 development.
(A) Schematic representation of lncIrf8 and Irf8 promoter repression (top and middle, respectively) by CRISPR interference with dCas9-KRAB. gRNAs were positioned downstream of lncIrf8 and Irf8 TSS to block gene transcription. dCas9-KRAB cells with non-targeting gRNA were used as control (bottom). Green and purple wavy lines represent Irf8 and lncIrf8 RNA, respectively. The number of wavy lines indicates levels of gene transcription determined by RT-qPCR in (C). (B) Representative flow cytometry analysis of CRISPR interference targeting the lncIrf8 and Irf8 promoters at day 5 and 7 of Flt3L directed DC differentiation. Two non-targeting gRNAs were used as controls and one representative non-targeting gRNA is shown (Non-targeting-KRAB). cDC and pDC at day 5 and cDC1 and cDC2 at day 7 of Flt3L directed DC differentiation are shown similar to Figure 5B. (C) Gene expression of lncIrf8 and Irf8 in lncIrf8-KRAB, Irf8-KRAB and non-targeting-KRAB HoxB8 MPP on day 0, 3, 5, and 7 of Flt3L directed DC differentiation (n=3). Gene expression analysis was by RT-qPCR and data are normalized to GAPDH. Data represent mean ± SD of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA, Tukey’s multiple comparisons test. Data that have no difference (p>0.05) are not labeled. (D and E) Quantification of pDC and cDC1 in percent of living single cells as in panel (B) on various days of Flt3L directed DC differentiation (n=3). Non-targeting-KRAB refers to both non-targeting-KRAB controls (n=6). Data represent mean ± SD of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, two-way ANOVA, Tukey’s multiple comparisons test. Data that have no difference (p>0.05) are not labeled. (F) Representative phase-contrast microscopy image of lncIrf8-KRAB, Irf8-KRAB and non-targeting-KRAB on day 7 of Flt3L directed DC differentiation. Scale bar: 200 μm.
Targeted repression of the Irf8 promoter decreased Irf8 expression as expected but massively increased lncIrf8 expression compared to non-targeting control (Figure 6A and C). This is very much in line with Irf8 impacting lncIrf8 expression by a negative feedback loop. Positioning the dCas9-KRAB repressor in the IncIRF8 promoter led to downregulation of both Irf8 and lncIrf8 expression, which confirms the lncIrf8 promoter element acting on the Irf8 promoter (Figure 6A and C).
Interestingly, targeted repression of the Irf8 promoter severely compromised development of all DC subsets, including pDC, cDC1, and cDC2, compared to non-targeting controls and yielded CD11c+ cells with progenitor-like spherical morphology (Figure 6B and D–F, Figure 5—figure supplement 1F–H). This reemphasizes the pivotal role of IRF8 for DC development known from studies on Irf8 knockout mice (Murakami et al., 2021; Schiavoni et al., 2002; Sichien et al., 2016; Tsujimura et al., 2003). In addition, targeted repression of the lncIrf8 promoter (lncIrf8-KRAB) also compromised pDC and cDC1 development (Figure 6B, D and E). This result is very similar to the lncIrf8 promoter KO analyzed above (Figure 2A–C).
All these findings support a model of Irf8 regulating its own expression by a negative feedback loop acting on the Irf8 +32 kb enhancer to limit Irf8 autoactivation (Figure 7). This regulatory Irf8 +32 kb enhancer element is marked by lncIrf8. Irf8 expression starts in CDP and further increases in pDC and cDC1, with particularly high expression in pDC (Figure 1A, Figure 7A and B, Figure 1—figure supplement 1). The increase in Irf8 expression is proposed to be due to an increase in the interactions of the Irf8 promoter with upstream and downstream sequences. In pDC, the Irf8 promoter-enhancer interactions are more with upstream chromatin regions (Figure 1C and Figure 7A), which relates to high Irf8 expression. In cDC1, the Irf8 promoter interactions are stronger with the regions downstream of Irf8 (Figure 1C and Figure 7A) and the Irf8 +32 kb enhancer marked by lncIrf8 confers transcriptional repression.
Figure 7

Negative feedback loop of Irf8 through +32 kb enhancer governs DC differentiation.
(A) Schematic representation of Irf8 gene regulation during DC differentiation. Irf8 transcription is induced at the CDP stage by its flanking enhancers, including the +32 kb enhancer, and is further increased in pDC and cDC1 (green wavy lines; the number of wavy lines indicates levels of gene transcription as in Figure 5A and Figure 6A). The increase in Irf8 expression is due to an increase in the interactions of the Irf8 promoter with upstream and downstream sequences. Irf8 promoter interactions with upstream sequences are stronger in pDC and Irf8 promoter interactions with downstream sequences are stronger in cDC1. In pDC the +32 kb enhancer marked by lncIrf8 is less repressed by IRF8 repressor complex compared to cDC1, resulting in particularly high Irf8 expression and lncIrf8 transcription in pDC (purple wavy lines). This negative feedback inhibition of IRF8 on the +32 kb enhancer allows Irf8 to regulate its own expression and thus DC differentiation. (B) Negative feedback regulation of Irf8 through the +32 kb enhancer in cDC1 and pDC specification as described in panel (A). The RNA-seq and IRF8 ChIP-seq data are shown as in Figure 1A, Figure 1—figure supplement 1 and Figure 2—figure supplement 2A. (C) Recapitulating Irf8 and lncIrf8 transcription by repression and activation of the Irf8 +32 kb enhancer in HoxB8 MPP by targeted dCas9-KRAB and dCas9-VP64, respectively. Green and purple wavy lines represent gene expression as described in Figure 5A and Figure 6A.
In our model, we propose an IRF8 repressor complex that differentially acts on the Irf8 +32 kb enhancer element in a DC subset specific manner to limit Irf8 autoactivation. In cDC1 the +32 kb enhancer is repressed by the IRF8 repressor complex through negative feedback inhibition (prominent IRF8 binding in cDC by ChIP-seq; Figure 7A and B), which limits Irf8 autoactivation and expression. Conversely, in pDC there is less IRF8 repressor complex binding to the +32 kb enhancer, which results in high Irf8 and lncIrf8 transcription (Figure 7A and B). Recapitulating the IRF8 repressor complex with dCas9-KRAB and targeting lncIrf8 promoter in the +32 kb enhancer reduced Irf8 expression (Figure 7C). Conversely, activation of the +32 kb enhancer boosted lncIrf8 and Irf8 expression (Figure 7C). Thus, an intricate feedback loop of IRF8 on the +32 kb enhancer orchestrates Irf8 expression and thus DC differentiation.
Discussion
Hematopoiesis is a particularly well studied stem cell system and therefore provides an excellent model for studying TF in lineage commitment and cell differentiation, and the molecular principles involved (Belz and Nutt, 2012; Graf and Enver, 2009; Laurenti and Göttgens, 2018; Nutt and Chopin, 2020). This work revealed a previously unrecognized negative feedback loop of Irf8 in DC differentiation and shows how Irf8 autoactivation is controlled and ultimately limited. IRF8 is crucial for DC lineage specification both in humans and mice (Anderson et al., 2021; Belz and Nutt, 2012; Cabeza-Cabrerizo et al., 2021; Cytlak et al., 2020; Kurotaki et al., 2019; Nutt and Chopin, 2020). Irf8 is upregulated by autoactivation via the +32 kb enhancer (Grajales-Reyes et al., 2015; Lin et al., 2015). However, how Irf8 expression is controlled at late stages of DC differentiation and eventually limited is not known. Here we demonstrate that Irf8 expression is limited by a negative feedback loop via a sequence element marked by lncIrf8 in the Irf8 +32 kb enhancer.
Irf8 expression in hematopoiesis is regulated by its flanking enhancers, which determine lineage specification and DC subset development (Bagadia et al., 2019; Durai et al., 2019; Grajales-Reyes et al., 2015; Murakami et al., 2021; Schönheit et al., 2013). Frequently, enhancers are identified by ATAC-seq and ChIP-seq (Durai et al., 2019; Murakami et al., 2021), and here we embarked on a different approach and searched for eRNA and enhancer-associated lncRNA by RNA-seq. We identified a novel pDC-specific lncRNA (lncIrf8) transcribed from the Irf8 +32 kb enhancer. lncIrf8 itself lacks activities in DC differentiation but a lncIrf8 promoter element is crucial for pDC and cDC1 development. Upon deletion of this lncIrf8 promoter element pDC and cDC1 development was severely compromised, demonstrating that this sequence is important for both pDC and cDC1 development.
We propose a model where Irf8 expression during DC differentiation is in a first step initiated and activated through flanking enhancers, including the +32 kb enhancer by autoactivation (Grajales-Reyes et al., 2015; Lin et al., 2015; Figure 7). In a second step, the lncIrf8 promoter element confers feedback inhibition, which limits Irf8 expression. This feedback inhibition is different for pDC and cDC1, both of which express high levels of Irf8 (Bornstein et al., 2014; Grajales-Reyes et al., 2015; Lin et al., 2015). In cDC1, Irf8 expression is attributed to Irf8 autoactivation through the +32 kb enhancer driven by the BATF3-IRF8 complex (Durai et al., 2019; Grajales-Reyes et al., 2015). In pDC, Irf8 expression is even higher than in cDC1 (Bornstein et al., 2014; Lin et al., 2015), which we propose is due to less feedback inhibition at late stages of DC differentiation.
A candidate for mediating Irf8 feedback inhibition is IRF8 itself, since IRF8 works as transcriptional activator or repressor, depending on context. IRF8 activator or repressor function depends largely on the heterodimers (or even heterotrimers) with partner TF that bind to specific DNA sequences (Chang et al., 2018; Huang et al., 2016; Humblin et al., 2017; Tamura et al., 2015; Yoon et al., 2014). Modifications on IRF8 protein, such as phosphorylation and small molecule conjugation, also alter IRF8 activity (Chang et al., 2012; Konieczna et al., 2008). Potential IRF8 heterodimer partners, to form repressor complexes, are ETV6 or IRF2 (Huang et al., 2016; Humblin et al., 2017; Lau et al., 2018). The IRF8 repressor complex is proposed to bind to the +32 kb enhancer in cDC but not in pDC. This notion is in line with a prominent IRF8 signal at the +32 kb enhancer in cDC, which is absent in pDC (Durai et al., 2019; Grajales-Reyes et al., 2015; Figure 1A, Figure 7A and B, Figure 1—figure supplement 1 and Figure 2—figure supplement 2A).
Further support of our model stems from our CRIPSR activation/interference experiments (Figure 7C). CRIPSR activation of Irf8 promoter by dCas9-VP64 mimics Irf8 up-regulation during DC differentiation and causes an increase in pDC and cDC1 (Figure 5). CRIPSR interference of lncIrf8 promoter by dCas9-KRAB recapitulates transcriptional repressor binding to the +32 kb enhancer and causes Irf8 promoter inhibition (Figure 6).
We extended our study to delineate the chromatin conformation of the Irf8 promoter with flanking sequences by Capture-C. The Irf8 promoter was found to interact with its flanking enhancers already at the CDP stage and then interactions with specific upstream and downstream sequences are proposed to guide pDC and cDC specification, respectively. This is in accord with previous studies where some of these Irf8 flanking enhancers were required to maintain high levels of Irf8 expression (Anderson et al., 2021; Murakami et al., 2021).
We demonstrate that deletion of the lncIrf8 promoter element severely decreased Irf8 expression and abolished both pDC and cDC1 development in vitro and in vivo upon cell transplantation. These results are very similar to a previous study on cDC1 specific +32 kb enhancer knockout mice, which demonstrates the impact of +32 kb enhancer for cDC1 development in vivo (Durai et al., 2019). The lncIrf8 promoter is located in close proximity to the cDC1 specific +32 kb enhancer and thus can be expected to have overlapping functions. Indeed, deletion of the cDC1 +32 kb enhancer in HoxB8 MPP showed some similar activities on DC differentiation as the deletion of the lncIrf8 promoter element, including regulation of lncIrf8 and Irf8 expression, but mainly affected cDC1 differentiation. These observations indicate that the lncIrf8 promoter element has further functions compared to the cDC1 specific +32 kb enhancer, for example for pDC development, and the underlying mechanisms warrant further investigation.
eRNA and enhancer-associated lncRNA are indicative of enhancer activity, however whether the process of their transcription, the transcripts themselves, or both are functionally linked to enhancer activity, remains unclear (Sartorelli and Lauberth, 2020; Statello et al., 2021). Previous studies revealed that some eRNA and enhancer-associated lncRNA are indeed functionally connected with expression of the respective target genes (Sartorelli and Lauberth, 2020; Statello et al., 2021). Here, we found no apparent activity of lncIrf8 on its own in pDC and cDC1 development, as demonstrated by lncIrf8 overexpression and rescue experiments. lncIrf8 appears to rather serve as an indicator for Irf8 +32 kb enhancer activity. However, lncIrf8 might have additional functions in DC biology, which are not revealed in the current study and remain to be identified.
In conclusion, by analyzing the gene expression and epigenetic profiles of the Irf8 locus, we identified an enhancer element marked by lncIrf8 that regulates Irf8 and controls DC differentiation through a negative feedback loop. Our results suggest that Irf8 regulates itself by its flanking enhancers in DC fate determination: First, Irf8 induces its expression by autoactivation via flanking enhancers, including the Irf8 +32 kb enhancer, to initiate DC differentiation, and second limits its expression at late stages via the lncIrf8 promoter element within the same +32 kb enhancer by feedback inhibition. This molecular principle of feedback inhibition is expected to also apply to other TF and cell differentiation systems.
Materials and methods
Mice
Wild type C57BL/6, Mx-Cas9-GFP knock-in mice (Kühn et al., 1995; Platt et al., 2014; Xu et al., 2022), and CD45.1 recipient C57BL/6 mice were used in this study. Mice were kept under specific pathogen-free conditions in the central animal facility of RWTH Aachen University Hospital, Aachen, Germany. All the animal experiments were approved by the local authorities of the German State North Rhine-Westphalia, Germany according to the German animal protection law (reference number: 81–02.04.2018 .A228).
Cell culture
Request a detailed protocolMultipotent progenitors (MPP) were obtained from mouse bone marrow and expanded in vitro with a two-step protocol as described in Felker et al., 2010. Conditionally immortalized HoxB8 MPP were generated by retrovirus infection of bone marrow cells from wild-type or Mx-Cas9-GFP knock-in mice with an estrogen (E2) inducible HoxB8 estrogen receptor (ER) fusion gene (HoxB8-ER) (Redecke et al., 2013; Xu et al., 2022). MPP were grown in RPMI 1640 medium with 10% FCS (Gibco, 10270106), 100 U/ml penicillin/streptomycin, 2 mM L-glutamine and 50 μM β-ME with a four-cytokine cocktail of SCF, Flt3 ligand (Flt3L), IGF-1, and IL-6/soluble IL-6 receptor fusion protein (hyper-IL-6) as before (referred to as MPP growth medium) (Felker et al., 2010; Lutz et al., 2022; Xu et al., 2022) (Appendix 1-key resources table). E2 (1 µM) was added to activate HoxB8-ER and to maintain the conditionally immortalized state of HoxB8 MPP. Cell density was adjusted to 1.5 million cells/ml every day. HEK293T cells for retrovirus and lentivirus production were grown in DMEM supplemented with 10% FCS (PAA, A01125-499), 100 U/ml penicillin/streptomycin, 2 mM L-glutamine (Appendix 1-key resources table).
In vitro DC differentiation with HoxB8 MPP
Request a detailed protocolHoxB8 MPP were differentiated into DC using a two-step protocol modified from Felker et al., 2010 and described in Lutz et al., 2022; Xu et al., 2022. In brief, 0.75 million cells/ml were grown in MPP growth medium with 50 ng/ml Flt3L (Peprotech, 300–19) and reduced E2 (0.01 μM) for two days and cell density was kept to 0.75 million cells/ml. To induce DC differentiation, HoxB8 MPP were then washed with PBS to remove cytokines and E2, and cultured in RPMI 1640 medium supplemented with FCS, penicillin/streptomycin, L-glutamine, β-ME (same concentrations as above), and Flt3L (50 ng/ml, Peprotech) (referred to as DC differentiation day 0). Partial medium changes were performed on differentiation day 3 and 6. Spontaneous DC differentiation of HoxB8 MPP was achieved simply by removing E2 from growth medium (SCF, Flt3L, IGF1 and hyper-IL6), and culturing the cells at 1.5 million cells/ml (Lutz et al., 2022; Xu et al., 2022).
Nuclear-Titrated (NuTi) Capture-C
Request a detailed protocolWild-type bone marrow cell-derived MPP, CDP, cDC1, cDC2, and pDC were generated in vitro with the two-step protocol as described in Felker et al., 2010. Cell populations were sorted by BD FACSAria IIu or BD FACSMelody, MPP are Gr1- CD117hi CD135low/-, CDP are Gr1- CD117int CD135+ CD115+, cDC1 are CD11c+ CD11blow/- XCR1+, cDC2 are CD11c+ CD11b+ XCR1-, pDC are CD11c+ CD11b- B220+.
The chromatin conformation capture (3 C) library preparation protocol used in this study was modified from Downes et al., 2021; Downes et al., 2022 with the reagents listed in Appendix 1-key resources table. MPP, CDP, cDC1, cDC2 and pDC were fixed with formaldehyde (final concentration 2%) and subjected to nuclear isolation according to the protocol in Downes et al., 2022; Li et al., 2019. Nuclei (15–20 million per sample) were digested with DpnII and DNA fragments were ligated by T4 DNA HC ligase. DNA was extracted and purified with Phenol-Chloroform-Isoamyl alcohol (PCI, 25:24:1). DpnII digestion efficiency was determined by SYBR qPCR with the primers listed in Appendix 1-key resources table and the quality of 3 C libraries was investigated by agarose gel (1%) electrophoresis; 3 C samples were used only if the DpnII digestion efficiency was more than 70%.
For Irf8 promoter viewpoint 2 oligonucleotide probes targeting Irf8 promoter were designed with the design tool Oligo (https://oligo.readthedocs.io/en/latest/index.html). Oligonucleotide probes are positioned adjacent to the DpnII cut sites on a restriction fragment spanning the Irf8 promoter (chr8:123,259,948–123,260,530) and 70 bp ssDNA biotinylated oligonucleotides were synthetized by Sigma-Aldrich (listed in Appendix 1-key resources table).
To enrich for fragments containing ligation events with Irf8 promoter viewpoint, NuTi Capture-C was performed as previously described (Downes et al., 2021; Downes et al., 2022). Briefly, the 3 C libraries prepared from MPP, CDP, cDC1, cDC2 and pDC were sonicated using Covaris S220 to an average size of ~200 bp using standard settings recommended by the manufacturer (time: 180 s, duty cycle: 10%, peak incident power: 175 W, cycles per burst: 200, temperature: 5–9°C). End repair was performed with the NEBNext Ultra II kit (New England Biolabs, E7645S) using 2 µg of sonicated 3 C library in duplicate for each sample. Illumina NEBNext Indices (New England Biolabs, E7500S, and E7335S) were added with a total of 6 cycles of amplification to allow for multiplexing. After this step, duplicates were pooled to increase sample complexity. We enriched samples as per NuTi Capture-C protocol, with two capture rounds in a multiplexed reaction, using 2 µg of each indexed sample as an input for the first capture. The hybridization with biotinylated probes was performed with the KAPA Hyper Capture Reagent Kit (Roche, 9075828001). Each ssDNA biotinylated probe was used at a concentration of 2.9 nM, with a final pool concentration of 5.8 nM, and 4.5 µl of the pooled oligonucleotides were used per sample. Captured DNA was pulled-down with M-270 Streptavidin Dynabeads (Invitrogen, 65305), washed and amplified off the beads with a total of PCR 14 cycles. The DNA obtained after the first capture was used as an input in the second capture round. The experiments were performed for the first and the second biological replicate separately, and then sequenced with NextSeq 550 Illumina System with 300 paired-end or 150 paired-end, respectively.
NuTi Capture-C data analysis
Request a detailed protocolThe Capture-C data was analyzed with CapCruncher (v0.1.1a1) pipeline (https://github.com/sims-lab/CapCruncher; sims-lab, 2022) in capture mode (Downes et al., 2022). The reads were aligned to the mm9 genome assembly with Bowtie2 (Langmead and Salzberg, 2012), with specific options: -p6--very-sensitive. Viewpoint coordinates used were: chr8:123,259,948–123,260,530, 1000 bp around viewpoint was excluded. The data were normalized to ~300 kb region around the viewpoint (chr8:123,132,865–123,433,117).
The Capture-C profiles in Figure 1 and Figure 1—figure supplement 1 show the mean number of unique interactions in two biological replicates, normalized to ~300 kb region around the viewpoint. Differential tracks were created by subtraction of the mean normalized tracks.
ATAC-seq, ChIP-seq, RNA-seq, and scRNA-seq
Request a detailed protocolATAC-seq analysis of MPP, CDP, cDC1, cDC2, and pDC was performed by Omni-ATAC-seq (Corces et al., 2017) with minor modifications as described in Li et al., 2019. RNA-seq analysis and ChIP-seq analysis was done as previously described (Allhoff et al., 2016; Lin et al., 2015).
To determin lncRNA expression in mouse ex-vivo pDC, scRNA-seq data of BM and splenic pDC were downloaded from GSE114313 (Rodrigues et al., 2018) and BAM files were converted back into FASTQ files using bamtofastq (https://support.10xgenomics.com/docs/bamtofastq). For visualization of lncRNA expression, we created a custom reference genome of mm9 by following the tutorial: available here. Cellranger pipeline was then processed to generate an expression count matrix of pDC. We next used scanpy (Wolf et al., 2018) to analyze the scRNA-seq data. We filtered cells based on the number of detected genes (<200 or>3500), and the proportion of mitochondrial genes (>10%). After data quality control, we retained 7044 cells for splenic pDC and 8158 cells for BM pDC, respectively. We then log-normalized the count matrix using a scaling factor of 10,000 and selected the top 3000 highly variable genes, which were used for dimensionality reduction based on principle component analysis. The cells were visualized using uniform manifold approximation and projection (UMAP) (Becht et al., 2019).
Plasmids
psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) for lentiviral packaging and envelope expressing plasmids were kind gifts from Didier Trono, EPFL, Lausanne, Switzerland. The gRNA expressing vector pLKO5.sgRNA.EFS.tRFP (Addgene #57823) and pLKO5.sgRNA.EFS.tRFP657 (Addgene #57824) were kind gifts from Benjamin Ebert, Harvard Medical School, Boston, USA (Heckl et al., 2014).
For lncIrf8 overexpression and rescue, lncIrf8 cDNA was introduced into polyA containing lentivirus vector pGFP-pA to generate plncIrf8-pA. pGFP-pA was constructed from pCIG3 (Addgene #78264; Caviness et al., 2014) by Gibson Assembly (New England Biolabs, E5510S; Gibson et al., 2009). In brief, the WPRE element was removed and a polyA signal ‘AATAAA’ was inserted at the 3’ end of GFP to construct pGFP-pA. lncIrf8 cDNA was sub-cloned into pGFP-pA using XhoI and SalI with the primers shown in Appendix 1-key resources table to obtain plncIrf8-pA. CRISPR activation and repression of lncIrf8 and Irf8 promoters were achieved by dCAS9-VP64_GFP (Addgene #61422) (Konermann et al., 2015) and pTet-KRAB-dCas9-GFP (Xu et al., 2022), respectively.
Lentivirus infection
Request a detailed protocolgRNAs, lncIrf8, dCas9-VP64-GFP, and KRAB-dCas9-GFP were delivered into HoxB8 MPP by lentiviral infection. Briefly, lentiviral particles were produced by calcium phosphate transfection (Graham and van der Eb, 1973) of HEK293T cells with psPAX2, pMD2.G, and the lentiviral transfer vector. At 48 and 72 hours after HEK293T cell transfection, supernatant containing virus particles was collected and concentrated using chondroitin sulfate sodium salt (CSS) and polybrene precipitation (Landazuri et al., 2007; Appendix 1-key resources table). HoxB8 MPP were infected twice with concentrated virus and subjected to Ficoll (Pancoll) purification to remove precipitate and dead cells.
Genetically modified HoxB8 MPP cell lines
lncIrf8 promoter and cDC1 specific +32 kb enhancer KO:
Request a detailed protocolMx-Cas9-GFP HoxB8 MPP were used to generate lncIrf8 promoter and cDC1 specific +32 kb enhancer knockout (KO) HoxB8 MPP by CRISPR/Cas9. Briefly, pairs of gRNAs each for lncIrf8 promoter KO and cDC1 specific +32 kb enhancer KO (Appendix 1-key resources table) were designed with the IDT online gRNA design tool. For cDC1 specific +32 kb enhancer KO we additionally used the same gRNA sequences from Durai et al., 2019 listed in Appendix 1-key resources table. gRNAs were cloned into pLKO5.sgRNA.EFS.tRFP and pLKO5.sgRNA.EFS.tRFP657 vectors (Heckl et al., 2014) using BsmBI-v2 (Appendix 1-key resources table), respectively.
One 10 cm dish (Bio-One) with 1.8 million HEK293T cells (70%–80% confluency) in 10 ml DMEM plus supplements (see above) was used to produce gRNA expressing lentivirus particles. HEK293T cells were transfected with 10 μg gRNA vector, 7.5 μg psPAX2, and 2.5 μg pMD2.G per gRNA by calcium phosphate transfection and lentivirus particles were harvested 48 and 72 h after transfection.
The gRNA expressing lentiviral particles were used to infect 3 million Mx-Cas9-GFP HoxB8 MPP. Cas9 and GFP expression were induced by mIFNα (1000 IU/ml; Appendix 1-key resources table) for 4 h, followed by FACS sorting for cells expressing the two gRNAs and Cas9 (GFP+ RFP+ RFP657+ cells). Single-cell clones were obtained by single-cell FACS sorting or limiting dilution.
Single-cell clones were genotyped by genomic PCR with primers listed in Appendix 1-key resources table. PCR products were sequenced by Eurofins Genomics and sequences were analyzed by SnapGene. Potential off-targets were routinely predicted by online software tools such as CRISPR-Cas9 gRNA checker (https://eu.idtdna.com/site/order/designtool/in-dex/CRISPR _SEQUENCE). For lncIrf8 promoter KO, 5 out of 71 single-cell colonies with homozygous deletions were subjected to off-target analysis. The top 2–5 predicted coding or non-coding targets were selected and HoxB8 MPP clones without off-target effects, or where off-target effects occurred in genes that were not expressed in MPP, CDP and DC subsets, were used for further studies (Table 1). For cDC1 specific +32 kb enhancer KO, 5 out of 165 single-cell clones with homozygous deletions were subjected to DC differentiation and further analyzed. Potential off-target genes of self-designed gRNAs for cDC1 +32 kb enhancer KO were not expressed in MPP, CDP, and DC subsets, and thus these gRNAs were used in the study.
Table 1
Off-target analysis.
| Potential off-targets | KO bulk(100 cells) | KO single-cell clones | Gene expression | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 7 | 19 | 21 | 24 | MPP | CDP | cDC | pDC | ||||
| gRNA1 | Chr 13: –48032179 | Gm36101 | No | No | No | No | No | No | No | No | No | No |
| Chr 2: –131293076 | Non-coding | No | No | No | No | No | No | No | No | No | No | |
| gRNA2 | Chr 1: –136050990 | AsCl5 | 16 bp deletion | 16 bp deletion | 16 bp deletion | No | 1 bp insertion | No | No | No | No | No |
| Chr 6:+119352090 | CACNA2D4 | No | No | No | No | No | No | No | No | No | Yes | |
| Chr 3:+33982811 | Non-coding | 3–29 bp deletion; 1–3 bp insertion | 3–29 bp deletion | 3 bp deletion | 1–2 bp insertion | No | No | No | No | No | No | |
| Chr 2: –132060887 | Non-coding | No | No | No | No | No | No | No | No | No | No | |
| Chr 2: –150624255 | Non-coding | No | No | No | No | No | No | No | No | No | No | |
-
Top potential off-targets of gRNA1 and gRNA2 predicted by CRISPR-Cas9 gRNA checker (see Materials and methods) were analyzed in lncIrf8 promoter KO bulk culture and KO single-cell clones by PCR analysis of genomic DNA. Potential off-target genes (coding) and non-coding sequences are listed. The absence of off-targets (No) and off-target deletions/insertion are shown.
lncIrf8 overexpression and lncIrf8 knockout rescue
Request a detailed protocollncIrf8 overexpression was performed in wild-type HoxB8 MPP. Lentiviral particles expressing lncIrf8 were produced from ten 6 cm dishes (Bio-One), each consisting of 0.75 million HEK293T cells (70–80% confluency) in 5 ml DMEM with supplements (see above). HEK293T cells were transfected with 5 μg plncIrf8-pA or pGFP-pA, 2.5 μg psPAX2, and 2.5 μg pMD2.G. Lentivirus particles were concentrated as above and used to infect 0.5 million HoxB8 MPP; single-cell clones were generated by limiting dilution and genotyped using the primers listed in Appendix 1-key resources table. Two out of 47 HoxB8 MPP colonies with plncIrf8-pA and 3 out of 19 HoxB8 MPP colonies with pGFP-pA (control) were expanded and subjected to Flt3L-directed DC differentiation.
lncIrf8 knockout rescue was carried out in lncIrf8 promoter KO HoxB8 MPP. FACS sorted cells that genotyped as lncIrf8 promoter KO homozygous deletion cells, were infected with lentiviral particles expressing lncIrf8. Lentiviral infection conditions were the same as for lncIrf8 overexpression in wild-type HoxB8 MPP (see above).
CRISPR activation and CRISPR interference
Request a detailed protocolCRISPR activation and CRISPR interference were performed by infecting wild-type HoxB8 MPP with lentiviral particles expressing dCAS9-VP64_GFP and pTet-KRAB-dCas9-GFP, respectively. The virus particles were produced as in the lncIrf8 overexpression experiments. In brief, virus particles from ten 6 cm dishes were used to infect 0.5 million wild-type HoxB8 MPP, followed by FACS sorting for GFP+ cells expressing dCas9-VP64 or dCas9-KRAB. Doxycycline (1 μg/ml) was used to induce dCas9-KRAB expression 2 days before cell sorting.
gRNAs targeting lncIrf8 and Irf8 promoters were cloned into pLKO5.sgRNA.EFS.tRFP as above. The dCas9-VP64-GFP and dCas9-KRAB-GFP HoxB8 MPP were then infected with specific gRNAs for gene activation and repression. The conditions for gRNA infection were the same as in lncIrf8 promoter KO experiments. Doxycycline (1 μg/ml) was given to the sorted dCas9-KRAB-GFP cells every 3 days to ensure sustained dCas9-KRAB expression.
Flow cytometry analysis and cell sorting
Request a detailed protocolDC subsets were analyzed by flow cytometry using FACS Canto II or LSR Fortessa (both from BD Biosciences). The information for flow cytometry antibodies is shown in Appendix 1-key resources table. For live/dead staining, cells were incubated with 3 μl 7-AAD per test for 5–10 min before flow cytometry measurement. Cells were sorted by FACS Aria IIu or FACS Melody, and flow cytometry data were analyzed by FlowJo V10 (all from BD Biosciences). Data on DC frequencies were subjected to the hierarchical clustering and represented in heatmap format using the online tool MORPHEUS (https://software.broadinstitute.org/morpheus/).
Cell transplantation
Request a detailed protocolCD45.1 recipient mice were sublethal irradiated (6.0 Gy, CP-160 Faxitron) 1 day before transplantation. lncIrf8 promoter KO and control HoxB8 MPP (single-cell clones) were expanded in vitro as described above. Cells were injected into recipient mice via the tail vein (5 million cells in 100 μl PBS per mouse). Donor cells from bone marrow and spleen were subjected to flow cytometry analysis at 7 and 14 days after cell transplantation.
RT-qPCR
Request a detailed protocolRNA was isolated by using the NucleoSpin RNA kit (Macherey-Nagel, 740955.250) according to the manufacturer’s instructions. RNA was subjected to cDNA synthesis using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814) with Murine RNase Inhibitor (New England Biolabs, M0314S) (Appendix 1-key resources table). RT-qPCR was performed by a StepOnePlus Real-Time PCR system (Applied Biosystems) with SYBR-green fluorescence (Applied Biosystems, 4385610). The primers for qPCR were listed in Appendix 1-key resources table. Mouse GAPDH was used for normalization of lncIrf8 and Irf8 gene expression.
Identification of lncIrf8
Request a detailed protocolDe-novo transcript assembly of RNA-seq data was used to search for unknown transcripts with no coding potential and this identified the pDC specific lncRNA Tcons_00190250 (referred to as lncIrf8) and the cDC1-specific lncRNA Tcons_00190258. In brief, paired end 2×100 bps reads from RNA-seq of MPP, CDP, cDC1, cDC2, and pDC were aligned to mm9 genome using Star aligner (version 2.4) (Dobin et al., 2013) and run for Cufflinks (version 2.0; Trapnell et al., 2012). Data were subjected to lenient filtering with the parameters: min isoform fraction 0.1% and pre-RNA-fraction of 0.1%, and ribosomal genes were also filtered. Next, all the predicted transcripts were merged with cuffmerge and all transcripts with no overlap with known transcripts in mouse GENCODE were selected for further analysis (Frankish et al., 2019). The coding potential and conservation of coding elements of the lncRNAs were checked with CPAT (Wang et al., 2013) and PhyloCSF (Lin et al., 2011), respectively. lncIrf8 (Tcons_00190250) and Tcons_00190258 show low scores in both analyses, which faithfully supports their role as non-coding genes and exhibit low cross-species conservation.
To further characterize the major transcripts of lncIrf8, 3' and 5' end Rapid Amplification of cDNA Ends (RACE) PCR was performed using template-switching RT enzyme mix (New England Biolabs, M0466) and TA cloning kit (Thermo Fisher Scientific, K202020) (Appendix 1-key resources table). The primers (listed in Appendix 1-key resources table) used for RACE PCR were synthesized by Eurofins Genomics except for 5' RACE-TSO, which was from IDT.
3' RACE PCR
Request a detailed protocolReverse transcript (RT) and template-switching: 4 μl (10 ng to 1 μg) total RNA (from DC differentiation day 5) were incubated at 80°C for 3 min and cooled rapidly on ice. RNA was then incubated with template-switching RT buffer, 1 mM dNTP, 5 mM DTT, 10 μM QT primer (Scotto-Lavino et al., 2006) and 1 μl RT enzyme in 10 μl at room temperature for 5 min, followed by 1 hr at 42°C, 10 min at 50°C, and 85°C for 5 min to inactive the RT enzyme mix and sample was then kept at 4°C and diluted with 20 μl MilliQ water.
First-round PCR: 1 μl of diluted sample was subjected to the first round PCR with Q0 primer and 3' RACE-GSP-lncIrf8-F1 (10 μM) using Q5 high fidelity DNA polymerase. Second-round PCR: the PCR products from the first round PCR (1:20 dilution) was then used as template for the second round of PCR by using Q1 primer (10 μM), 3' RACE-GSP-lncIrf8-F2 (10 μM). Q0 and Q1 primers and the PCR programs are described in Scotto-Lavino et al., 2006. Products from the second-round PCR were purified using the PCR clean-up kit (Macherey-Nagel, 740609.50).
A-tailing and TA cloning: a reaction containing 5 μl of PCR clean-up product in Taq buffer, 1 mM MgCl2, 0.4 mM dATP and 1 μl Taq DNA polymerase in 25 μl was prepared and incubated at 70°C for 20 min for A-tailing of PCR products. Five μl of the A-tailed products were subjected to TA cloning into pCR2.1 vector according to the manufacturer’s instruction followed by Sanger sequencing.
5' RACE PCR
Request a detailed protocolIn order to identify the TSS of lncIrf8, template RNA from bone marrow cell-derived pDC was used to perform 5' RACE PCR using template-switching RT enzyme mix (New England Biolabs, M0466) according to the manufacturer’s instructions. Template switching was by 5' RACE-GSP-lncIrf8-R2 primer (10 μM) and template switching oligo (TSO) (75 μM). Similar to the 3' RACE PCR, two rounds of PCR were used to improve PCR specificity. In brief, 5' RACE-GSP-lncIrf8-R2 (10 μM) and TSO-specific primer (10 μM) were used to perform the first-round PCR, 5' RACE-GSP-lncIrf8-R1 primer (10 μM) and the TSO-specific primer (10 μM) were used to perform the second round PCR, followed by fragments A-tailing, TA cloning, and Sanger sequencing as described above for 3' RACE PCR.
Identification of transcription factor binding sites
Request a detailed protocolTranscription factor binding sites (TFBS) in the Irf8 +32 kb enhancer were predicted using a motif matching tool based on the MOODS (Korhonen et al., 2009) and position weight matrixes (PWMs) were obtained from the JASPAR database (Fornes et al., 2020). The bit-score cut-off thresholds were determined by applying the dynamic programming approach as described in Wilczynski et al., 2009 with an FPR of 0.0001. DC TF were considered and are depicted.
Statistical analysis
Request a detailed protocolStatistical analyses were performed using Prism (GraphPad). Unpaired t test and Multiple t-tests were used to compare data from two groups, two-way ANOVA with Tukey’s multiple comparisons test was used to analyze data from more than two groups.
Appendix 1
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (M. musculus) | C57BL/6 mice (CD45.1 and CD45.2) | Jackson Laboratory | ||
| Genetic reagent (M. musculus) | Mx-Cas9-GFP mice | Medical Faculty, RWTH Aachen University Xu et al., 2022 | ||
| Cell line (H. sapiens) | HEK293T | ATCC | https://www.atcc.org | Lentivirus and retrovirus production |
| Cell line (M. musculus) | lncIrf8 promoter KO HoxB8 MPP; Control HoxB8 MPP | This paper | lncIrf8 promoter KO | |
| Cell line (M. musculus) | plncIrf8-pA HoxB8 MPP; pGFP-pA HoxB8 MPP | This paper | lncIrf8 overexpression and rescue | |
| Cell line (M. musculus) | Irf8-VP64 HoxB8 MPP; lncIrf8-VP64 HoxB8 MPP; Non-targeting-VP64-1 HoxB8 MPP; Non-targeting-VP64-2 HoxB8 MPP | This paper | Irf8 and lncIrf8 promoters activation | |
| Cell line (M. musculus) | Irf8-KRAB HoxB8 MPP; lncIrf8-KRAB HoxB8 MPP; Non-targeting-KRAB-1 HoxB8 MPP; Non-targeting-KRAB-2 HoxB8 MPP | This paper | Irf8 and lncIrf8 promoters repression | |
| Antibody | APC/Cyanine 7 anti-mouse/human B220 (rat monoclonal) | Biolegend | Cat# 103223; RRID: AB_313006 | FACS (1:400) |
| Antibody | Brilliant Violet 510 anti-mouse/human CD11b (rat monoclonal) | Biolegend | Cat# 101245; RRID: AB_2561390 | FACS (1:400) |
| Antibody | PE/Cyanine 7 anti-mouse CD11c (Armenian hamster monoclonal) | Biolegend | Cat# 117317; RRID: AB_493569 | FACS (1:400) |
| Antibody | APC anti-mouse CD115 (rat monoclonal) | eBioscience | Cat# 17-1152-80; RRID: AB_1210789 | FACS (1:400) |
| Antibody | PE/Cyanine 7 anti-mouse CD117 (rat monoclonal) | eBioscience | Cat# 25-1172-82; RRID: AB_469646 | FACS (1:400) |
| Antibody | PE anti-mouse CD135 (rat monoclonal) | eBioscience | Cat# 12-1351-82; RRID: AB_465859 | FACS (1:400) |
| Antibody | Biotin anti-mouse CD19 (rat monoclonal) | Biolegend | Cat# 115503; RRID: AB_313638 | FACS (1:800) |
| Antibody | Biotin anti-mouse CD3e (Armenian hamster monoclonal) | eBioscience | Cat# 13-0031-82; RRID: AB_466319 | FACS (1:800) |
| Antibody | APC/Cyanine 7 anti-mouse CD45.2 (mouse monoclonal) | Biolegend | Cat# 109823; RRID: AB_830788 | FACS (1:400) |
| Antibody | Biotin anti-mouse F4/80 (rat monoclonal) | Biolegend | Cat# 123105; RRID: AB_893499 | FACS (1:800) |
| Antibody | Alexa Fluor 700 anti-mouse Gr1 (rat monoclonal) | Biolegend | Cat# 108421; RRID: AB_493728 | FACS (1:400) |
| Antibody | PerCP/Cyanine 5.5 anti-mouse Gr1 (rat monoclonal) | eBioscience | Cat# 45-5931-80; RRID: AB_906247 | FACS (1:400) |
| Antibody | Alexa Fluor 700 anti-mouse Ly6C (rat monoclonal) | Biolegend | Cat# 128023; RRID: AB_10640119 | FACS (1:400) |
| Antibody | Biotin anti-mouse Ly6G (rat monoclonal) | Biolegend | Cat# 127603; RRID: AB_1186105 | FACS (1:800) |
| Antibody | Brilliant Violet 785 anti-mouse MHCII (rat monoclonal) | Biolegend | Cat# 107645; RRID: AB_2565977 | FACS (1:2000) |
| Antibody | Biotin anti-mouse NK1.1 (mouse monoclonal) | eBioscience | Cat# 14-5941-82; RRID: AB_467736 | FACS (1:800) |
| Antibody | Super Bright anti-mouse Siglec-H (rat monoclonal) | Invitrogen | Cat# 63-0333-82 RRID: AB_2784853 | FACS (1:400) |
| Antibody | PE/Dazzle 594 Streptavidin | Biolegend | Cat# 405247 | FACS (1:1000) |
| Antibody | Biotin anti-mouse Ter119 (rat monoclonal) | eBioscience | Cat# 14-5921-82; RRID: AB_467727 | FACS (1:800) |
| Antibody | Brilliant Violet 421 anti-mouse/rat XCR1 (mouse monoclonal) | Biolegend | Cat# 148216; RRID: AB_2565230 | FACS (1:400) |
| Antibody | 7-Aminoactinomycin D (7-AAD) | Thermo Fisher Scientific | Cat# A1310 | FACS (3 μl per test) |
| Chemical compound, drug | β-estradiol (E2) | Sigma-Aldrich | Cat# E2758 | |
| Chemical compound, drug | β-mercaptoethanol (β-ME) | Gibco | Cat# 31350010 | |
| Chemical compound, drug | BsmBI-v2 | New England Biolabs | Cat# R0739S | |
| Chemical compound, drug | Chondroitin sulfate sodium salt from shark cartilage (CSS) | Sigma-Aldrich | Cat# C4384 | |
| Chemical compound, drug | cOmplete Mini | Roche | Cat# 11836153001 | |
| Chemical compound, drug | dATP | New England Biolabs | Cat# N0440S | |
| Chemical compound, drug | Dimethysulfoxide (DMSO) | Sigma-Aldrich | Cat# D8418 | |
| Chemical compound, drug | Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 | |
| Chemical compound, drug | DpnII | A kind gift from A. Marieke Oudelaar, Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany. DpnII enzyme with a similar activity is also available from New England Biolabs. | Cat# R0543M | |
| Chemical compound, drug | DMEM | Gibco | Cat# 41965039 | |
| Chemical compound, drug | DTT | Thermo Fisher Scientific | Cat# 20290 | |
| Chemical compound, drug | EDTA | Gibco | Cat# 15575–038 | |
| Chemical compound, drug | Fetal calf serum (FCS) | PAA | Cat# A01125-499 | |
| Chemical compound, drug | Fetal calf serum (FCS) | Gibco | Cat# 10270106 | |
| Chemical compound, drug | Formaldehyde (37%) | AppliChem | Cat# A0877 | |
| Chemical compound, drug | Recombinant human Flt3-Ligand (Flt3L) | Peprotech | Cat# 300–19 | |
| Chemical compound, drug | Recombinant murine stem cell factor (SCF) | Peprotech | Cat# 250–03 | |
| Chemical compound, drug | Human IGF-1 long range | Sigma-Aldrich | Cat# 85,580 C | |
| Chemical compound, drug | Recombinant IL-6/soluble IL-6 receptor fusion protein | A kind gift from S. Rose-John, Kiel, Germany Fischer et al., 1997. R&D Systems provides a similar product with the same activity. | Cat# 9038 SR | |
| Chemical compound, drug | HEPES | Sigma-Aldrich | Cat# H4034 | |
| Chemical compound, drug | L-glutamine | Gibco | Cat# 25030081 | |
| Chemical compound, drug | M-270 Streptavidin Dynabeads | Invitrogen | Cat# 65305 | |
| Chemical compound, drug | Mouse interferon α (mIFNα) | Miltenyi Biotec | Cat# 130-093-131 | |
| Chemical compound, drug | Murine RNase Inhibitor | New England Biolabs | Cat# M0314S | |
| Chemical compound, drug | Pancoll human, density 1.077 g/ml (Ficoll) | PAN-Biotech | Cat# P04-601000 | |
| Chemical compound, drug | Penicillin/streptomycin | Gibco | Cat# 15140122 | |
| Chemical compound, drug | Phenol-Chloroform-Isoamyl alcohol (PCI) | Sigma-Aldrich | Cat# 77617 | |
| Chemical compound, drug | Phosphate buffered saline (PBS) | Gibco | Cat# 10010023 | |
| Chemical compound, drug | Polybrene (PB, Hexadimethrine bromide) | Sigma-Aldrich | Cat# H9268 | |
| Chemical compound, drug | Q5 high fidelity DNA polymerase | New England Biolabs | Cat# M0491L | |
| Chemical compound, drug | RPMI 1640 | Gibco | Cat# 31870025 | |
| Chemical compound, drug | SalI | New England Biolabs | Cat# R0138S | |
| Chemical compound, drug | Supernatant from Flt3L-producing B16F1 cells (1%) | Homemade. Flt3L from Peprotech has the same activity (1:1000) | Cat# 300–19 | |
| Chemical compound, drug | Supernatant from CHO KLS C6 cells expressing soluble murine SCF (1%) | Homemade. Peprotech provides a similar product with the same activity (1:1000). | Cat# 250–03 | |
| Chemical compound, drug | SYBR-green fluorescence | Applied Biosystems | Cat# 4385610 | |
| Chemical compound, drug | T4 DNA HC ligase | Life Tech | Cat# EL0013 | |
| Chemical compound, drug | Taq DNA polymerase | Homemade | ||
| Chemical compound, drug | Taq buffer (10 x) | Thermo Fisher Scientific | Cat# B33 | |
| Chemical compound, drug | Template-switching RT enzyme mix | New England Biolabs | Cat# M0466 | |
| Chemical compound, drug | XhoI | New England Biolabs | Cat# R0146S | |
| Commercial assay or kit | Gibson Assembly kit | New England Biolabs | Cat# E5510S | |
| Commercial assay or kit | High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 | |
| Commercial assay or kit | KAPA Hyper Capture Reagent Kit | Roche | Cat# 9075828001 | |
| Commercial assay or kit | NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S | |
| Commercial assay or kit | NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) | New England Biolabs | Cat# E7335S | |
| Commercial assay or kit | NEBNext Multiplex Oligos for Illumina (Index Primers Set 2) | New England Biolabs | Cat# E7500S | |
| Commercial assay or kit | NucleoSpin RNA kit | Macherey-Nagel | Cat# 740955.250 | |
| Commercial assay or kit | PCR clean-up kit | Macherey-Nagel | Cat# 740609.50 | |
| Commercial assay or kit | TA cloning kit | Thermo Fisher Scientific | Cat# K202020 | |
| Sequence-based reagent | 5'RACE-TSO | New England Biolabs | 5’ RACE PCR primers | GCTAATCATTGCAAGCAGTGGTATC AACGCAGAGTACATrGrGrG |
| Sequence-based reagent | 5'RACE-TSO-Specific | New England Biolabs | 5’ RACE PCR primers | CATTGCAAGCAGTGGTATCAAC |
| Sequence-based reagent | 5'RACE-GSP-lncIrf8-R1 | New England Biolabs | 5’ RACE PCR primers | TGTCAGTGATGGGGGCTGGAGAAAT |
| Sequence-based reagent | 5'RACE-GSP-lncIrf8-R2 | New England Biolabs | 5’ RACE PCR primers | GCTCAGGATGCCAGGTCCCTTCTT |
| Sequence-based reagent | 3'RACE-QT | Scotto-Lavino et al., 2006 | 3’ RACE PCR primers | CCAGTGAGCAGAGTGACGAGGACTC GAGCTCAAGCTTTTTTTTTTTTTTTTT |
| Sequence-based reagent | 3'RACE-Q0 | Scotto-Lavino et al., 2006 | 3’ RACE PCR primers | CCAGTGAGCAGAGTGACG |
| Sequence-based reagent | 3'RACE-QI | Scotto-Lavino et al., 2006 | 3’ RACE PCR primers | GAGGACTCGAGCTCAAGC |
| Sequence-based reagent | 3'RACE-GSP-lncIrf8-F1 | This paper | 3’ RACE PCR primers | ATTTCTCCAGCCCCCATCACTGACA |
| Sequence-based reagent | 3'RACE-GSP-lncIrf8-F2 | This paper | 3’ RACE PCR primers | AAGAAGGGACCTGGCATCCTGAGC |
| Sequence-based reagent | lncIrf8-F | This paper | Genotyping primers | TCCTGAAGGGACAGGCAAG |
| Sequence-based reagent | lncIrf8-R | This paper | Genotyping primers | CTTGGACATTGAGGACGCC |
| Sequence-based reagent | cDC1 +32 kb-F1 | This paper | Genotyping primers | GTGACTGCAAGTAAGTTCTTCGG |
| Sequence-based reagent | cDC1 +32 kb-F2 | This paper | Genotyping primers | AAGTAGAGATTCCCTTTCTAAGCC |
| Sequence-based reagent | cDC1 +32 kb-R | This paper | Genotyping primers | ATCAGGCTGGGTGGTGGTT |
| Sequence-based reagent | Sc-lncIrf8-F | This paper | Cloning primers | ACACTCGAGACTGTCAGATGCAGGGG; the underline sequences represent cloning sites |
| Sequence-based reagent | Sc-lncIrf8-R | This paper | Cloning primers | AAAAAAGTCGACGCATCAGATTTAATATA GAACTAGGACA; the underline sequences represent cloning sites |
| Sequence-based reagent | CMV-lncIrf8-F | This paper | Genotyping primers | TGGGCGTGGATAGCGGTTT |
| Sequence-based reagent | CMV-lncIrf8-R | This paper | Genotyping primers | CACTGAGACTTAGCAAGGGGGA |
| Sequence-based reagent | CMV-GFP-F | This paper | Genotyping primers | TGGGCGTGGATAGCGGTTT |
| Sequence-based reagent | CMV-GFP-R | This paper | Genotyping primers | TGGGGGTGTTCTGCTGGTAG |
| Sequence-based reagent | mlncIrf8-tQ-F | This paper | RT-qPCR primers | ACTGTCAGATGCAGGGG |
| Sequence-based reagent | mlncIrf8-tQ-R | This paper | RT-qPCR primers | TCACAATCGTCTGTAACTCCG |
| Sequence-based reagent | mIrf8-tQ-F | This paper | RT-qPCR primers | GAGCGAAGTTCCTGAGATGG |
| Sequence-based reagent | mIrf8-tQ-R | This paper | RT-qPCR primers | TGGGCTCCTCTTGGTCATAC |
| Sequence-based reagent | mGAPDH-tQ-F | This paper | RT-qPCR primers | ACCTGCCAAGTATGATGACATCA |
| Sequence-based reagent | mGAPDH-tQ-R | This paper | RT-qPCR primers | GGTCCTCAGTGTAGCCCAAGAT |
| Sequence-based reagent | m3C-F | Downes et al., 2021; Downes et al., 2022 | Capture-C qPCR primers | GGAGAAAGAAGGCTGGTGTTAT |
| Sequence-based reagent | m3C-R | Downes et al., 2021; Downes et al., 2022 | Capture-C qPCR primers | TATCTGAGTTGGACAGCATTGG |
| Sequence-based reagent | m3C-control-F | Downes et al., 2021; Downes et al., 2022 | Capture-C qPCR primers | TTATCTTGCATTTGCCAACTCG |
| Sequence-based reagent | m3C-control-R | Downes et al., 2021; Downes et al., 2022 | Capture-C qPCR primers | TGGGTTTCCCTGATTCTGAAA |
| Sequence-based reagent | Irf8_P_L | This paper | Capture probes | GATCCGTGCATCACCAGCCTCC TTGACCTTAGGCAGACGCCCCA GCCCCCCGGCCATTTTTGGGGCAGCC |
| Sequence-based reagent | Irf8_P_R | This paper | Capture probes | CCAAATGAACAAACACCTCTCCC TTTAAAATCTGCCTGATGGCCAA CTTCATAATGAAGAGAAATAGATC |
| Sequence-based reagent | gRNA-1-F | This paper | gRNAs | CACCGTCCATTATACTAAGATACCC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-1-R | This paper | gRNAs | AAACGGGTATCTTAGTATAATGGAC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-2-F | This paper | gRNAs | CACCGGTGCCGAGAAAGGACACGT; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-2-R | This paper | gRNAs | AAACACGTGTCCTTTCTCGGCACC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO1-5‘F | Durai et al., 2019 | gRNAs | CACCGTTGTGATCTTTGAGGTAGA; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO1-5’R | Durai et al., 2019 | gRNAs | AAACTCTACCTCAAAGATCACAAC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO1-3‘F | Durai et al., 2019 | gRNAs | CACCGAACTGGCCTGGGGCAGGTC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO1-3’R | Durai et al., 2019 | gRNAs | AAACGACCTGCCCCAGGCCAGTTC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO2-5’F | This paper | gRNAs | CACCGACATTCTGCACCCCAGTCA; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO2-5’R | This paper | gRNAs | AAACTGACTGGGGTGCAGAATGTC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO2-3’F | This paper | gRNAs | CACCGAGGATCGCACCTCACCTACT; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-KO2-3’R | This paper | gRNAs | AAACAGTAGGTGAGGTGCGATCCTC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-Irf8-VP64-F | This paper | gRNAs | CACCGACGGTCGCGCGAGCTAATTG; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-Irf8-VP64-R | This paper | gRNAs | AAACCAATTAGCTCGCGCGACCGTC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-Irf8-KRAB-F | This paper | gRNAs | CACCGCGGCAGGTAGGACGCGATG; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-Irf8-KRAB-R | This paper | gRNAs | AAACCATCGCGTCCTACCTGCCGC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-lncIrf8-VP64-F | This paper | gRNAs | CACCGGTGCCGAGAAAGGACACGT; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-lncIrf8-VP64-R | This paper | gRNAs | AAACACGTGTCCTTTCTCGGCACC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-lncIrf8-KRAB-F | This paper | gRNAs | CACCGAGTCACTCGTCCTTTGGGG; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-lncIrf8-KRAB-R | This paper | gRNAs | AAACCCCCAAAGGACGAGTGACTC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-non-targeting-1-F | Manguso et al., 2017 | gRNAs | CACCGCGAGGTATTCGGCTCCGCG; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-non-targeting-1-R | Manguso et al., 2017 | gRNAs | AAACCGCGGAGCCGAATACCTCGC; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-non-targeting-2-F | Manguso et al., 2017 | gRNAs | CACCGATGTTGCAGTTCGGCTCGAT; the underline sequences represent cloning sites |
| Sequence-based reagent | gRNA-non-targeting-2-R | Manguso et al., 2017 | gRNAs | AAACATCGAGCCGAACTGCAACATC; the underline sequences represent cloning sites |
| Software, algorithm | Bamtofastq | 10 x Genomics | https://support.10xgenomics.com/docs/bamtofastq | |
| Software, algorithm | Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net | |
| Software, algorithm | CapCruncher (v0.1.1a1) | Downes et al., 2022 | https://github.com/sims-lab/CapCruncher | |
| Software, algorithm | CPAT | Wang et al., 2013 | http://code.google.com/p/cpat/ | |
| Software, algorithm | Cufflinks (version 2.0) | Trapnell et al., 2012 | http://cufflinks.cbcb.umd.edu/ | |
| Software, algorithm | FlowJo V10 | BD Biosciences | ||
| Software, algorithm | IGV browser | Broad Institute | ||
| Software, algorithm | MOODS | Korhonen et al., 2009 | https://www.cs.helsinki.fi/group/pssmfind/ | |
| Software, algorithm | Oligo | Oudelaar et al., 2020 | https://oligo.readthedocs.io/en/latest/index.html | |
| Software, algorithm | PhyloCSF | Lin et al., 2011 | http://compbio.mit.edu/PhyloCSF | |
| Software, algorithm | Prism | GraphPad | ||
| Software, algorithm | Scanpy | Wolf et al., 2018 | ||
| Software, algorithm | Star aligner (version 2.4) | Dobin et al., 2013 | http://code.google.com/p/rna-star/ | |
| Software, algorithm | Snapgene | GSL Biotech | ||
| Software, algorithm | UMAP | Becht et al., 2019 |
Data availability
ATAC-seq (MPP, CDP and cDC2), Capture-C targeting Irf8 promoter (MPP, CDP, cDC1, cDC2 and pDC), IRF8 ChIP-seq (cDC and pDC), and RNA-seq (MPP, CDP, cDC1, cDC2 and pDC) data generated in this study have been deposited in Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE198651.ATAC-seq data of cDC1 and pDC (GSE118221) are published (Lin et al., 2015), and the ATAC-seq data of MPP, CDP and cDC2 of the same study are described here (GSE198651). ChIP-seq of CTCF in DC (GSE36099) (Garber et al., 2012), H3K27ac (GSE73143) (Allhoff et al., 2016), H3K4me1 and PU.1 (GSE57563) (Allhoff et al., 2014), H3K4me3 and H3K9me3 (GSE64767) (Lin et al., 2015) in MPP, CDP, cDC and pDC were re-analyzed in this study. scRNA-seq data were reanalyzed from GSE114313 (Rodrigues et al., 2018). Bulk RNA-seq data of splenic pDC, cDC1 and cDC2 were reanalyzed from GSE188992 (Pang et al., 2022).The sequence of the pDC specific lncRNA (lncIrf8 identified by RACE PCR) and the cDC1 specific lncRNA (Tcons_00190258 identified by RNA-seq) has been submitted to GenBank. The GenBank accession numbers for lncIrf8 and Tcons_00190258 are ON134061 and ON134062, respectively.
-
NCBI Gene Expression OmnibusID GSE198651. A lncRNA identifies Irf8 enhancer element in negative feedback control of dendritic cell differentiation.
-
NCBI NucleotideID ON134061. Mus musculus lncIRF8 lncRNA, partial sequence.
-
NCBI NucleotideID ON134062. Mus musculus TCONS_00190258 lncRNA, partial sequence.
-
NCBI Gene Expression OmnibusID GSE118221. Identification of Transcription Factor Binding Sites using ATAC-seq.
-
NCBI Gene Expression OmnibusID GSE36099. A high throughput in vivo protein-DNA mapping approach reveals principles of dynamic gene regulation in mammals (ChIP-Seq).
-
NCBI Gene Expression OmnibusID GSE73143. Differential Peak Calling of ChIP-Seq Signals with Replicates with THOR.
-
NCBI Gene Expression OmnibusID GSE57563. Detecting differential peaks in ChIP-seq signals with ODIN.
-
NCBI Gene Expression OmnibusID GSE64767. Epigenetic Program and Transcription Factor Circuitry of Dendritic Cell Development.
-
NCBI Gene Expression OmnibusID GSE114313. A distinct lineage of origin reveals heterogeneity of plasmacytoid dendritic cells II (scRNAseq).
-
NCBI Gene Expression OmnibusID GSE188992. Bulk RNAseq of Mouse Splenic Dendritic Cell Subsets.
References
-
Detecting differential peaks in ChIP-seq signals with ODINBioinformatics 30:3467–3475.https://doi.org/10.1093/bioinformatics/btu722
-
Differential peak calling of ChIP-seq signals with replicates with THORNucleic Acids Research 44:e153.https://doi.org/10.1093/nar/gkw680
-
Genetic models of human and mouse dendritic cell development and functionNature Reviews. Immunology 21:101–115.https://doi.org/10.1038/s41577-020-00413-x
-
Transition from cMyc to L-Myc during dendritic cell development coordinated by rising levels of IRF8The Journal of Experimental Medicine 219:e20211483.https://doi.org/10.1084/jem.20211483
-
Dimensionality reduction for visualizing single-cell data using UMAPNature Biotechnology 37:38–44.https://doi.org/10.1038/nbt.4314
-
Transcriptional programming of the dendritic cell networkNature Reviews. Immunology 12:101–113.https://doi.org/10.1038/nri3149
-
Dendritic cells revisitedAnnual Review of Immunology 39:131–166.https://doi.org/10.1146/annurev-immunol-061020-053707
-
Epigenetic aspects of DC development and differentiationMolecular Immunology 128:116–124.https://doi.org/10.1016/j.molimm.2020.10.011
-
STAR: ultrafast universal RNA-seq alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Cryptic activation of an Irf8 enhancer governs cDC1 fate specificationNature Immunology 20:1161–1173.https://doi.org/10.1038/s41590-019-0450-x
-
I. A bioactive designer cytokine for human hematopoietic progenitor cell expansionNature Biotechnology 15:142–145.https://doi.org/10.1038/nbt0297-142
-
JASPAR 2020: update of the open-access database of transcription factor binding profilesNucleic Acids Research 48:D87–D92.https://doi.org/10.1093/nar/gkz1001
-
GENCODE reference annotation for the human and mouse genomesNucleic Acids Research 47:D766–D773.https://doi.org/10.1093/nar/gky955
-
Expanding dendritic cell nomenclature in the single-cell eraNature Reviews. Immunology 22:67–68.https://doi.org/10.1038/s41577-022-00675-7
-
IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptorsNature Reviews. Immunology 6:644–658.https://doi.org/10.1038/nri1900
-
Constitutive activation of SHP2 in mice cooperates with ICSBP deficiency to accelerate progression to acute myeloid leukemiaThe Journal of Clinical Investigation 118:853–867.https://doi.org/10.1172/JCI33742
-
Retrovirus-polymer complexes: study of the factors affecting the dose response of transductionBiotechnology Progress 23:480–487.https://doi.org/10.1021/bp060336y
-
Fast gapped-read alignment with bowtie 2Nature Methods 9:357–359.https://doi.org/10.1038/nmeth.1923
-
Transcription factor ETV6 regulates functional differentiation of cross-presenting classical dendritic cellsThe Journal of Experimental Medicine 215:2265–2278.https://doi.org/10.1084/jem.20172323
-
Epigenetic program and transcription factor circuitry of dendritic cell developmentNucleic Acids Research 43:9680–9693.https://doi.org/10.1093/nar/gkv1056
-
Guidelines for mouse and human DC generationEuropean Journal of Immunology.https://doi.org/10.1002/eji.202249816
-
Potential autoregulation of transcription factor PU.1 by an upstream regulatory elementMolecular and Cellular Biology 25:2832–2845.https://doi.org/10.1128/MCB.25.7.2832-2845.2005
-
The relationship between genome structure and functionNature Reviews. Genetics 22:154–168.https://doi.org/10.1038/s41576-020-00303-x
-
Enhancer RNAs are an important regulatory layer of the epigenomeNature Structural & Molecular Biology 27:521–528.https://doi.org/10.1038/s41594-020-0446-0
-
ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8α+ dendritic cellsThe Journal of Experimental Medicine 196:1415–1425.https://doi.org/10.1084/jem.20021263
-
3’ end cDNA amplification using classic raceNature Protocols 1:2742–2745.https://doi.org/10.1038/nprot.2006.481
-
Gene regulation by long non-coding RNAs and its biological functionsNature Reviews. Molecular Cell Biology 22:96–118.https://doi.org/10.1038/s41580-020-00315-9
-
The IRF family transcription factors in immunity and oncogenesisAnnual Review of Immunology 26:535–584.https://doi.org/10.1146/annurev.immunol.26.021607.090400
-
Regulation of myelopoiesis by the transcription factor IRF8International Journal of Hematology 101:342–351.https://doi.org/10.1007/s12185-015-1761-9
-
Advances in understanding epigenetic impacts on dendritic cell regulation and functionClinical and Translational Discovery 2:e53.https://doi.org/10.1002/ctd2.53
-
Specific protein binding to the simian virus 40 enhancer in vitroMolecular and Cellular Biology 6:2098–2105.https://doi.org/10.1128/mcb.6.6.2098-2105.1986
-
CRISPR/Cas9 editing in conditionally immortalized HoxB8 cells for studying gene regulation in mouse dendritic cellsEuropean Journal of Immunology 52:1859–1862.https://doi.org/10.1002/eji.202149482
-
Interferon regulatory factor 8 (IRF8) interacts with the B cell lymphoma 6 (BCL6) corepressor BCORThe Journal of Biological Chemistry 289:34250–34257.https://doi.org/10.1074/jbc.M114.571182
Article and author information
Author details
Funding
German Research Foundation
- Martin Zenke
German Ministry of Science and Technology (Fibromap)
- Ivan G Costa
Interdisciplinary Center for Clinical Research Aachen
- Ivan G Costa
- Martin Zenke
China Scholarship Council (202008080170)
- Huaming Xu
CAPES-Alexander von Humboldt Foundation (99999.001703/2014-05)
- Marcelo AS de Toledo
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We acknowledge the support of the Interdisciplinary Center for Clinical Research Aachen (IZKF Aachen) FACS Core Facility and Genomics Facility. We thank Magdalena Karpinska and Marieke Oudelaar for help with the Capture-C experiments and analysis, Susanne Schmitz and Paul Wanek for assistance, Stefan Rose-John for hyper-IL-6, Lisa Weixler and Carmen Schalla for help with enzyme purification, and Thomas Hieronymus for valuable discussion and suggestions. Part of this work was supported in part by funds from German Research Foundation (DFG) to MZ and from the Germany Ministry of Science and Technology (BMBF - Fibromap) and the Interdisciplinary Center for Clinical Research Aachen (IZKF Aachen) from the RWTH Medical Faculty to IC. HX was supported by a fellowship of China Scholarship Council (CSC) (Grant number 202008080170). MAST was funded by CAPES-Alexander von Humboldt postdoctoral fellowship (99999.001703/2014–05) and donation by U Lehmann.
Ethics
All the animal experiments were approved by the local authorities of the German Federal State North Rhine-Westphalia, Germany according to the German animal protection law (reference number 81-02.04.2018.A228).
Copyright
© 2023, Xu et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,158
- views
-
- 154
- downloads
-
- 7
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A lncRNA identifies Irf8 enhancer element in negative feedback control of dendritic cell differentiation
eLife 12:e83342.
https://doi.org/10.7554/eLife.83342
Further reading
-
- Cell Biology
- Immunology and Inflammation
Macrophages are crucial in the body’s inflammatory response, with tightly regulated functions for optimal immune system performance. Our study reveals that the RAS–p110α signalling pathway, known for its involvement in various biological processes and tumourigenesis, regulates two vital aspects of the inflammatory response in macrophages: the initial monocyte movement and later-stage lysosomal function. Disrupting this pathway, either in a mouse model or through drug intervention, hampers the inflammatory response, leading to delayed resolution and the development of more severe acute inflammatory reactions in live models. This discovery uncovers a previously unknown role of the p110α isoform in immune regulation within macrophages, offering insight into the complex mechanisms governing their function during inflammation and opening new avenues for modulating inflammatory responses.
-
- Immunology and Inflammation
The incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) has been increasing worldwide. Since gut-derived bacterial lipopolysaccharides (LPS) can travel via the portal vein to the liver and play an important role in producing hepatic pathology, it seemed possible that (1) LPS stimulates hepatic cells to accumulate lipid, and (2) inactivating LPS can be preventive. Acyloxyacyl hydrolase (AOAH), the eukaryotic lipase that inactivates LPS and oxidized phospholipids, is produced in the intestine, liver, and other organs. We fed mice either normal chow or a high-fat diet for 28 weeks and found that Aoah-/- mice accumulated more hepatic lipid than did Aoah+/+ mice. In young mice, before increased hepatic fat accumulation was observed, Aoah-/- mouse livers increased their abundance of sterol regulatory element-binding protein 1, and the expression of its target genes that promote fatty acid synthesis. Aoah-/- mice also increased hepatic expression of Cd36 and Fabp3, which mediate fatty acid uptake, and decreased expression of fatty acid-oxidation-related genes Acot2 and Ppara. Our results provide evidence that increasing AOAH abundance in the gut, bloodstream, and/or liver may be an effective strategy for preventing or treating MASLD.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}