Discovery of lipid binding sites in a ligand-gated ion channel by integrating simulations and cryo-EM
- Science for Life Laboratory & Swedish e-Science Research Center, Department of Applied Physics, KTH Royal Institute of Technology, Sweden
- Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Sweden
Figures
Figure 1 with 2 supplements
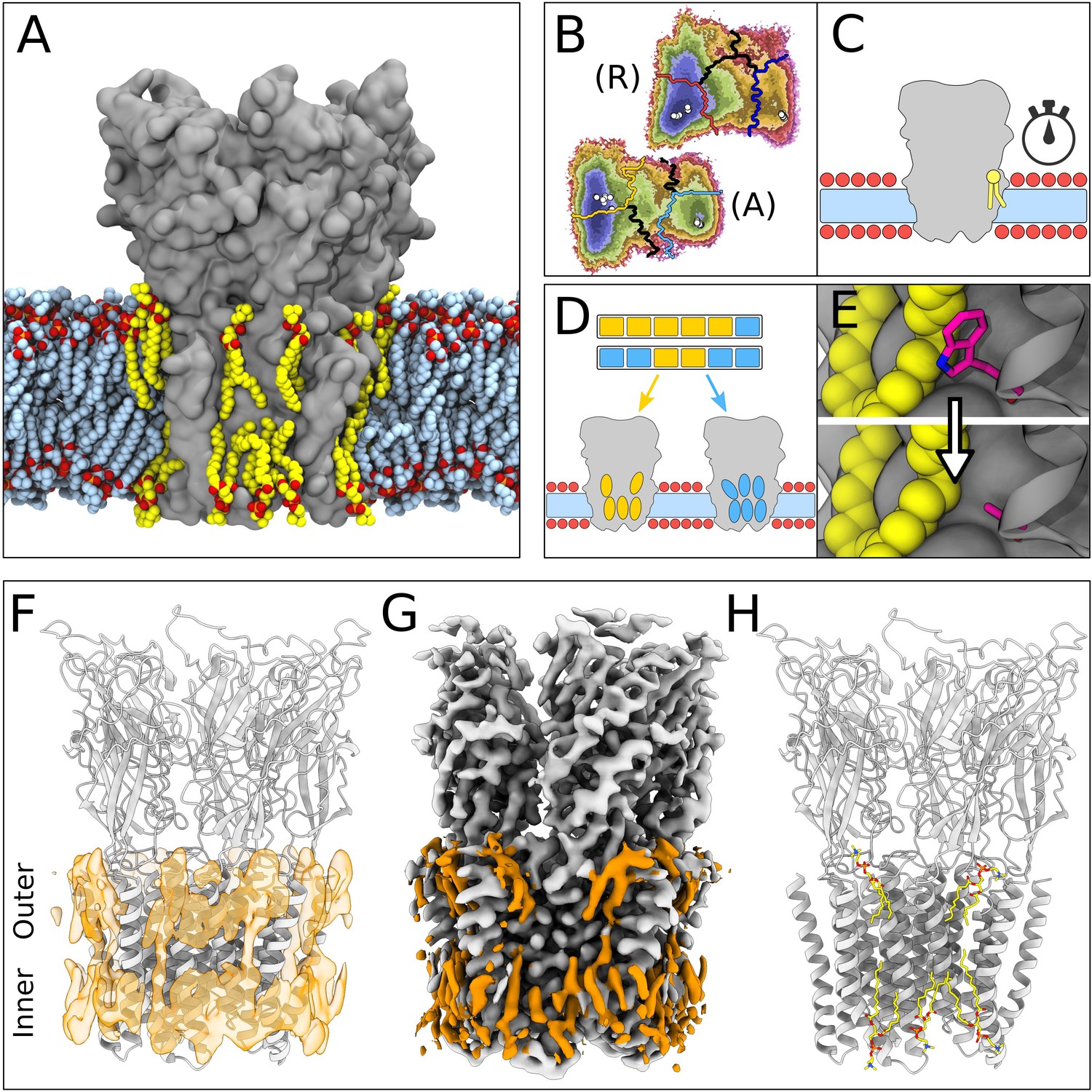
Lipid-protein interactions mapped by Markov state modeling (MSM) and cryo-electron microscopy (cryo-EM).
(A) Overview of the molecular dynamics (MD) simulation system with GLIC (gray surface) embedded in a lipid bilayer (light blue carbon atoms) with interacting lipids highlighted (yellow carbon atoms). Lipid oxygens are shown in red; phosphorus, orange; nitrogen, dark blue. Water molecules and ions have been omitted for clarity. (B) Markov state models were used to cluster simulations conducted under resting (R) or activating (A) conditions into five states, including closed (left of the light or dark orange lines) and open (right of the light or dark blue lines). Black lines mark edges of other state clusters derived from MSM eigenvectors. Experimental structures are highlighted as white circles. (C) To characterize the stability of lipid interactions, duration times were measured in individual, unclustered, trajectories. (D) To identify lipid binding sites, lipid-occupied densities were obtained from state-clustered frames for closed (orange) and open (blue) states. (E) Further simulations were performed to test the role of residues involved in extensive lipid interactions through single residue mutations around lipid binding sites. (F) View of a manually built cryo-EM model of closed GLIC (gray) with computationally derived lipid occupancies in orange (semi-transparent). (G) Cryo-EM reconstruction of closed GLIC (gray), with partly defined non-protein densities (orange). (H) Equivalent view of a manually built cryo-EM model as in F, with newly built lipids shown as sticks (yellow, heteroatom; phosphorus, orange; oxygen, red; nitrogen, blue).
Figure 1—figure supplement 1
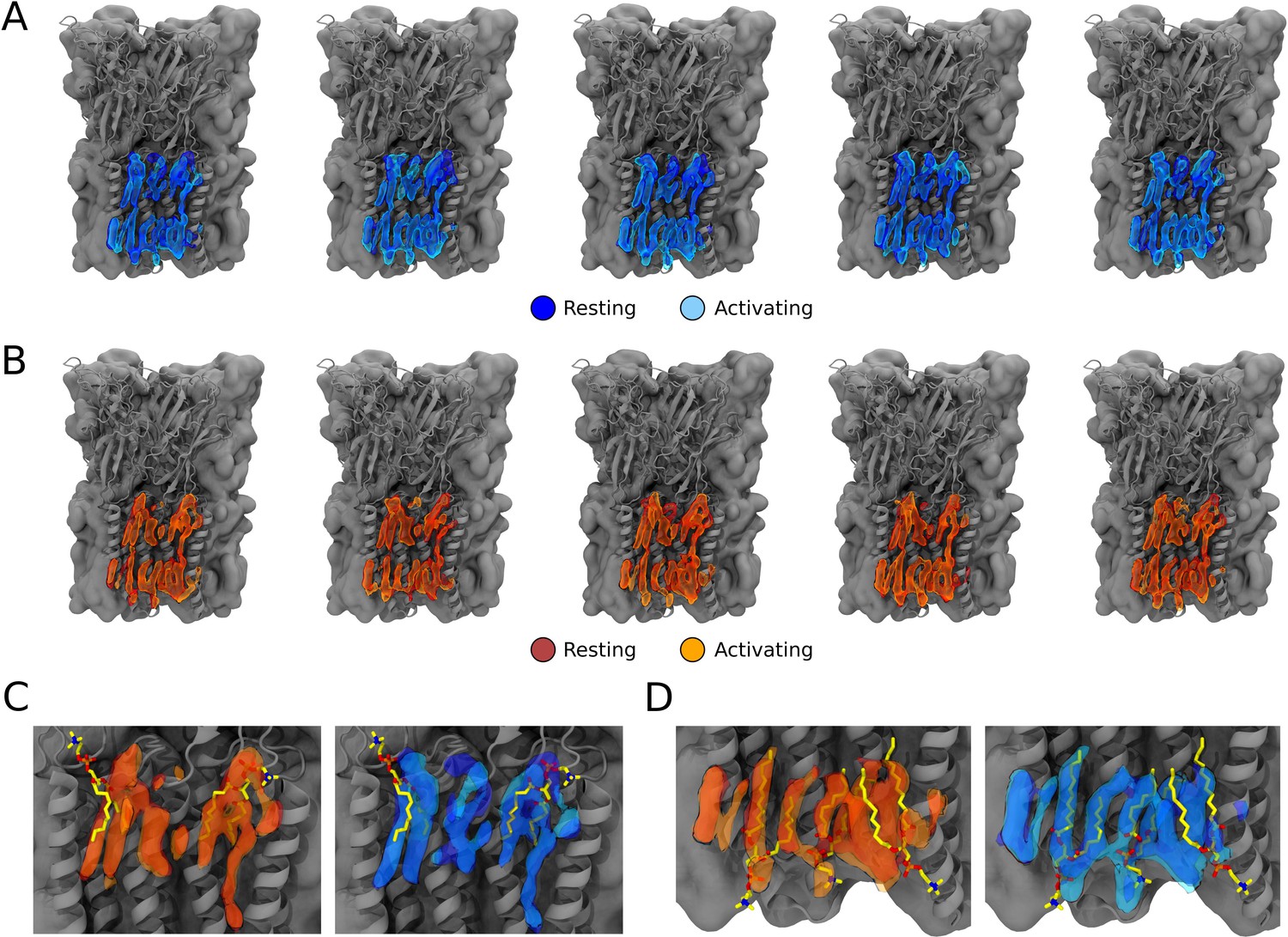
Occupational lipid densities from simulations for all five subunits and overalys with built cryogenic electron microscopy (cryo-EM) lipids.
(A) Lipid occupational densities obtained from simulation frames of open states at resting (dark blue) and activating (light blue) conditions for each of the five ion channel subunits. (B) Lipid occupational densities obtained from simulation frames of closed states at resting (red) and activating (orange) conditions for each of the five ion channel subunits. (C) Overlay of computational densities of the outer leaflet with built lipids from the cryo-EM structure (yellow sticks). (D) Overlay of computational densities in the inner leaflet with built lipids from the cryo-EM structure (yellow sticks).
Figure 1—figure supplement 2
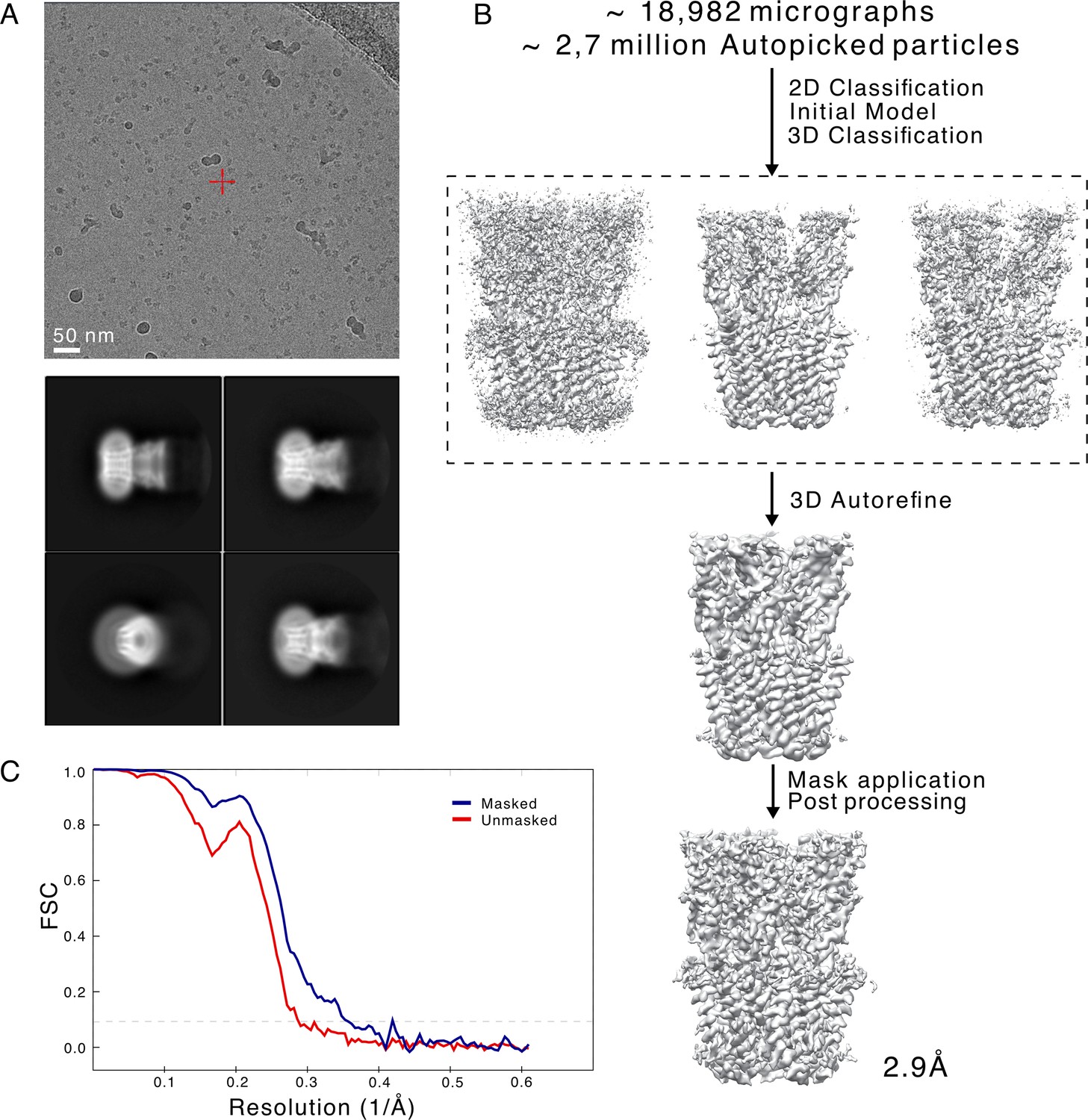
Cryogenic electron microscopy (cryo-EM) data and processing pipeline.
(A) Representative micrograph from a dataset collected on a Titan Krios, showing detergent-solubilized GLIC particles (top). Representative 2D class averages at 0.82 Å/px in a 256×256 pixel box and a 180 Å mask (bottom). (B) Overview of the cryo-EM processing pipeline for merged GLIC data. (C) FSC curves for unmasked (red) and masked (blue) map.
Figure 2 with 1 supplement
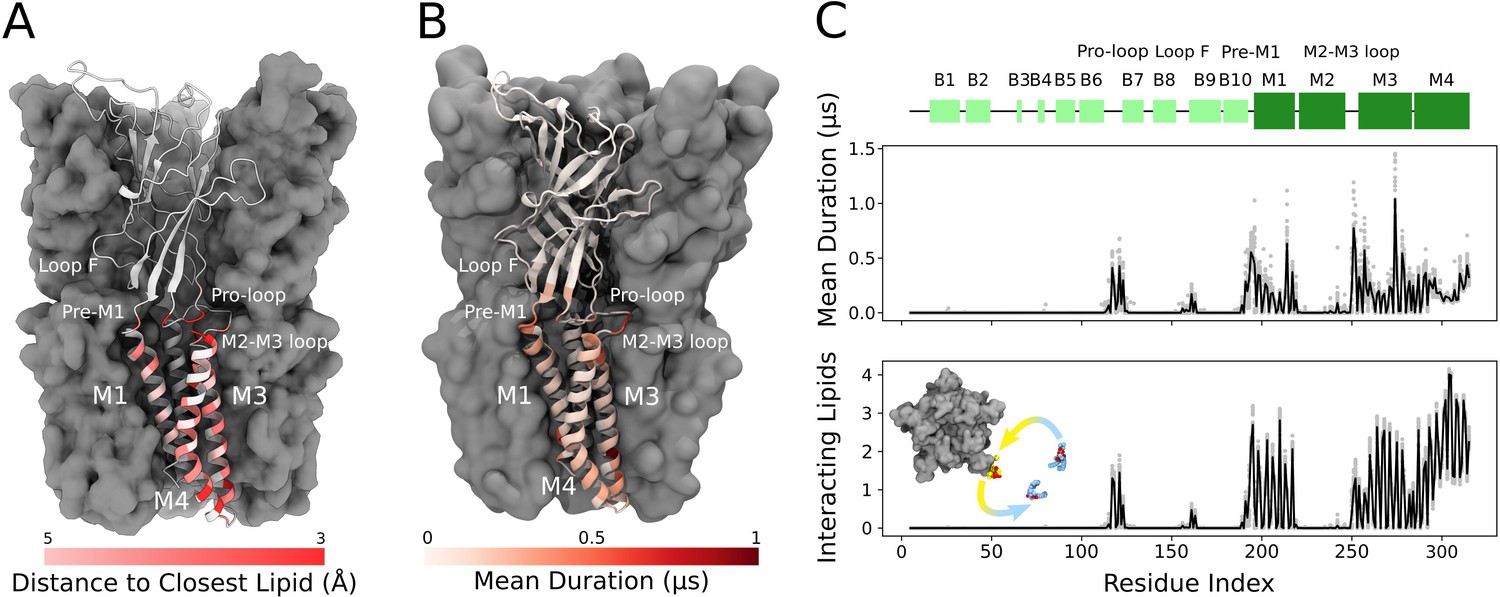
Cryogenic electron microscopy (cryo-EM) and simulations reveal lipid interaction sites.
(A) Cryo-EM reconstruction of closed GLIC with distance to the interacting lipids colored according to the bar below. (B) Mean duration time of lipid-residue interactions, scaled (white-red) according to the color bar below, projected onto the closed-state GLIC structure (PDB ID: 4NPQ). For both panels, a single subunit is displayed in cartoon. (C) Secondary structure schematic for GLIC (top), mean duration time of each lipid-residue interaction (middle), and number of lipids interacting with each residue during simulations (bottom). Light gray points represent numbers from individual trajectories and black lines ensemble averages (500 trajectories).
Figure 2—figure supplement 1
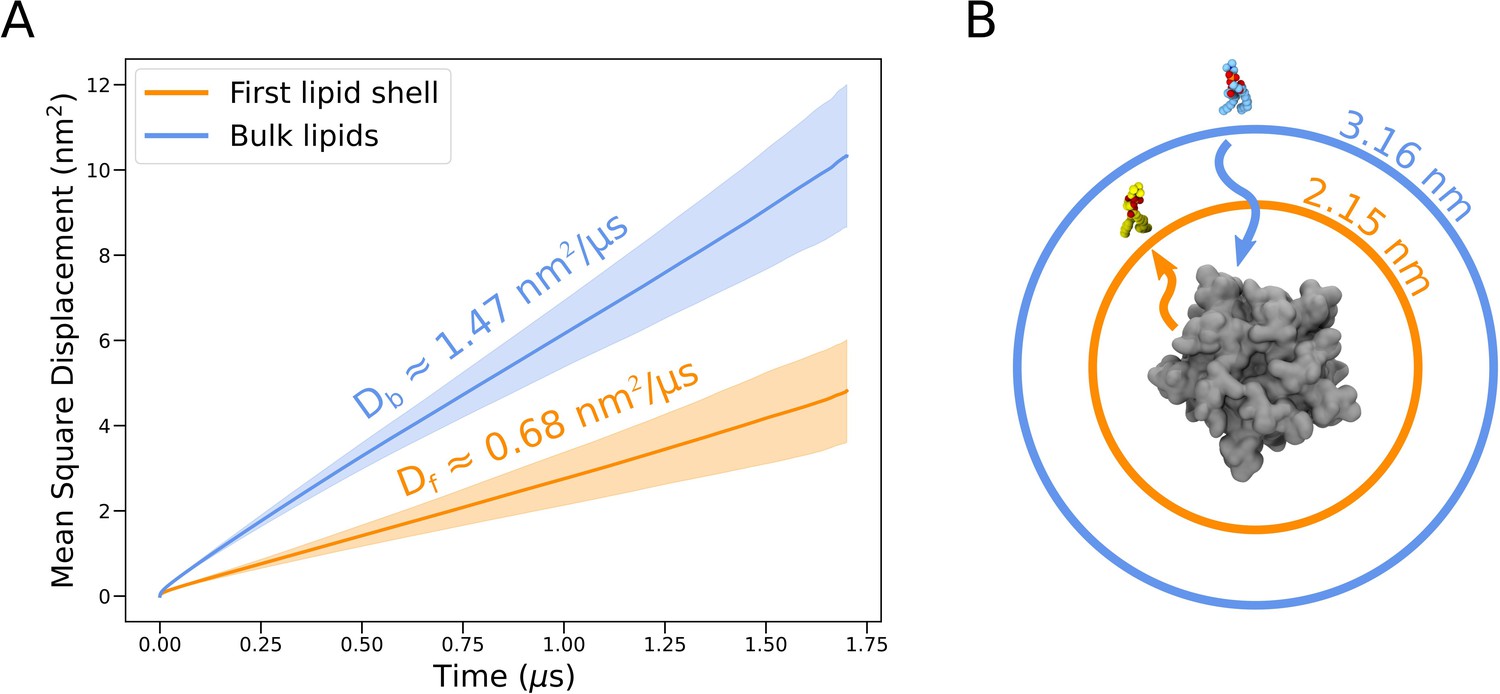
Lateral diffusion of lipids initially with and without contact to the protein surface.
(A) Mean square displacements for lipids initially in the first lipid shell (<4 Å from the protein surface, orange) and bulk lipids (>4 Å from the protein surface, blue), averaged over all trajectories. Lighter bands represent standard deviations. Self-diffusion coefficients for lipids of the first shell, Df , and bulk lipids, Db, are shown above each mean square displacement line. (B) Illustration of average distances traveled by lipids initially in the first lipid shell (orange) and bulk (blue) by the end of individual simulations (1.7 µs). Distances apply in all directions of the membrane plane but are here shown as radial displacements with respect to the surface of the protein.
Figure 3 with 1 supplement
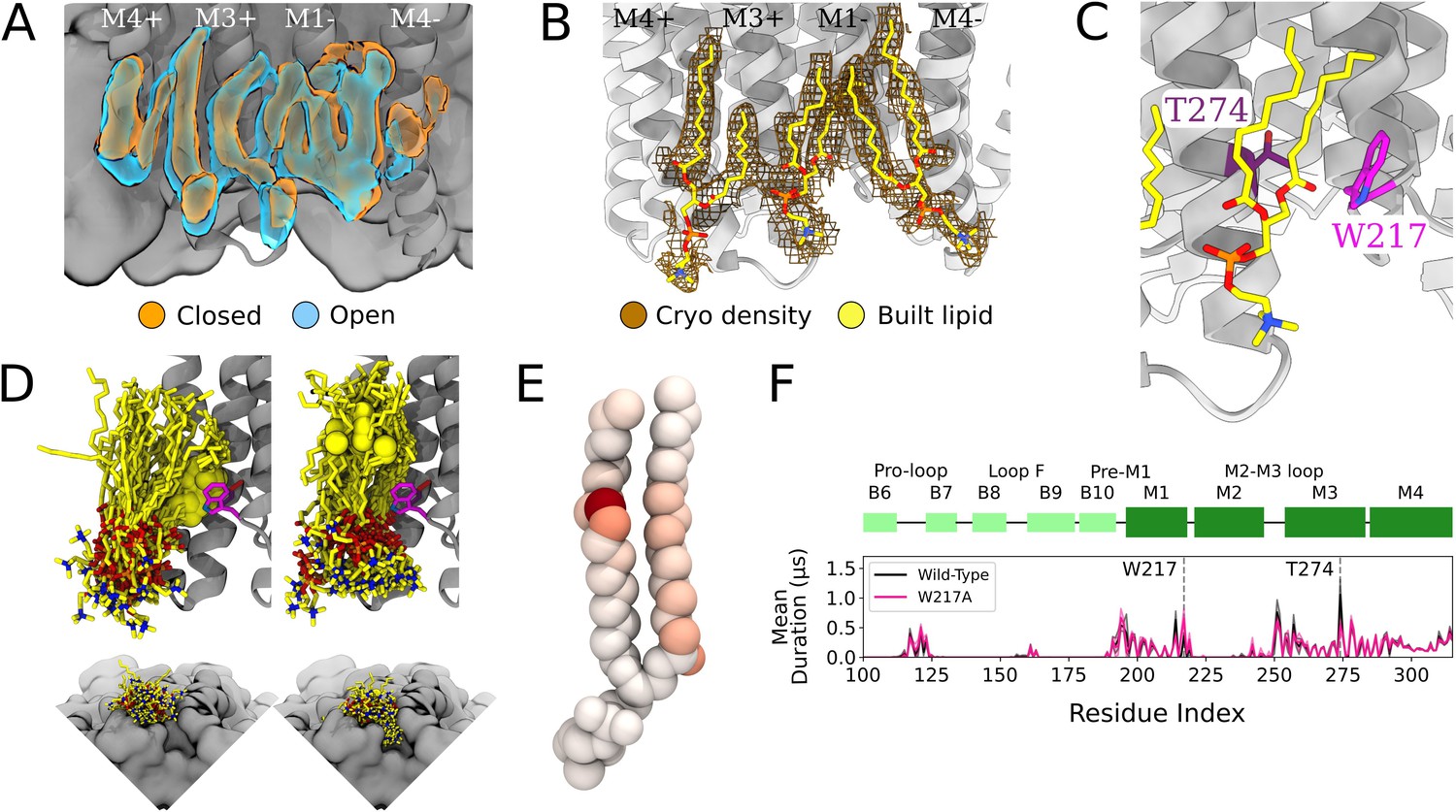
Protein and lipid determinants of state-independent inner-leaflet binding.
(A) Computationally derived densities from the open (blue) and closed (orange) states generally agree. (B) Modeled lower leaflet lipids (yellow, stick, heteroatom coloring) from cryogenic electron microscopy (cryo-EM) densities (brown mesh). (C) Zoom-in view of the lower leaflet buried lipid interaction site at the M1-M3 subunit interface. Lipids particularly interact with residues T274 (purple) and W217 (magenta). (D) The 100 simulation frames that best fit the computational densities clustered into two distinct binding poses at the T274 binding site. When the tail with a double bond (yellow sphere) is situated in the pocket lipid heads are directed out from the channel (left panel), while a larger variety of poses are sampled when the lipid tail without a double bond occupies the site, allowing the head to enter into a crevice on the bottom of the channel, approaching the pore (right panel). The M4 helix is not shown for clarity in the top panels. (E) Number of contacts made by specific 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid atoms with residue T274 in simulations at both resting and activating conditions (colors span 0 contacts, white, to 137,015 contacts, dark red). For the tail with an unsaturation, interactions are concentrated around the double-bond region while the saturated tail displays interactions interspersed along the tail. (F) Mutation of residue W217, lining this pocket, reveals shortened interactions at the T274 binding site (magenta).
Figure 3—figure supplement 1
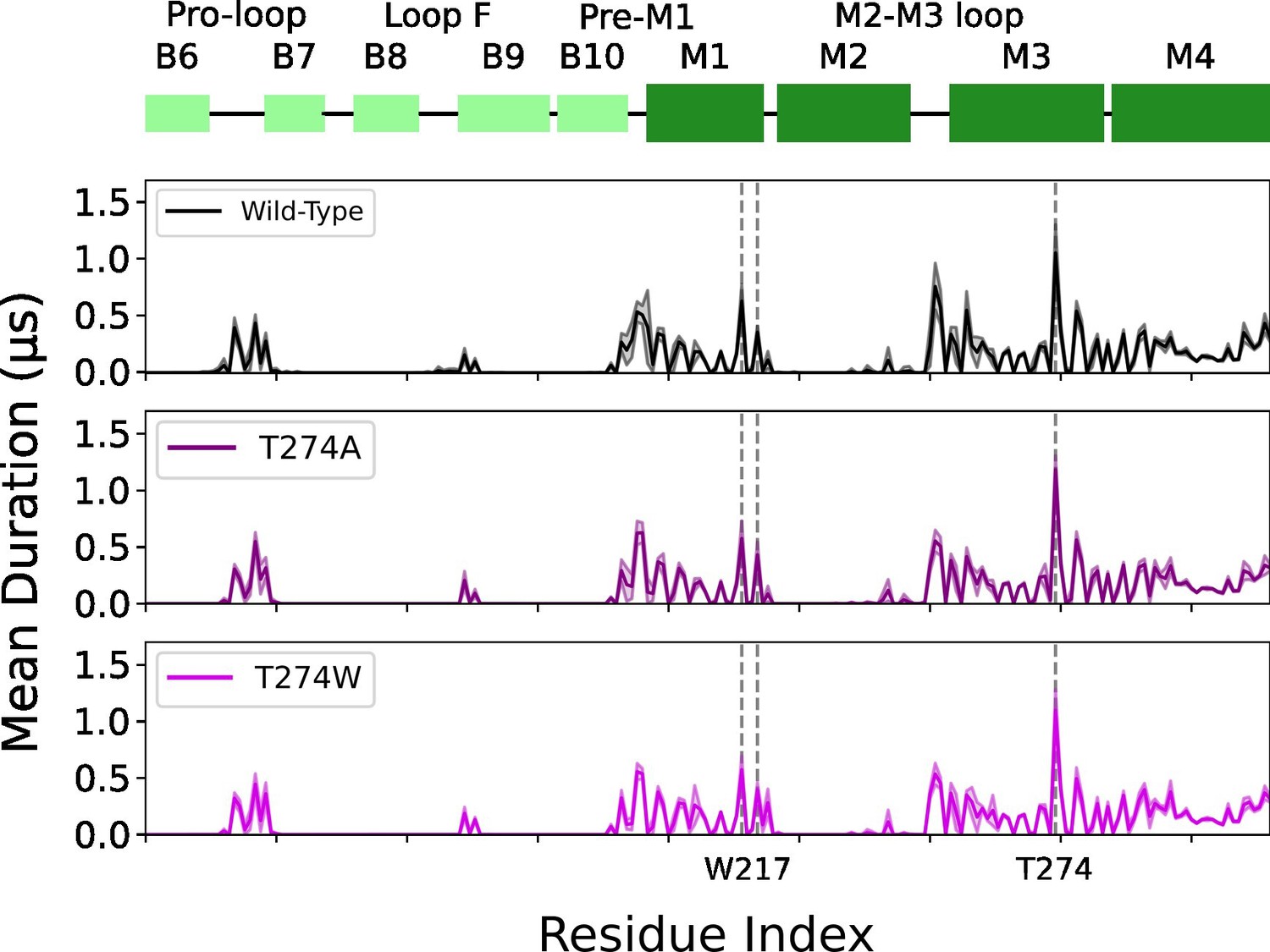
W217 and T274 mutants mean duration times compared to wild type.
Mean duration time (µs) of lipid interaction time for GLIC wild type (black, top row), T274A mutant (purple, middle row) and T274W mutant (light purple, bottom row).
Figure 4 with 4 supplements
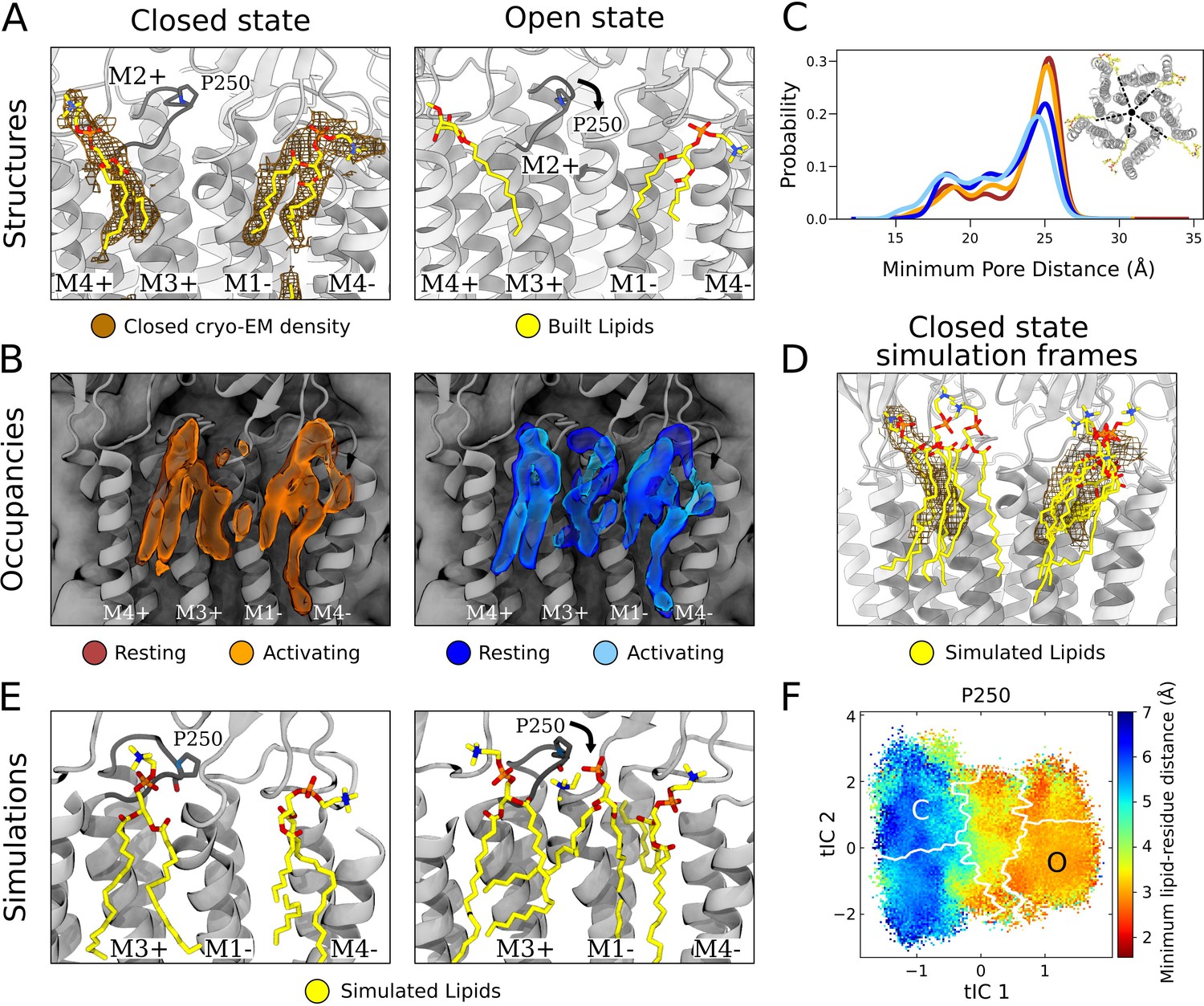
State-dependent protein-lipid interactions in the upper leaflet.
(A) Zoom view of the outer transmembrane domain of the closed GLIC cryogenic electron microscopy (cryo-EM) structure (left, gray), showing non-protein cryo-EM densities (brown mesh) overlaid with built lipids (yellow). For comparison, the same region is shown of an open-state X-ray structure (PDB ID: 6HZW) (Hu et al., 2018) with built lipid and detergent in equivalent positions (right). Conformational changes of the M2-M3 loop, including residue P250, in channel opening are highlighted. Lipids are colored by heteroatom (phosphorus, orange; oxygen, red; nitrogen, blue). (B) Same region as in (A) showing densities occupied by lipids (>40%) in simulation frames clustered as closed (left, orange) or open (right, blue) states (top). Open-state differences include an enhanced density on the right-hand side of the intersubunit cleft, and a more deeply penetrating density in the intrasubunit cleft (Figure 4—figure supplement 1). Overlaid densities for each state represent simulations conducted under resting (dark shades) or activating (light shades) conditions, which were largely superimposable within each state. (C) Radial distribution of the lipid atoms closest to the upper pore, showing closer association of lipids in the open (light blue, dark blue) versus closed (orange, red) states. (D) Same region and cryo-EM densities (mesh) as in (A) overlaid with lipids from a few simulation frames where the lipids had the highest correlation with the closed-state computational occupancies in (B). (E) Snapshots from simulation frames that had the highest correlation with the occupancies (activating conditions) in (B). The open states are characterized by an additional lipid at the intersubunit site, interacting with the oxygen of P250. (F) The minimum distance between P250 and the closest lipid projected onto the free energy landscapes obtained from Markov state modeling. P250-lipid interactions are possible in the open state, but not in the closed state.
Figure 4—figure supplement 1
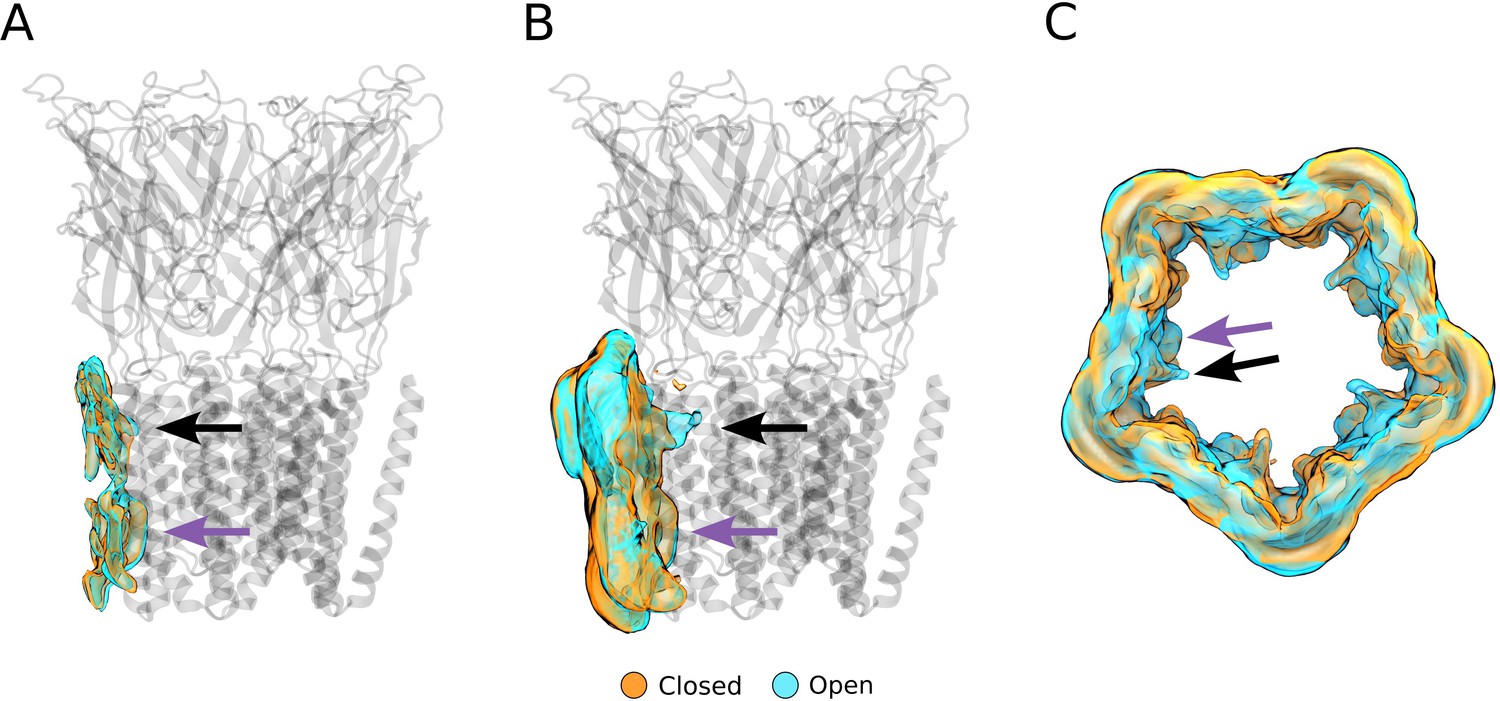
Alternative views of the computationally derived lipid densities at activating conditions.
(A) Side view of a single-subunit lipid density with >40% occupancy. (B) Side view of a single-subunit lipid density with >10% occupancy. (C) Top view of the full lipid density with >10% occupancy. Black arrows indicate the outer-leaflet complementary intrasubunit lipid site, while purple arrows indicate the inner-leaflet intersubunit site.
Figure 4—figure supplement 2
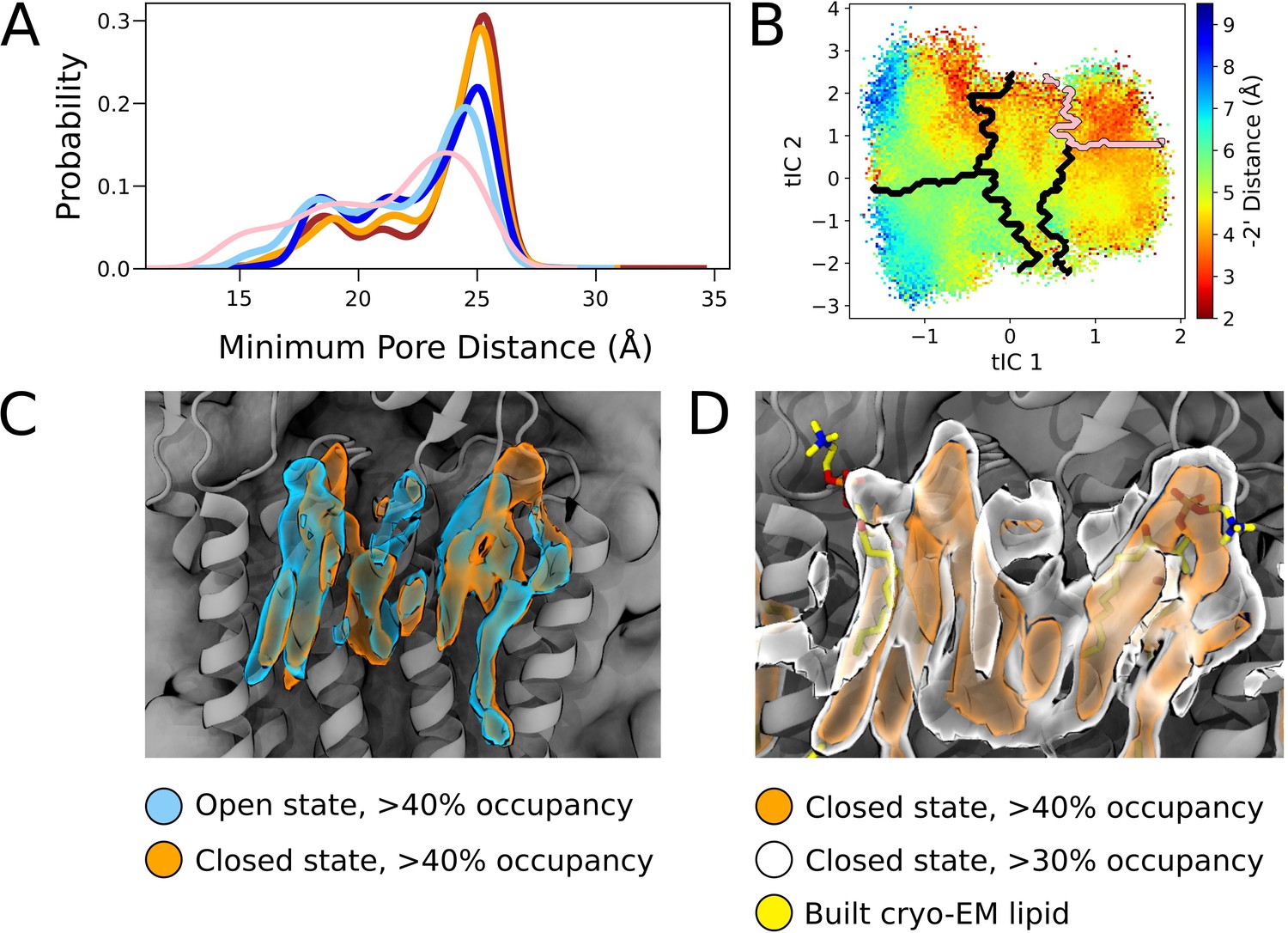
Comparisons of state-dependent lipid interactions in the outer transmembrane domain.
(A) Radial distributions of nearest lipid atom from the upper pore, with a presumed pre-desensitized state displaying the highest probability of lipid tail penetration (pink), followed by open states (dark blue, light blue) and closed states (red, orange). (B) Radial distance to the –2’ gate projected onto the tIC space at activating conditions. The presumed pre-desensitized state is marked in pink. (C) Lipid densities derived from simulations with >40% occupancies, representing open (blue) and closed (orange) states. (D) Lipid densities from closed-state simulation frames with >40% occupancy (orange) and >30% (white). Built cryo-EM lipids are shown in yellow.
Figure 4—figure supplement 3
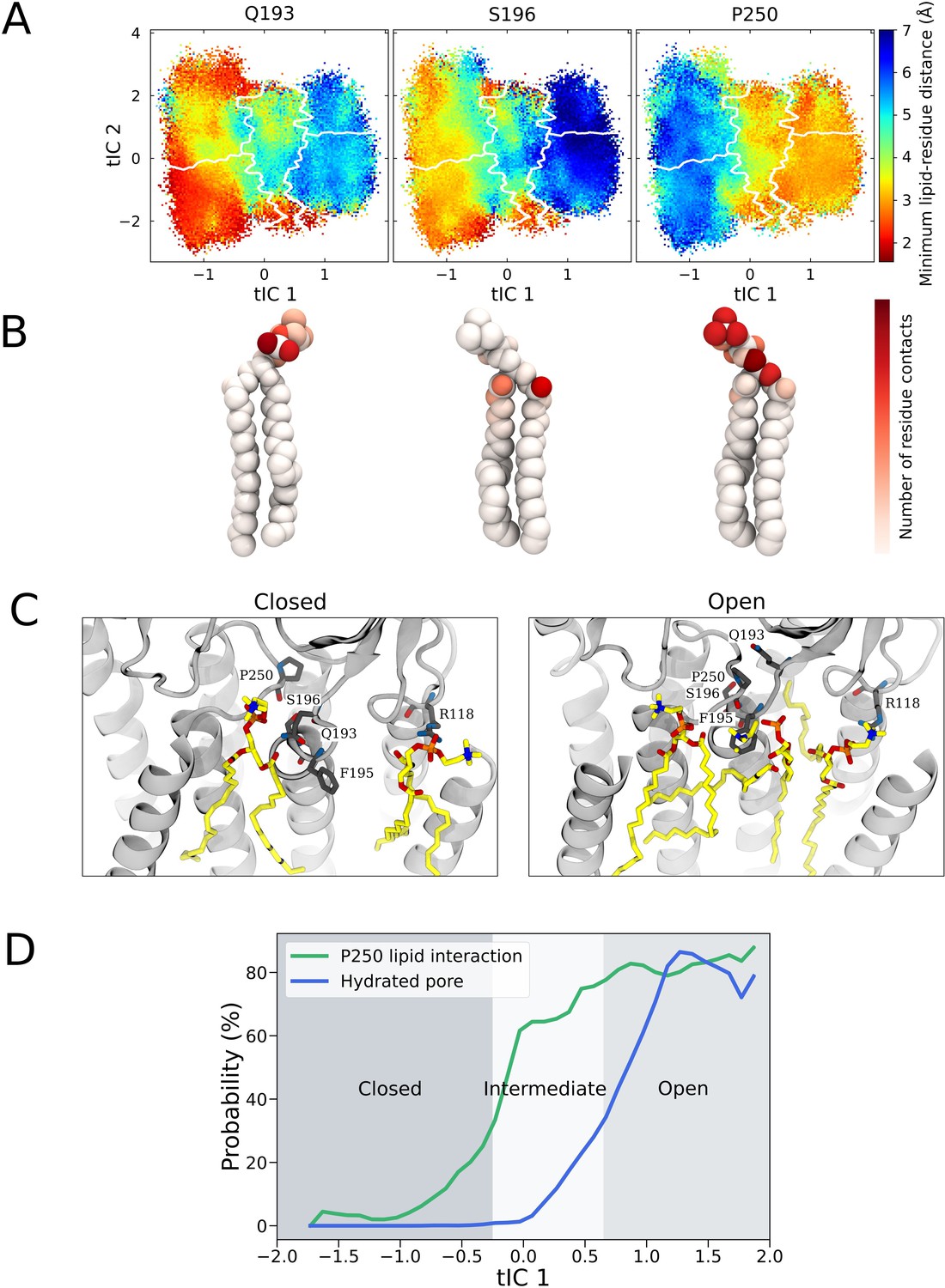
State-dependent lipid interactions and conformational changes in the upper transmembrane domain.
(A) Minimum residue-lipid distance projected onto the activating condition free energy landscape for residues Q193, S196, and P250. (B) The normalized number of residue-lipid atom contacts displayed for each of the residues in (A), with red areas highlighting atoms with close interactions with the residue. (C) Representative GLIC open and closed state simulation frames with the highest correlations between lipid positions and the computational occupancies (Figure 4B). (D) The probabilities of P250-lipid contact formation (<4 Å) and pore hydration (>20 water molecules in the pore) projected onto the main reaction coordinate describing the gating process.
Figure 4—video 1
Morph between open and closed conformations showcasing changes in lipid interactions of the outer leaflet.
A linear interpolation between open and closed ion channel conformations and representative binding poses of lipids in the outer leaflet. P250 is marked by green carbon atoms, S193 by blue carbon atoms, and F195 in gray space-filling representation. Lipids are shown as yellow carbon atoms in a stick representation, and the protein as a gray cartoon representation. Oxygens are red, phosphorus orange, nitrogen blue, and hydrogens white.
Figure 5
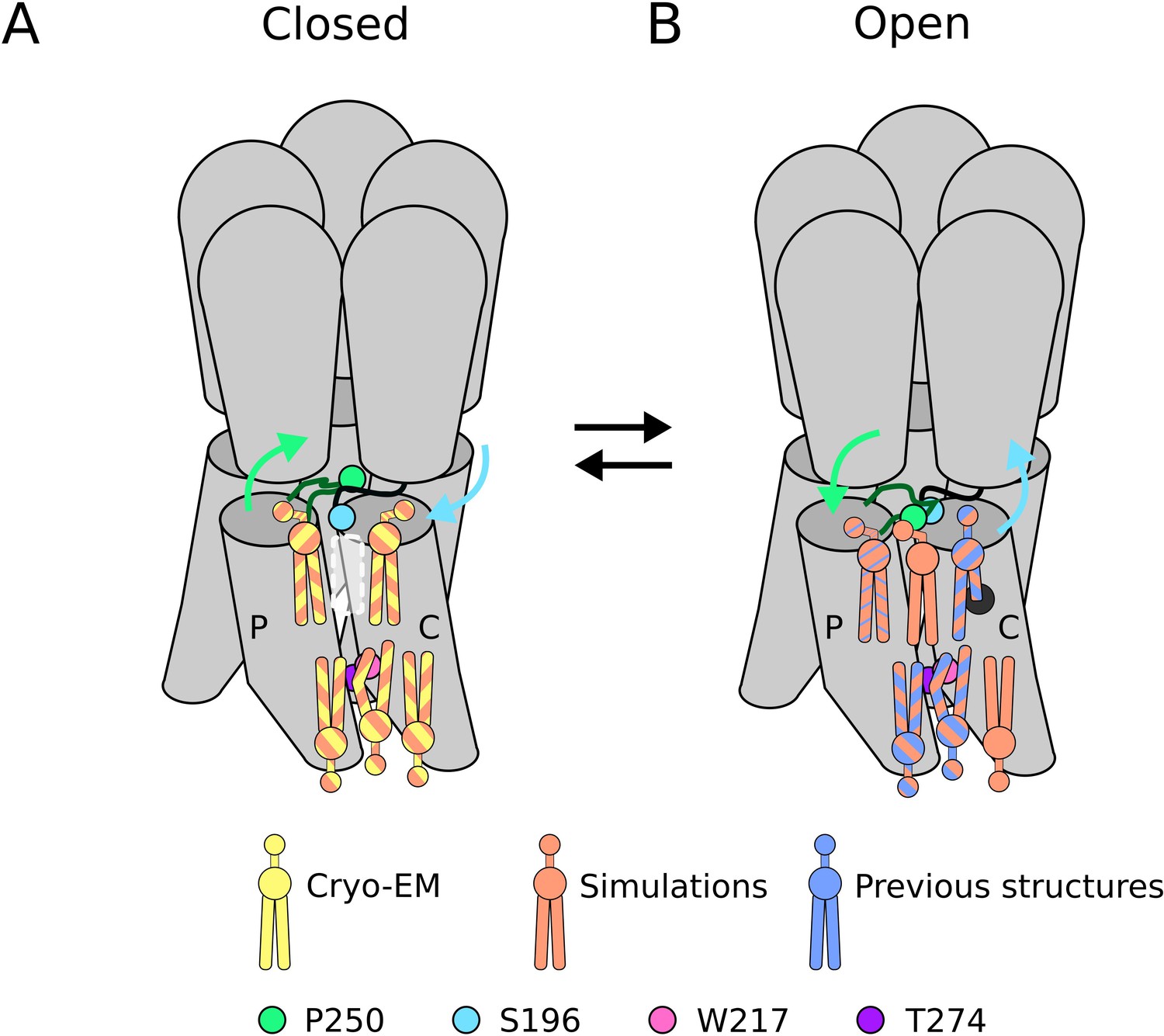
Lipid sites identified by cryogenic electron microscopy (cryo-EM) and simulations.
(A) In the closed state, five lipid interaction sites were independently identified through both cryo-EM and simulations. In the outer leaflet, two intrasubunit sites are situated at the principal (P) and complementary (C) subunits, with the lipid at the principal subunit site interacting with S196 of the pre-M1 loop. Particularly, no lipid site could be found at the subunit interface. In the inner leaflet, a buried lipid pose was found at the subunit interface, determined by interactions with T274 and W217 of the M3 and M1 helices, respectively. (B) In the open state, six lipid interaction sites were identified from simulations, out of which three sites were known from previous structures and one presumably occupied by a detergent molecule (Hu et al., 2018) (thin blue stripes). In the outer leaflet, an additional lipid site could be found at the subunit interface in the open state, interacting with P250 on the M2-M3 loop. Additionally, the site at the principal intrasubunit site is slightly closer to the M4 helix compared to the closed state, and the lipid site at the principal intrasubunit site has an increased likelihood of tail penetration into an allosteric pocket compared to the closed state. Lipid sites in the inner leaflet are at positions equivalent to those in the closed state.
Tables
Table 1
Cryogenic electron microscopy (cryo-EM) data collection and model refinement statistics.
| Data collection | |
|---|---|
| Microscope | FEI Titan Krios |
| Magnification | 165,000 |
| Voltage (kV) | 300 |
| Electron exposure (e-/Å2) | ~40 |
| Defocus range (µm) | –2.6 to –3.8 |
| Pixel size (Å) | 0.82 |
| Symmetry imposed | C5 |
| Number of images | ~18,982 |
| Particles picked | ~2.7 million |
| Particles refined | 16,586 |
| Refinement | |
| Resolution (Å) | 2.9 |
| FSC threshold | 0.143 |
| Map sharpening B-factor | –80 |
| Model composition | |
| Non-hydrogen protein atoms | 12,010 |
| Protein residues | 1555 |
| Ligands | 0 |
| B-factor (Å2) | 144 |
| RMSD | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.584 |
| Validation | |
| Molprobity score | 1.84 |
| Clashscore | 6.82 |
| Poor rotamers (%) | 0 |
| Ramachandran plot | |
| Favored (%) | 92.6 |
| Allowed (%) | 7.4 |
| Outliers (%) | 0 |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Discovery of lipid binding sites in a ligand-gated ion channel by integrating simulations and cryo-EM
eLife 12:RP86016.
https://doi.org/10.7554/eLife.86016.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}