Cancers adapt to their mutational load by buffering protein misfolding stress
- Department of Biology, Stanford University, United States
- Department of Medicine, Division of Oncology, Stanford University School of Medicine, United States
- Department of Genetics, Stanford University School of Medicine, United States
- Stanford Cancer Institute, Stanford University School of Medicine, United States
Figures
Figure 1 with 2 supplements
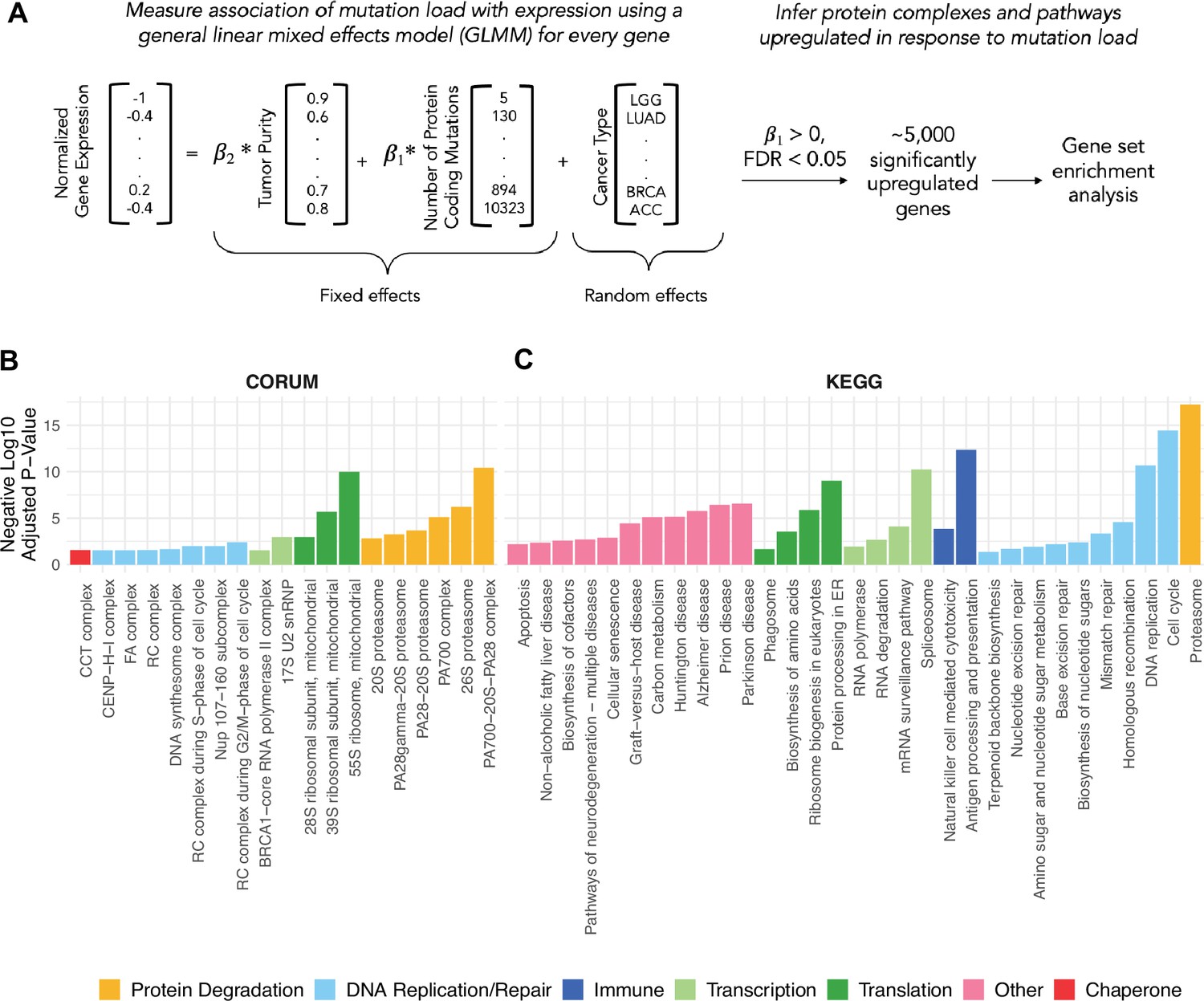
General linear mixed effects model (GLMM) identifies protein complexes and pathways up-regulated in response to mutational load in human tumors.
(A) Overview of the GLMM used to measure the association of mutation load with gene expression while controlling for potential co-variates (purity and cancer type). Genes with a significant, positive regression coefficient and false discovery rate (FDR)<0.05 are used for gene set enrichment analysis. (B, C) Bar plots of protein complexes from the CORUM database (left) and pathways from the KEGG database (right) that are significantly enriched (p<0.05) in response to mutational load. Length of bars denotes negative log10 of adjusted p-value and colors denote broad functional groups enriched in both databases.
Figure 1—figure supplement 1
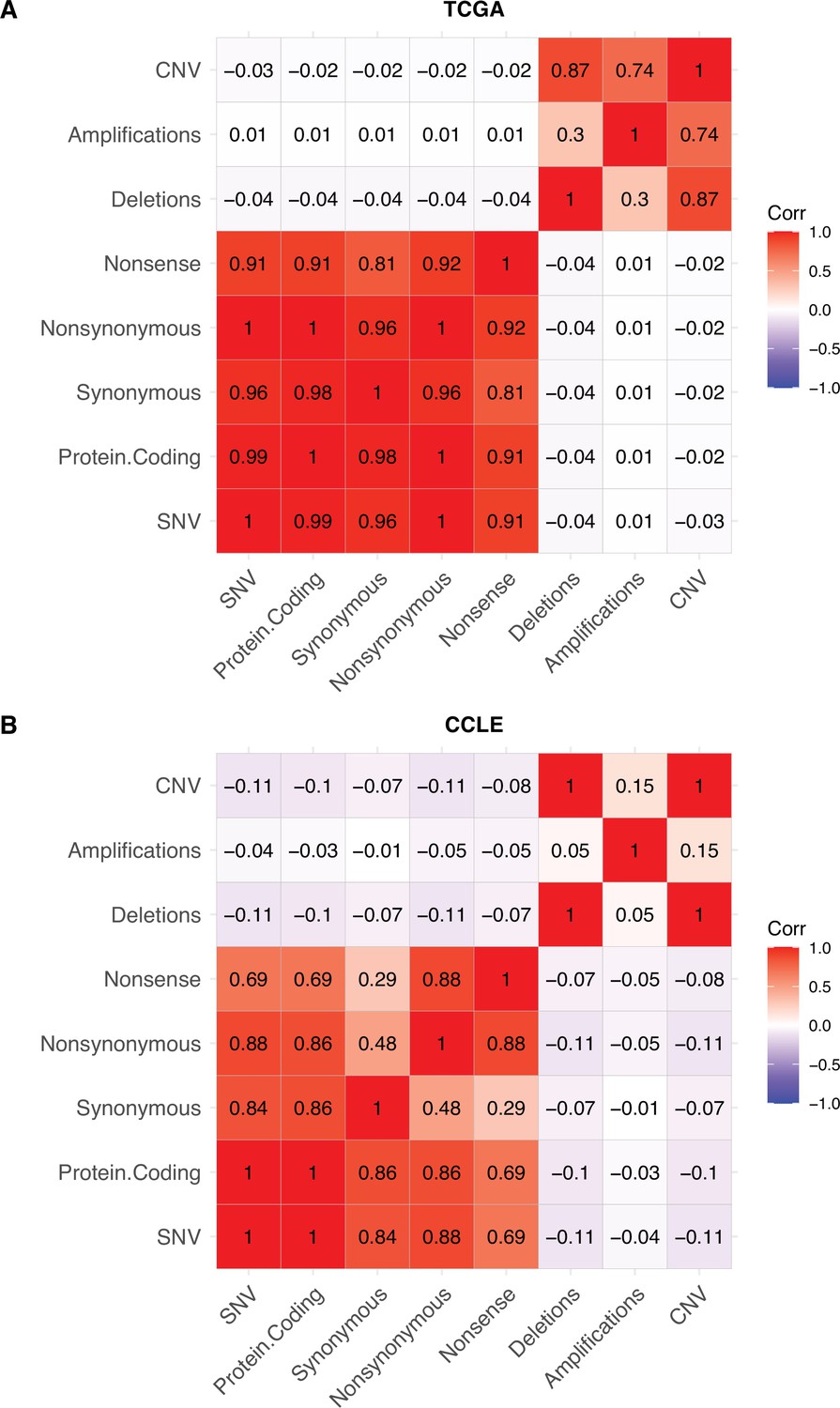
No collinearity of point mutations and copy number alterations in human tumors (TCGA) and cancer cell lines (CCLE).
Heatmap of Pearson’s correlation coefficients between different classes of mutations in (A) CCLE (cancer cell lines) and (B) TCGA (human tumors). Colors denote the magnitude of correlation coefficients and whether the relationship is positive (red), negative (blue), or negligible (white). Copy number alterations (CNAs) are defined as the combined number of amplifications and deletions, while SNVs are the combined number of all point mutations.
Figure 1—figure supplement 2
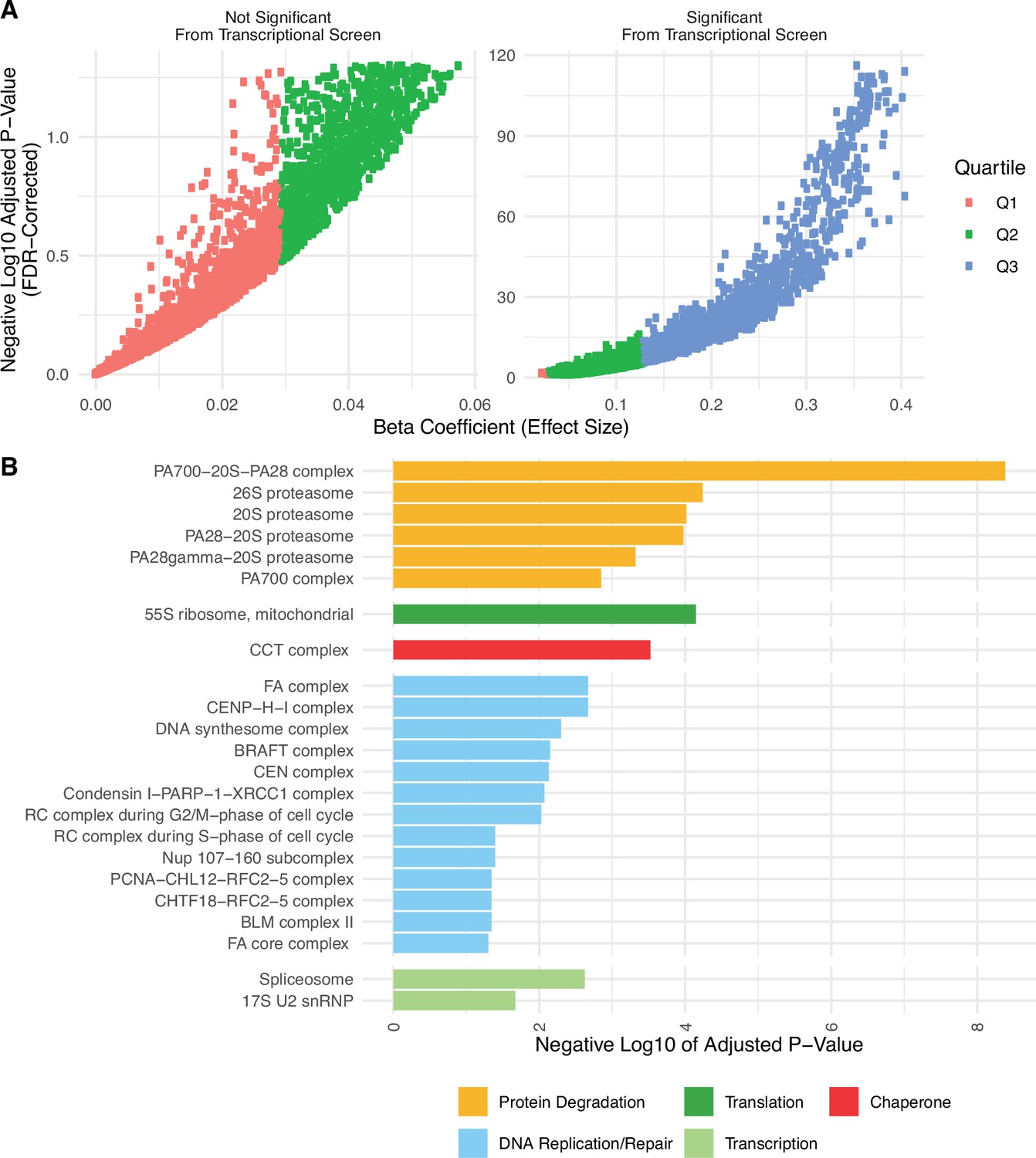
Genes significantly expressed from the transcriptional screen mostly fall into the upper quartile of effect sizes, which are enriched for proteostasis complexes.
(A) Volcano plot of positive regression coefficients and negative log10 adjusted p-values measuring the association of mutation load and the expression of individual genes from the transcriptional screen in Figure 1A. Colors denote the lower (Q1 in red), median (Q2 in green), and upper (Q3 in red) quartiles of each positive beta coefficient from the regression model. Genes that are significantly expressed from the transcriptional screen mostly fall into the upper quartile. (B) Barplot of significant protein complexes in the CORUM database identified using gene set enrichment analysis only on genes that fall into the upper quartile of effect sizes. Genes in the upper quartile of effect sizes contain half of the genes that were identified as significant previously (n=2152 vs n=5330), yet still identify protein degradation, translation, and chaperones as the top significant protein complexes.
Figure 2 with 2 supplements
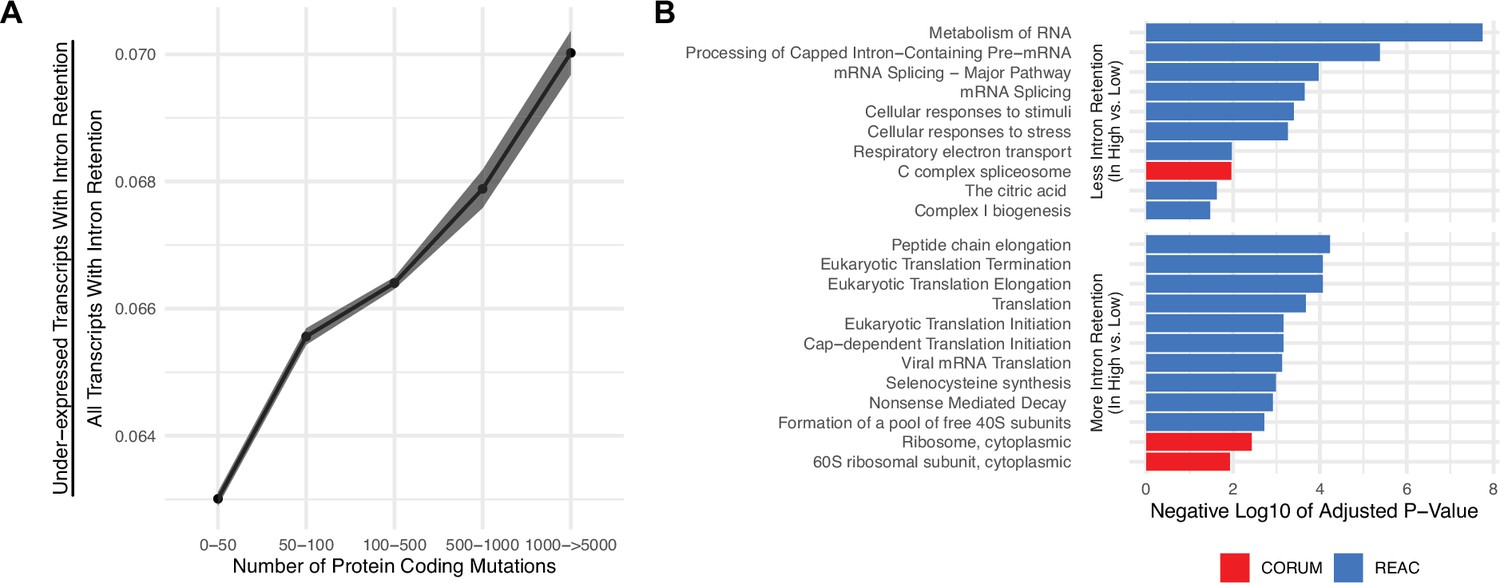
Gene silencing is elevated in high mutational load tumors likely through the coupling of intron retention with mRNA decay.
(A) Counts of the number of under-expressed transcripts with intron retention events, relative to counts of all intron retention events in tumors binned by the total number of protein-coding mutations. Intron retention events with PSI>80% are counted. Error bars are 95% confidence intervals determined by bootstrap sampling. (B) Barplot of significant protein complexes in the CORUM database (in red) and Reactome pathway database (in blue) with more (bottom) and less (top) intron retention events in high mutational load tumors compared to low mutational load tumors.
Figure 2—figure supplement 1
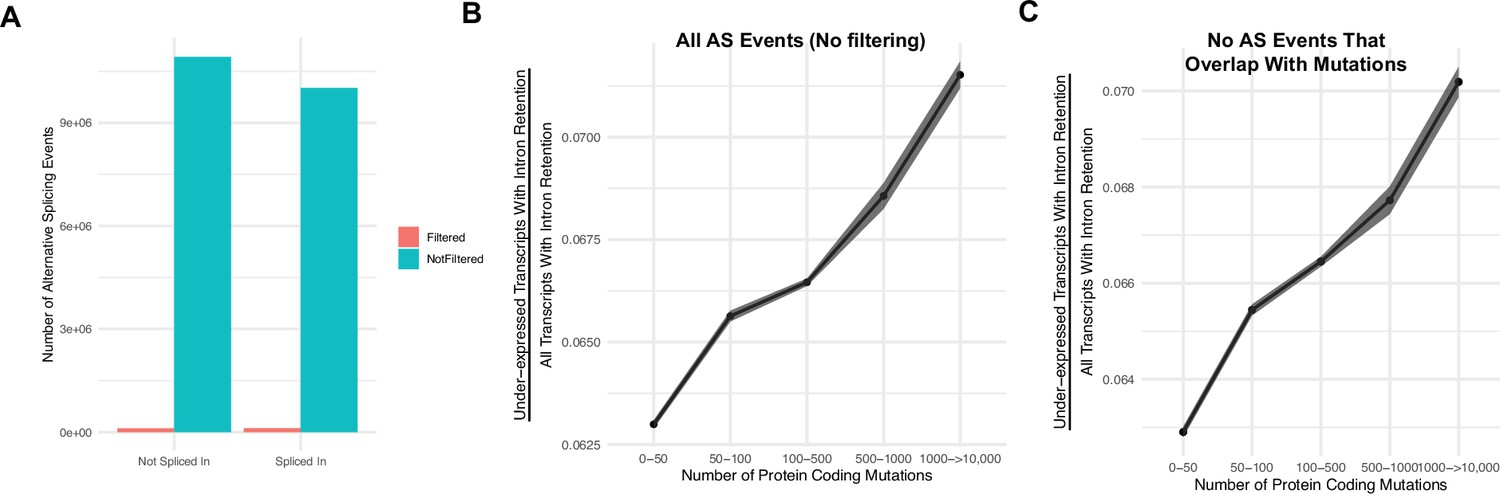
Intron retention events that overlap with mutations do not account for the association of gene silencing in high mutational load tumors.
(A) Counts of the number of intron retention events filtered (in red) due to overlap with a mutation present in the same gene (and thus corresponding to potential eQTLs) compared the number of remaining alternative splicing events with no overlap with a mutation (in blue). Alternative splicing events filtered represent ~1% of all alternative splicing events across all tumors. (B, C) Counts of the number of under-expressed transcripts with intron retention events, relative to counts of all intron retention events in tumors binned by the total number of protein-coding mutations. Shown are when trends when (B) not filtering alternative splicing events due to overlap with mutations and (C) when events are filtered (same as Figure 2A). Intron retention events with PSI >80% are counted. Error bars are 95% confidence intervals determined by bootstrap sampling. These results further support the prediction that gene silencing is elevated in high mutational load tumors and likely mediated by the coupling of intron retention with mRNA decay.
Figure 2—figure supplement 2
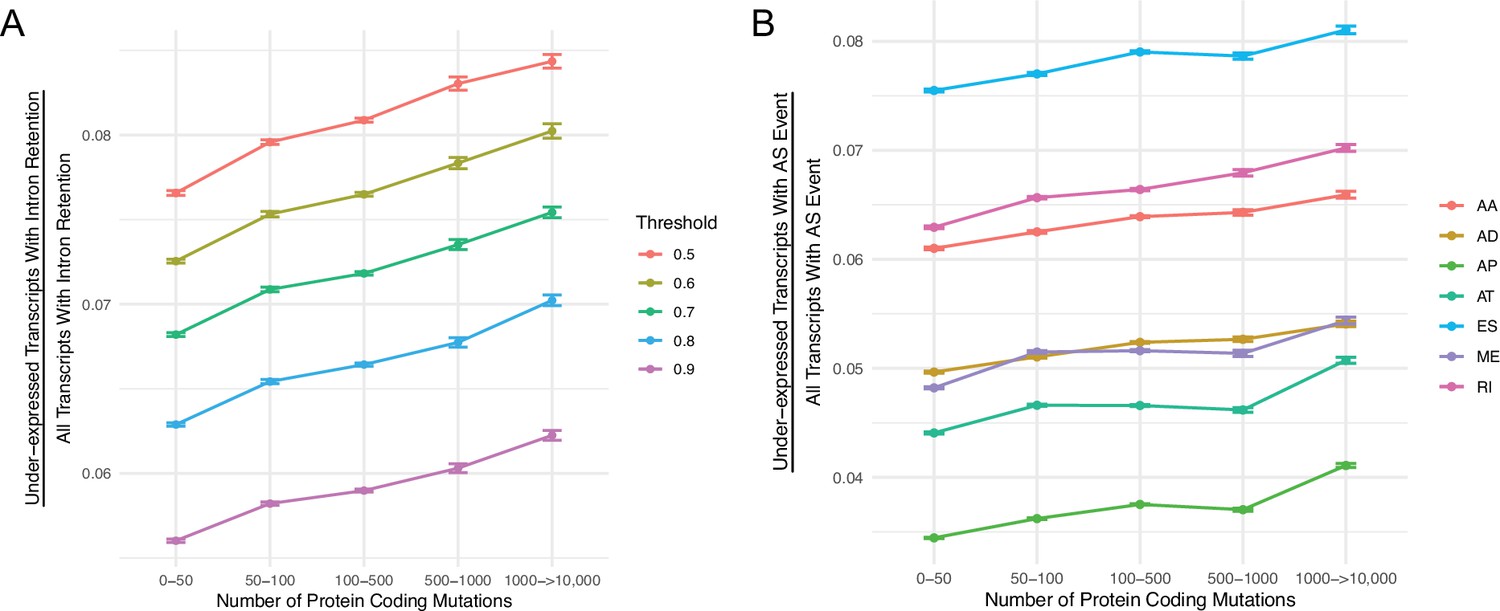
The number of under-expressed transcripts increases with the mutational load of tumors for different PSI value thresholds and alternative splicing events.
(A) Counts of the number of under-expressed transcripts with intron retention events, relative to counts of all intron retention events in tumors binned by the total number of protein-coding mutations. Intron retention events with different PSI thresholds are shown in color. (B) Counts of the number of under-expressed transcripts that contain different classes alternative of splicing events, relative to counts of all alternative splicing events of the same class in tumors binned by the total number of protein-coding mutations. Alternative splicing events of different classes are shown colored (AA = Alternate Acceptor Sites, AD = Alternate Donor Sites, AP = Alternate Promoter, AT = Alternate Terminator, ES = Exon Skip, ME = Mutually Exclusive Exons, RI = Retained Intron). Error bars are 95% confidence intervals determined by bootstrap sampling.
Figure 3 with 5 supplements
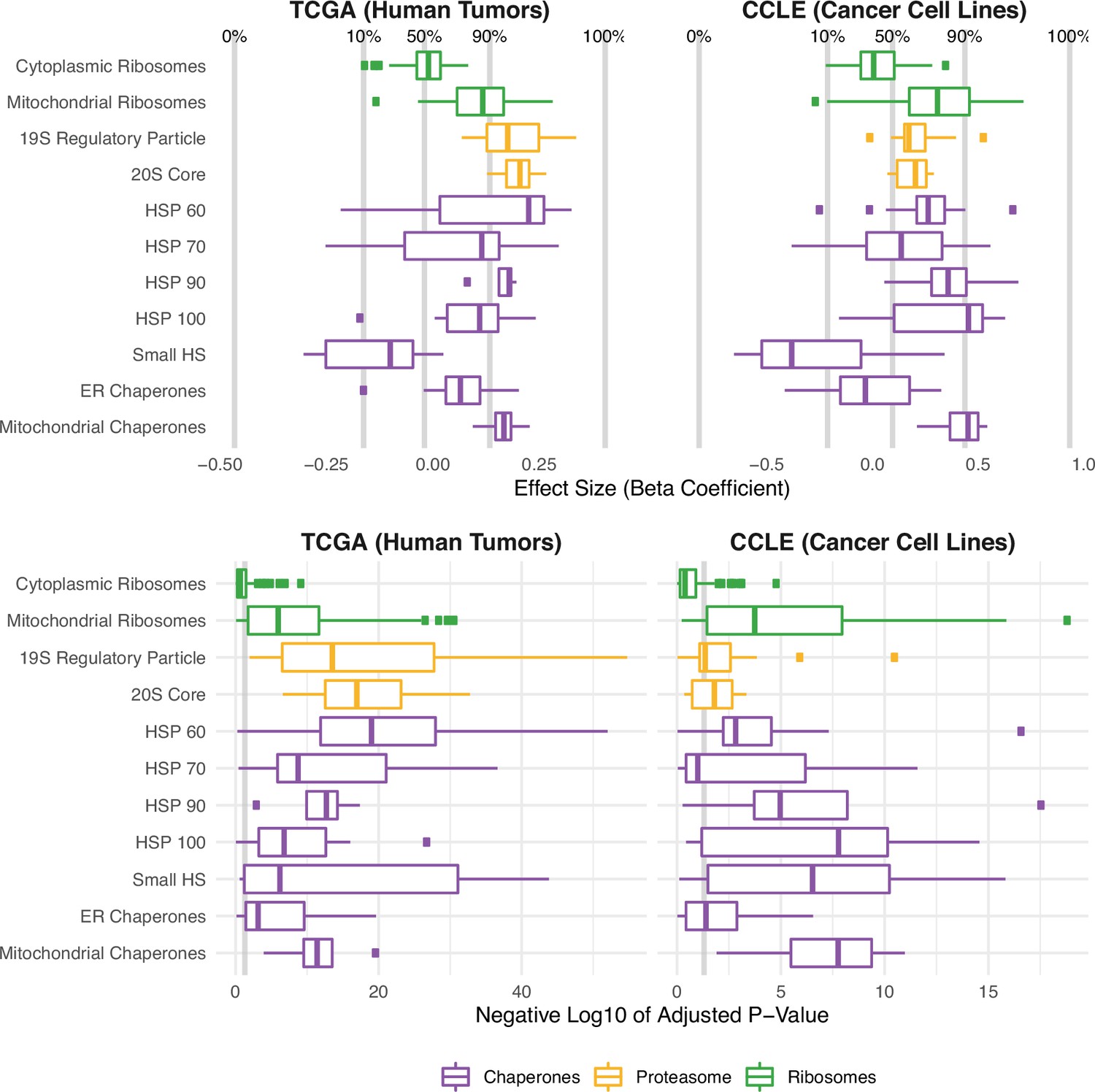
Protein folding, degradation, and synthesis are regulated in both high mutational load tumors (TCGA) and cell lines (CCLE).
Box plots of regression coefficients (top panels) and negative log10 adjusted p-values (bottom panels) measuring the association of mutation load and the expression of individual genes in chaperone (purple), proteasome (yellow), and ribosome (green) complexes. Shown are regression coefficients from human tumors (TCGA) on the left and cell lines (CCLE) on the right. Percentages and gray lines on top panels show the quantile distribution of regression coefficients measuring the association of mutational load and expression for all genes in the genome within each dataset. Vertical gray line on the bottom panels shows the threshold of significance (p=0.05).
Figure 3—figure supplement 1
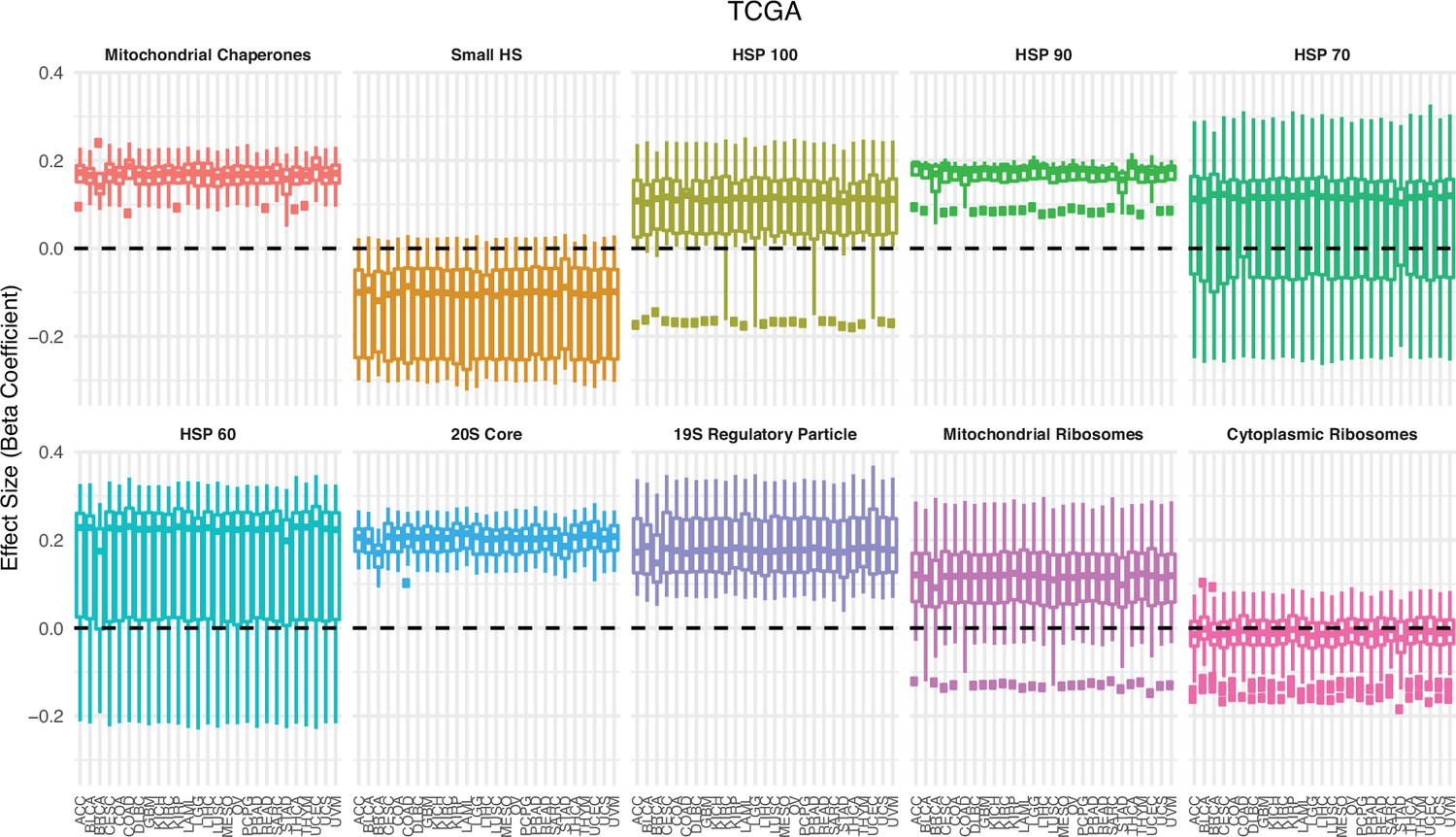
Association between expression in proteostasis complexes and mutational load is not driven by a single cancer type in The Cancer Genome Atlas (TCGA).
Box plots of regression coefficients from the generalized linear mixed model (GLMM) measuring the association of the expression of each individual gene with the mutational load of tumors in TCGA colored by different proteostasis complexes. Shown are regression estimates after removing each individual cancer type (x-axis) and re-running the GLMM.
Figure 3—figure supplement 2
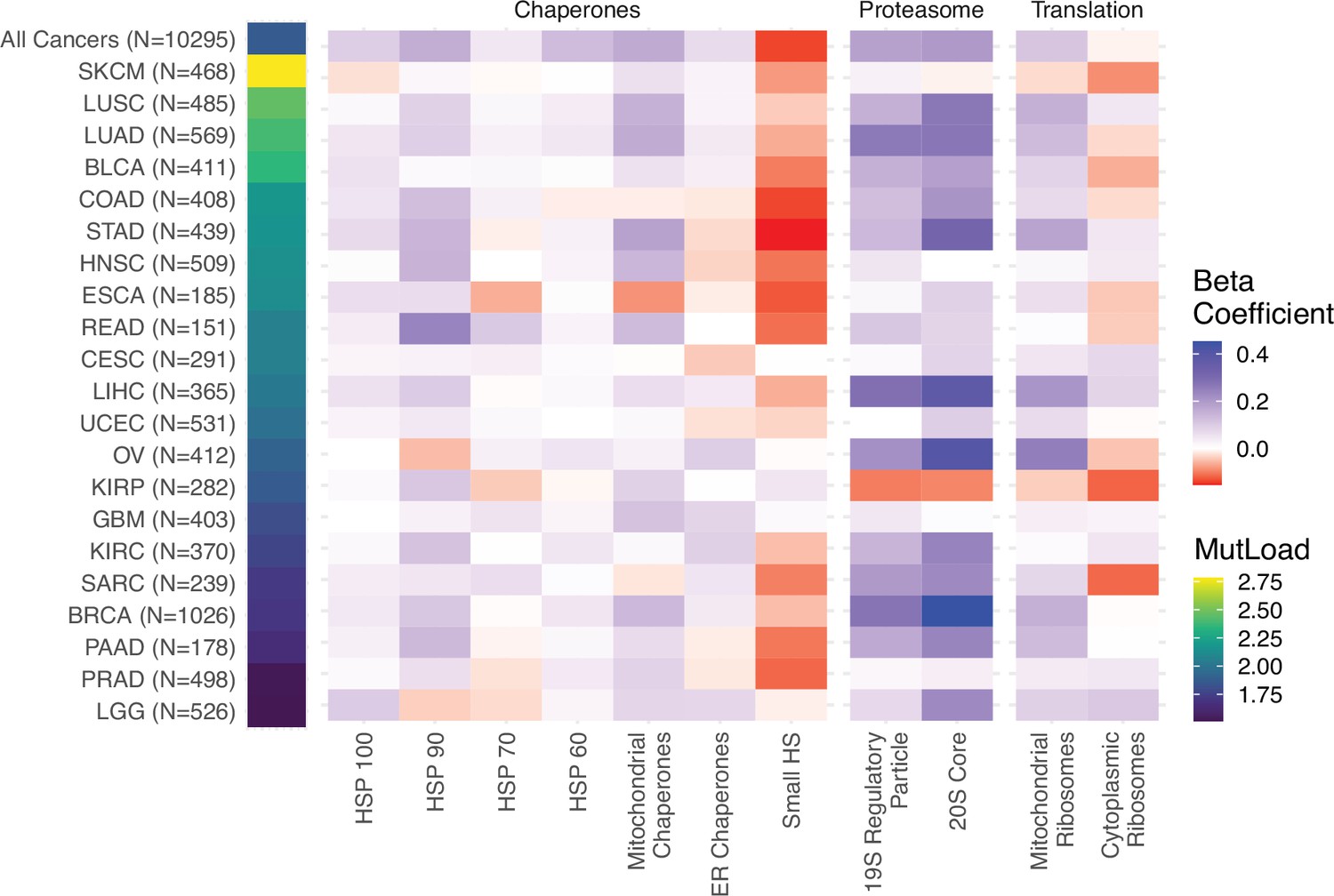
Linear regression analysis within cancer types in The Cancer Genome Atlas (TCGA) captures similar expression responses to mutational load across proteostasis complexes.
Heatmap of regression coefficients measuring the effect of mutational load on gene expression in proteostasis complexes while controlling for tumor purity within cancer types which have enough samples to accurately measure effect sizes (n>150) and contain a sufficiently large enough mutational load to potentially generate a proteostasis response (median protein-coding mutations >25). ‘MutLoad’ shows log10 of the median number of protein-coding mutations for each cancer type.
Figure 3—figure supplement 3
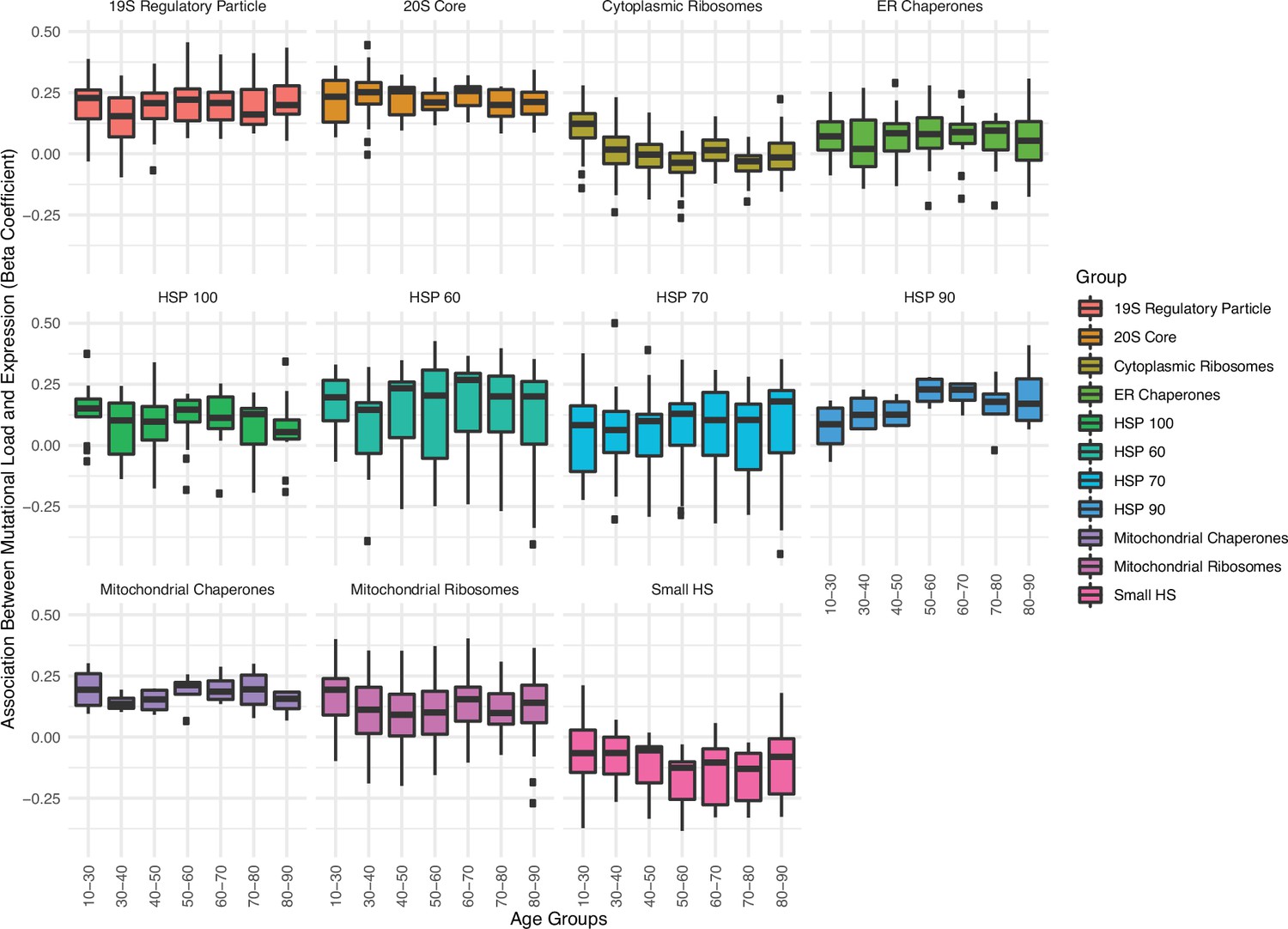
Association between the expression in proteostasis complexes and mutational load is not driven by patient age.
Boxplots of regression coefficients from the generalized linear mixed model (GLMM) measuring the association of the expression of each individual gene with the mutational load of tumors from The Cancer Genome Atlas (TCGA) colored by different proteostasis complexes. Shown are regression coefficients when running the GLMM on tumors stratified by different age groups (x-axis).
Figure 3—figure supplement 4
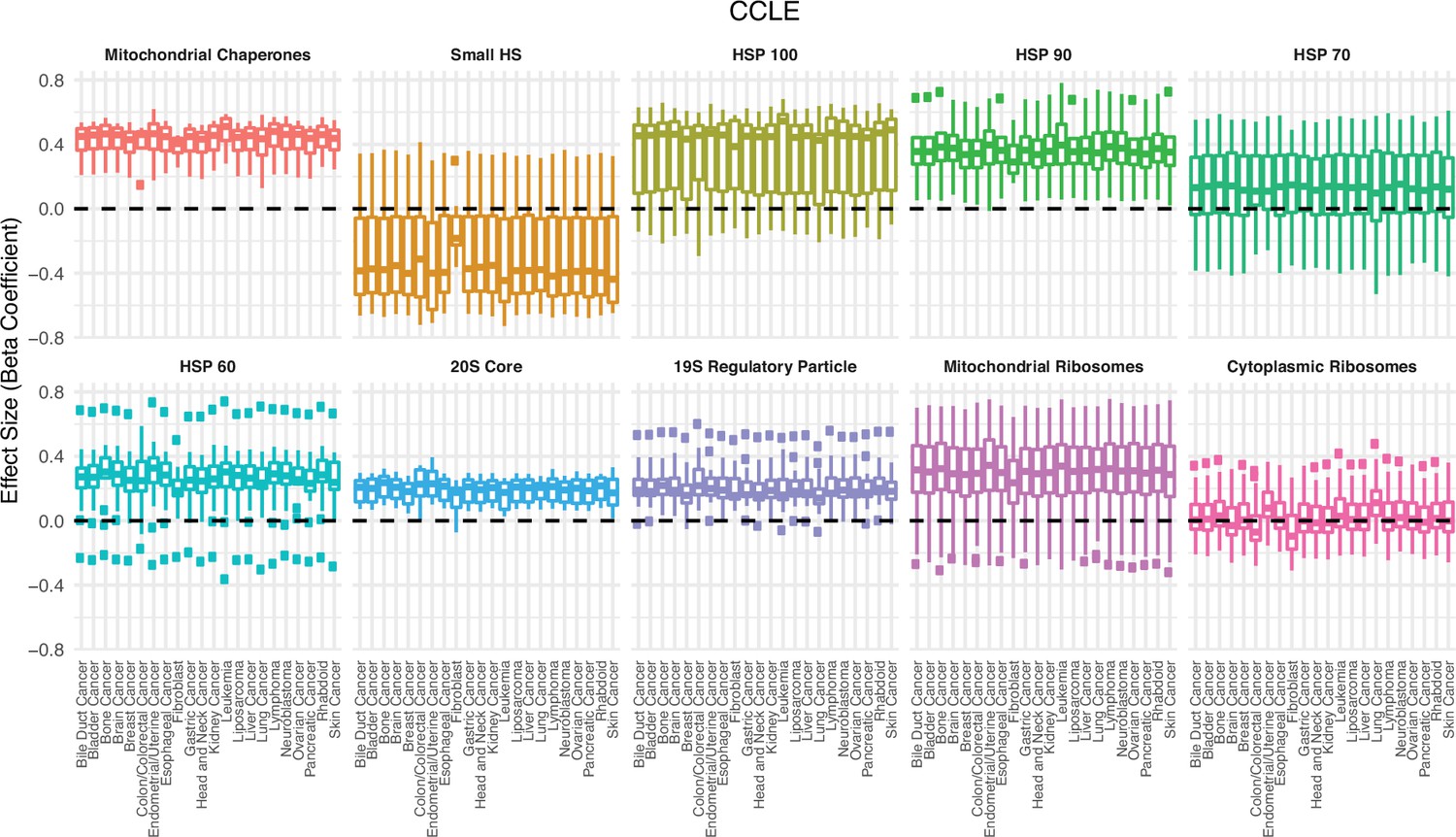
Association between the expression in proteostasis complexes and mutational load is not driven by a single cancer type in cancer cell lines (CCLE).
Box plots of regression coefficients from the generalized linear model (GLM) measuring the association of the expression of each individual gene with the mutational load of tumors colored by different proteostasis complexes. Shown are regression estimates after removing each cancer type in CCLE (x-axis) and re-running the GLM.
Figure 3—figure supplement 5
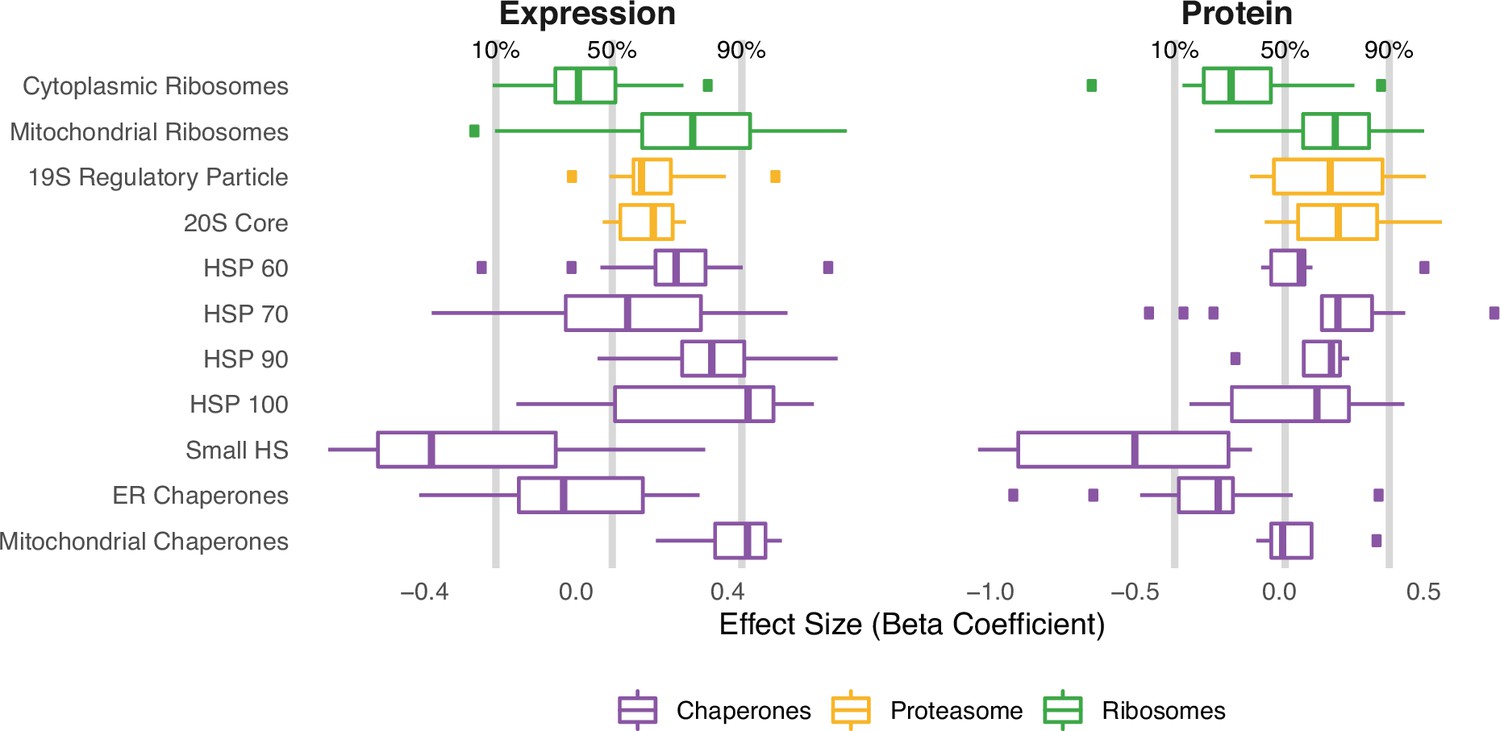
Similar patterns of expression and protein abundances in response to mutational load in cancer cell lines (CCLE) within genes that regulate protein folding, degradation, and synthesis.
Box plots of regression coefficients measuring the association of mutation load and protein abundance (right) or gene expression (left) of individual genes in chaperone (purple), proteasome (yellow), and ribosome (green) complexes. Shown are regression coefficients from cancer cell lines (CCLE), which contain the largest dataset available of RNA (n=1377) and protein (n=373) abundances which are harmonized across samples. Percentages and gray lines on top panels show the quantile distribution of regression coefficients measuring the association of mutational load and expression for all genes in the genome within each dataset.
Figure 4
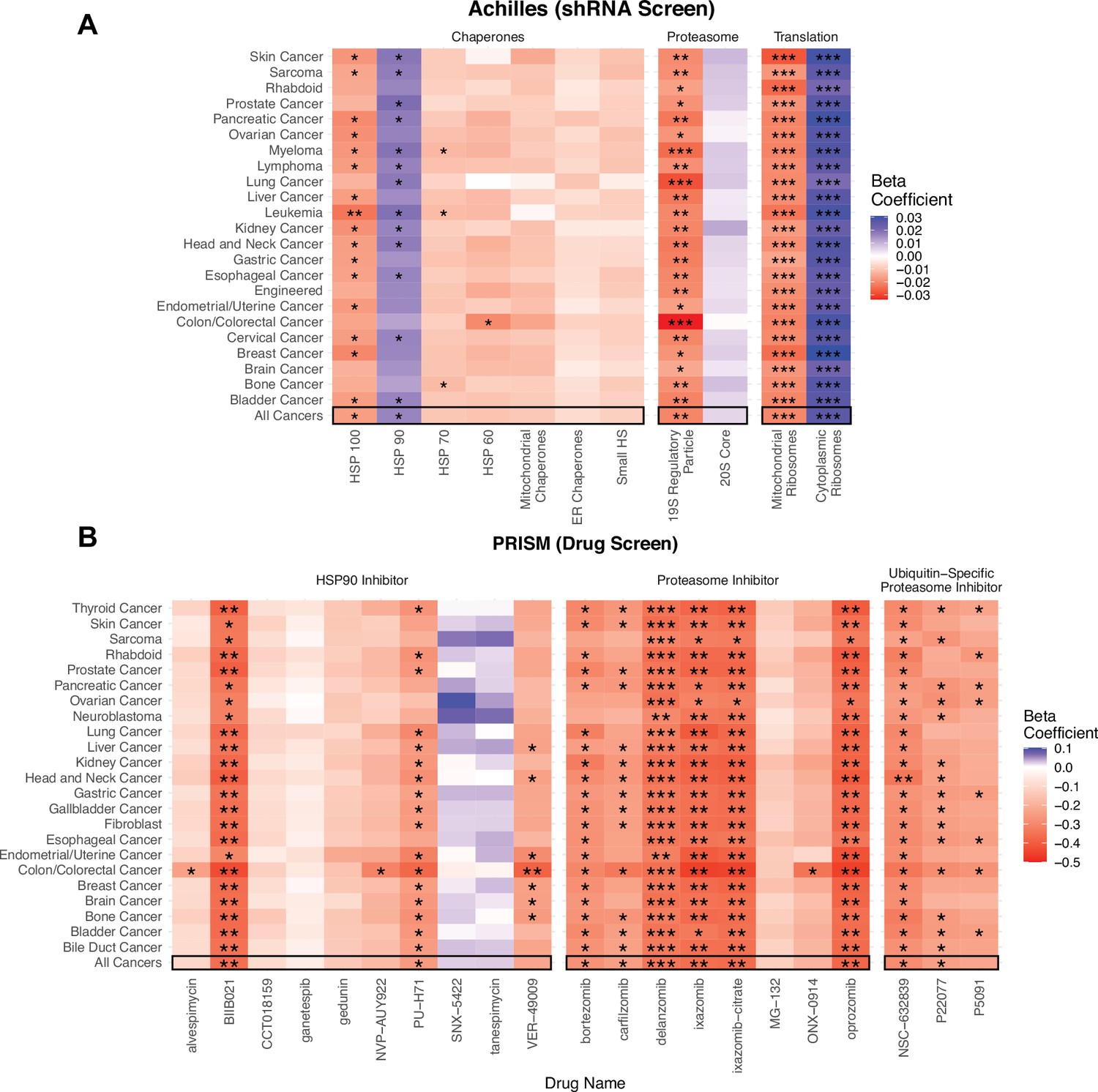
Viability in high mutational load cell lines decreases when proteostasis machinery is disrupted.
(A) Heatmap of regression coefficients jointly measuring the association of mutational load and cell viability after expression knockdown of individual genes in proteostasis complexes. (B) Heatmap of regression coefficients measuring the association mutational load and cell viability after inhibition of proteostasis machinery via drugs. Both panels show how stable regression estimates are when including all cancer types (‘All Cancers’) shown in black boxes and when removing each individual cancer type on the y-axis. Colors denote a positive (blue), zero (gray), or negative (red) relationship between mutational load and cell viability after expression knockdown or drug inhibition. Stars denote whether the relationship is significant (*p<0.05; **p<0.005; ***p<0.0005).
Figure 5
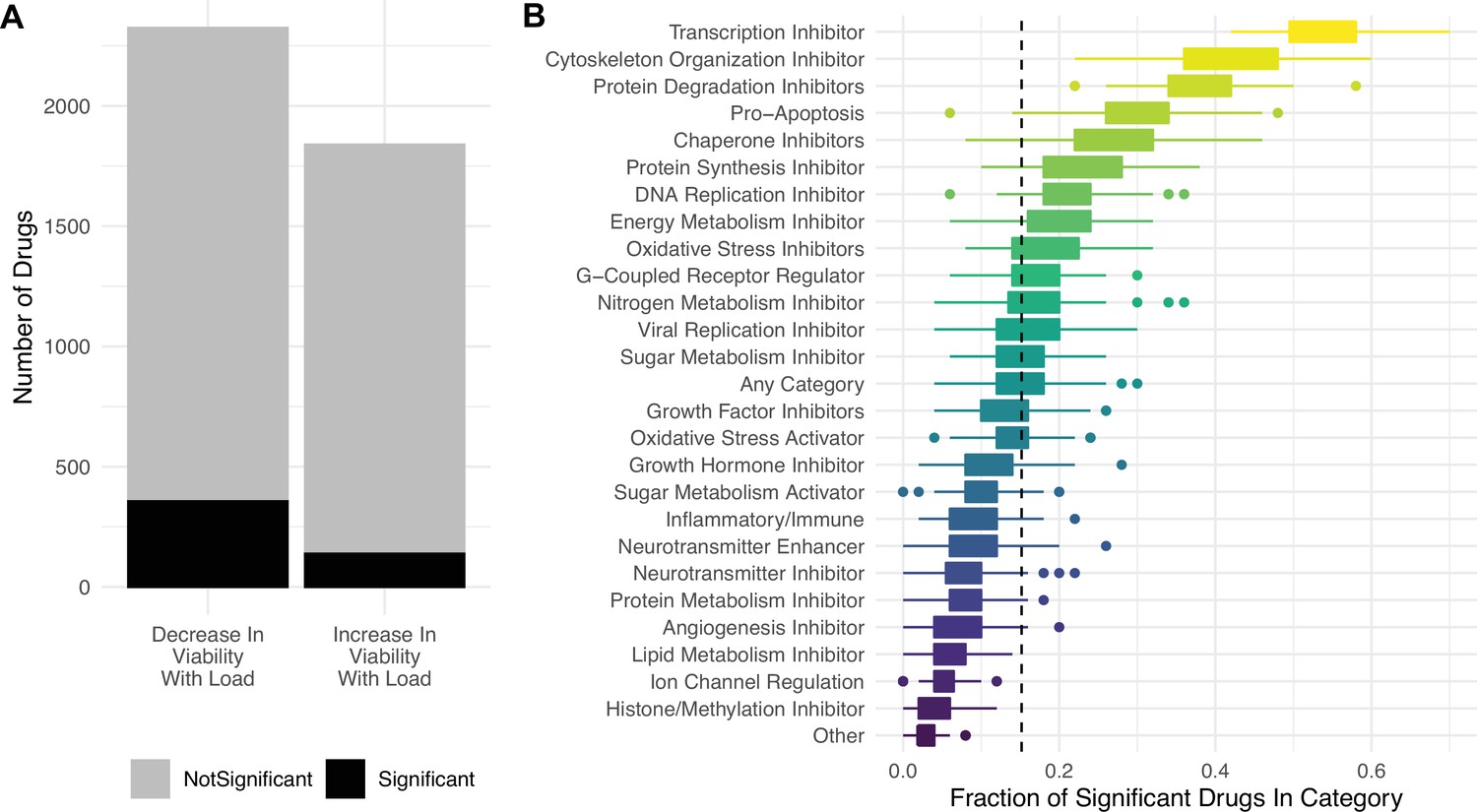
Targeting proteostasis machinery is a key vulnerability in high mutational load cell lines.
(A) Bar plot of the number of drugs in the PRISM database significantly (black) and not significantly (gray) associated with mutational load and cell viability using a simple generalized linear model (GLM). (B) Fraction of drugs in broad functional categories significantly negatively associated with mutational load and cell viability from the GLM. Confidence intervals were determined by randomly sampling 50 drugs in each functional category 100 times. Dashed line is the median of randomly sampled drugs across all categories.
Additional files
-
Supplementary file 1
The full set of enrichment terms for all analyses.
- https://cdn.elifesciences.org/articles/87301/elife-87301-supp1-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/87301/elife-87301-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cancers adapt to their mutational load by buffering protein misfolding stress
eLife 12:RP87301.
https://doi.org/10.7554/eLife.87301.2




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}