Spatial transcriptomics of meningeal inflammation reveals inflammatory gene signatures in adjacent brain parenchyma
- Division of Neuroimmunology, Department of Neurology, Johns Hopkins University School of Medicine, United States
- Department of Neurology, University of Pittsburgh, United States
- Solomon Snyder, Department of Neuroscience, Johns Hopkins University School of Medicine, United States
Figures
Figure 1 with 2 supplements
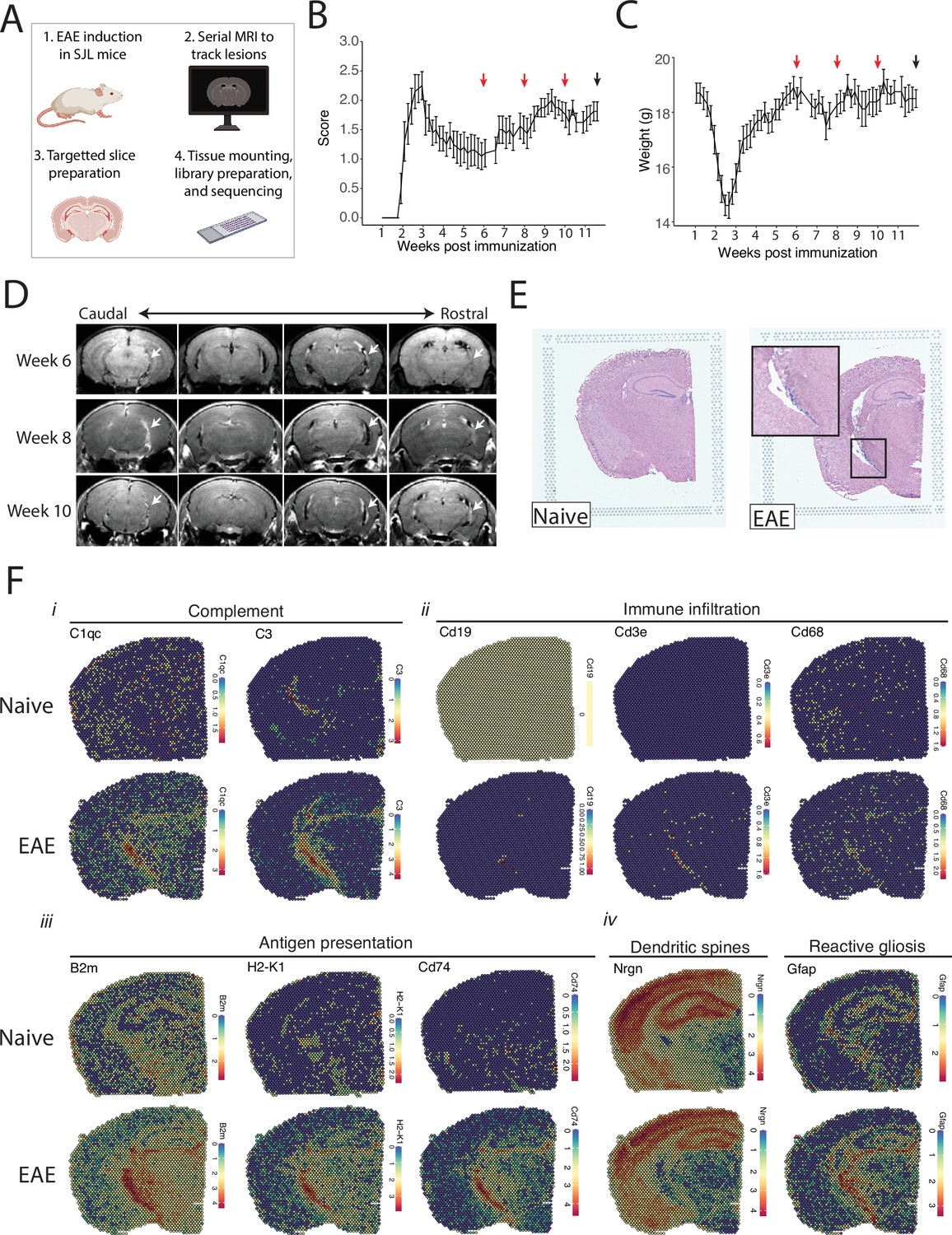
Magnetic resonance imaging (MRI)-guided spatial transcriptomics of meningeal-based inflammation in Swiss Jim Lambert (SJL) experimental autoimmune encephalomyelitis (EAE).
(A) Schematic describing the experimental paradigm. SJL mice underwent brain MRI 6, 8, and 10 weeks’ post-immunization with PLP139-151. Brain slices from regions with meningeal inflammation were collected and processed for spatial transcriptomics on the 10x Genomics platform. (B–C) Behavior scores (B) and mouse weights (C) of the EAE cohort. Red arrows indicate MRI time points, black arrow indicates time of tissue harvesting (N=4). (D) Representative post-contrast MRI brain images, white arrows indicate areas of meningeal-based inflammation. (E) Representative images of hematoxylin and eosin (H&E)-stained tissue sections mounted on spatial transcriptomics slides (left, naïve; right, EAE). (F) Spatial feature plots from naïve (top row) and EAE (bottom row) representative samples demonstrate altered expression of genes related to complement (i), immune infiltration (ii), antigen presentation (iii), dendritic spines, and astrocyte activation (iv).
Figure 1—figure supplement 1
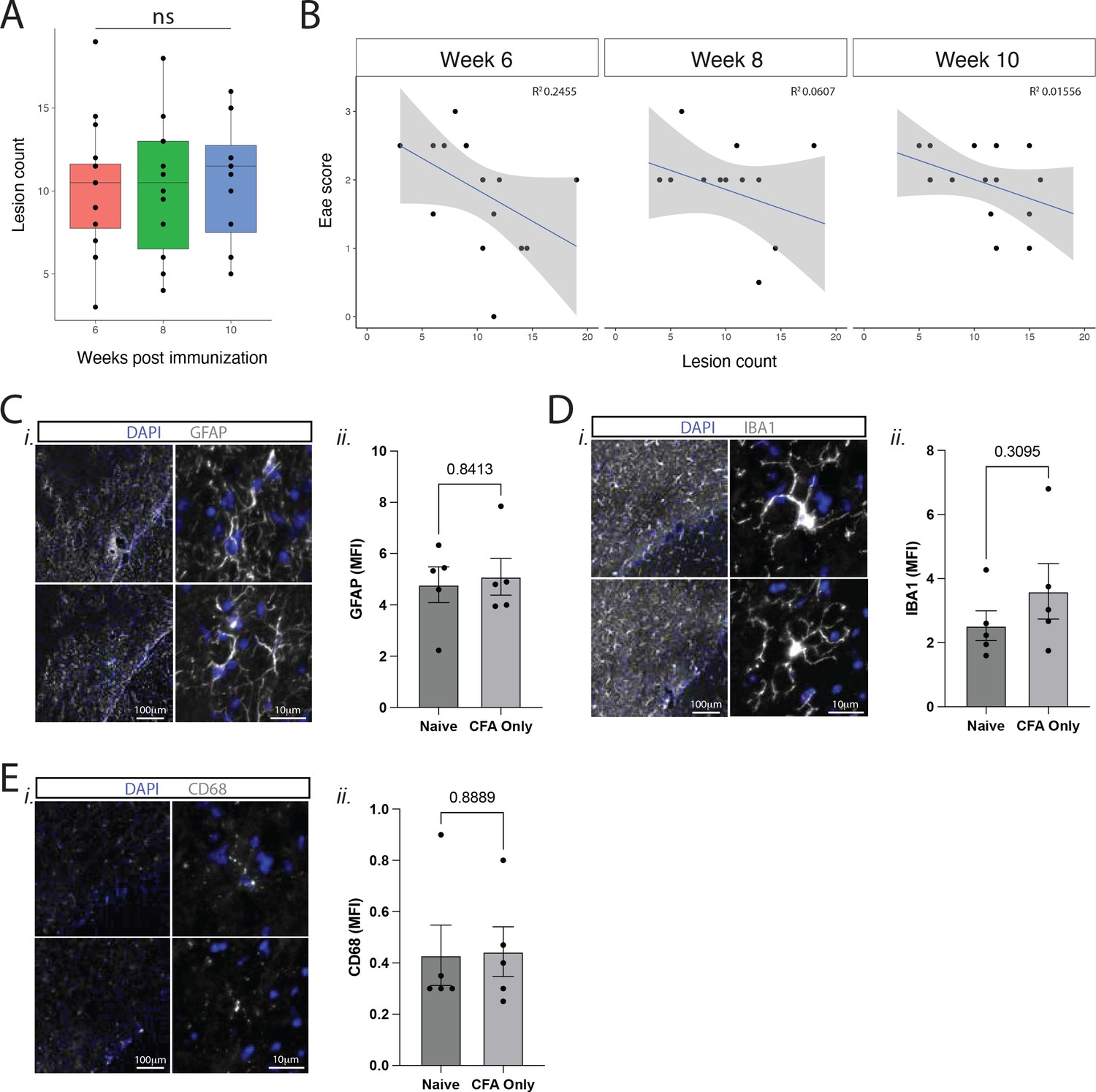
Contrast enhancing meningeal inflammation in Swiss Jim Lambert (SJL) experimental autoimmune encephalomyelitis (EAE) does not change at chronic time points or with clinical disease scores.
(A) Box plot of lesion number vs. week post-immunization. ANOVA with Tukey’s HSD post hoc test. (B) Lesion number plotted against EAE score demonstrates no strong correlation (R2<0.4 in all cases). N=16, data is representative of three pooled experiments. (C–E) Immunofluorescence analysis of glial reactivity in naïve and CFA only groups, 11 weeks after injection. Representative staining at low and high magnification and MFI (mean fluorescence intensity) calculations of (C) GFAP, (D) IBA1, (E) CD68. N=5, Mann-Whitney tests.
Figure 1—figure supplement 2
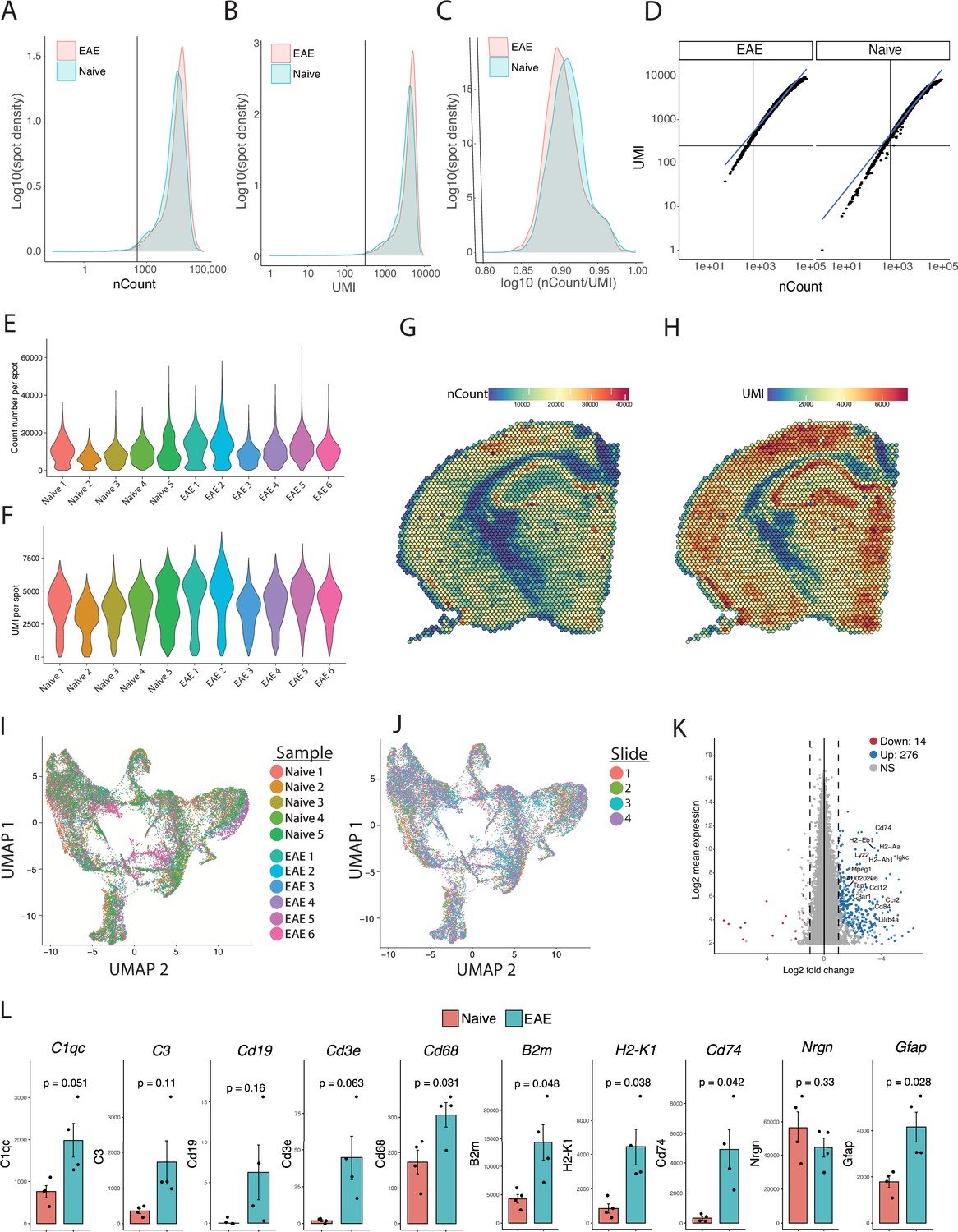
Quality control analysis of spatial transcriptomic data.
(A) Density of read counts, (B) number of unique molecular identifiers (UMI), (C) and the ratio of read count/UMI per spot for naïve and experimental autoimmune encephalomyelitis (EAE) samples. Most spots had >500 read count (A), >250 UMI (B), and >0.80 log(nCount/UMI) (C). (D) Dot plot shows most spots having linear correlation between UMI and read count. (E–F) Violin plots of read counts (E) and UMI (F) per sample. (G–H) Representative spatial feature plots of read count (G) and UMI (H) demonstrate expected anatomic variability in transcript amount and diversity. (I–J) UMAP dimensionality reduction plots, colored by mouse (I) or slide (J). (K) MA plot of genes enriched in EAE compared to naïve samples. Data was pseudobulked by sample and groups were compared using DESeq2 for gene enrichment (adjusted p-value<0.05, log 2 fold change >1). (L) Comparison of average expression for select transcripts in naïve and EAE samples. N=4, Student’s t-test.
Figure 2
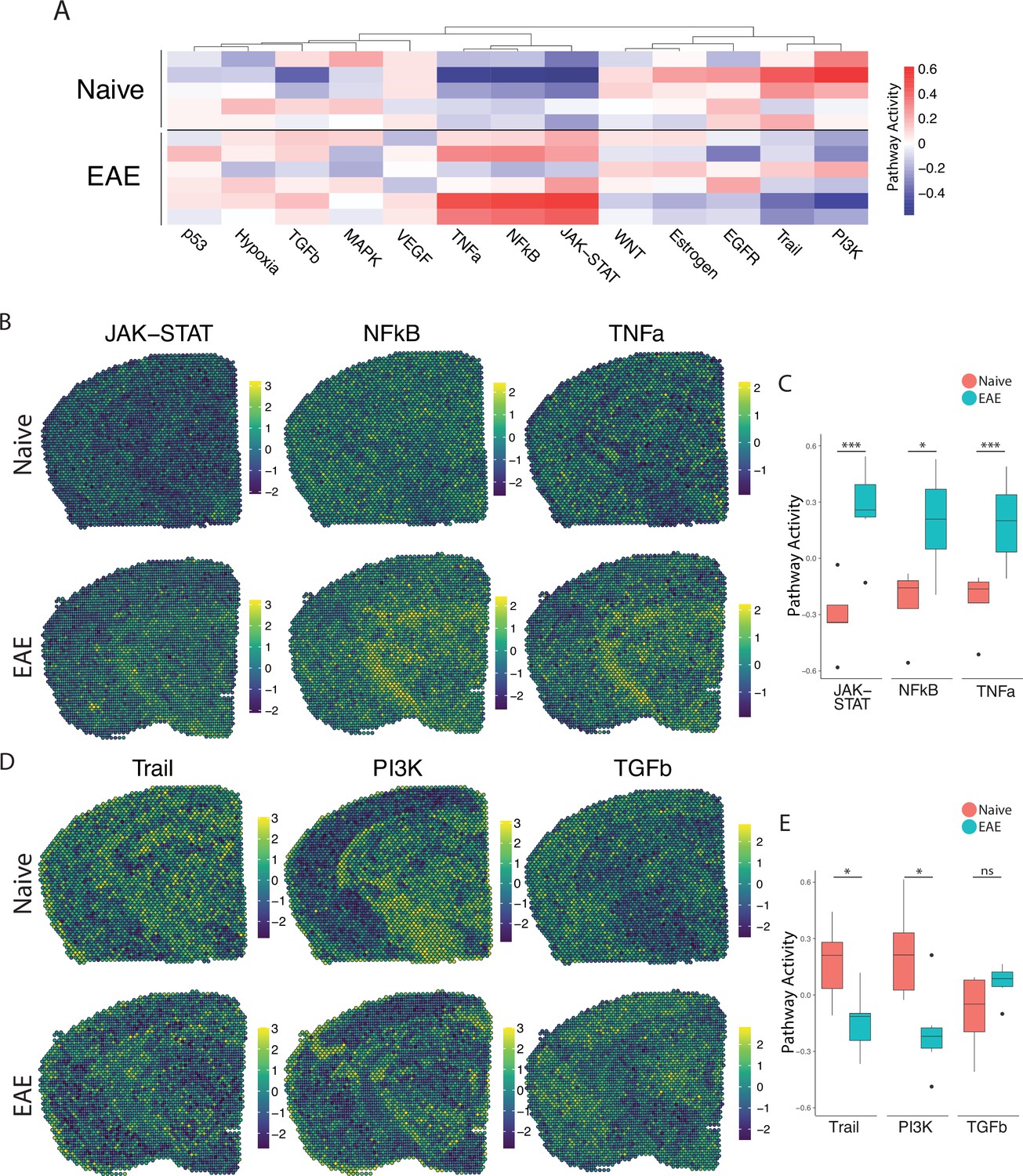
PROGENy analysis reveals spatially restricted pathway activity differences between naïve and experimental autoimmune encephalomyelitis (EAE).
(A) Heatmap displaying averaged PROGENy pathway analysis results. (B) Representative spatial plot showing activity of the JAK-STAT, NFkB, and TNFa signaling pathways. (C) Comparison of JAK-STAT, NFkB, and TNFa pathway activities between groups. (D) Representative spatial plot showing activity of the Trail, PI3K, and TGFb signaling pathways. (E) Comparison of Trail, PI3K, and TGFb pathway activities between groups. (Naïve mouse N=4, sample N=5; EAE mouse N=4, sample N=6; multiple t-tests corrected for multiple comparisons with the Benjamini, Krieger, and Yekutieli method; *p<0.05, **p<0.01, ***p<0.001.)
Figure 3 with 1 supplement
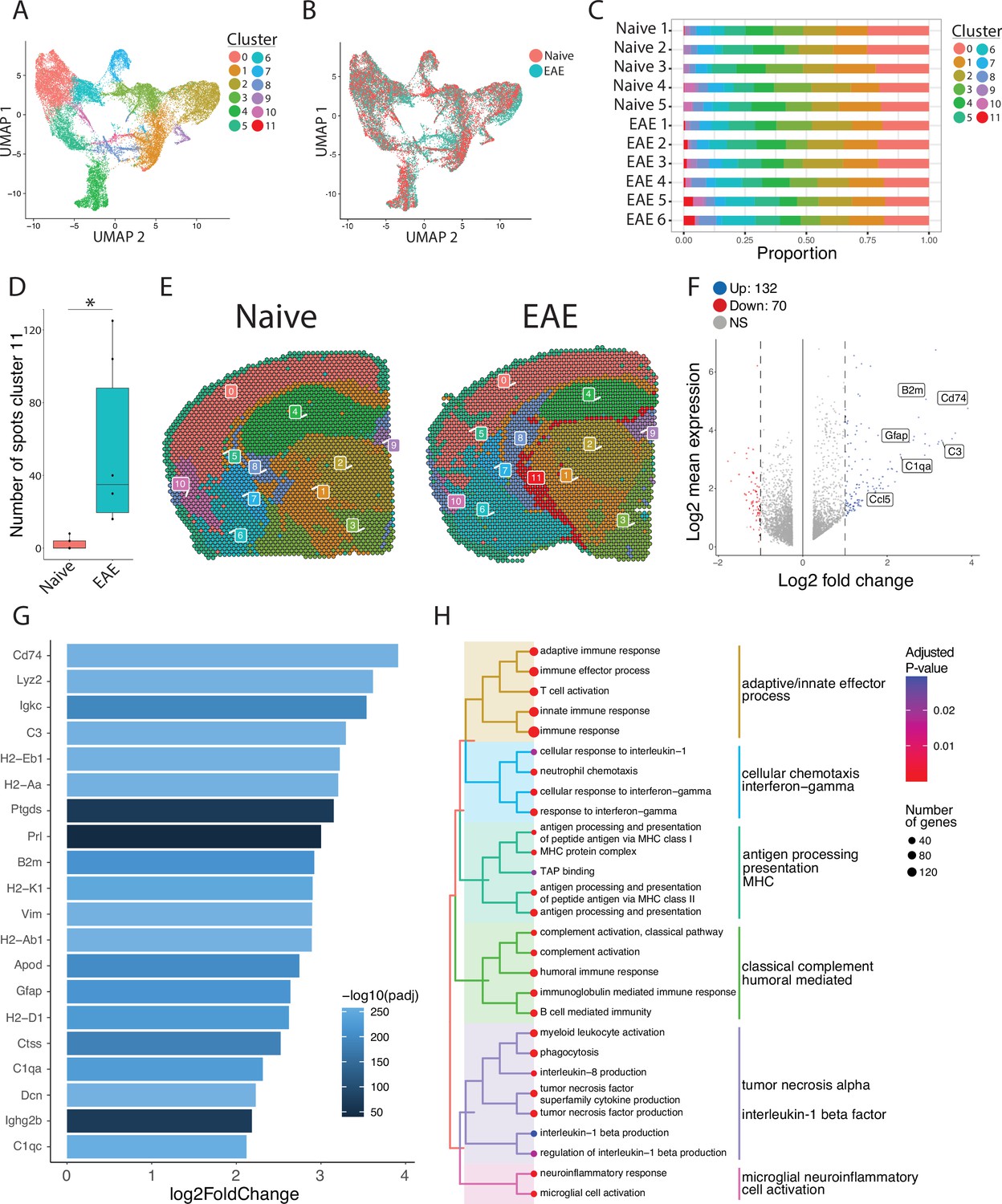
Unbiased clustering reveals a group of spots enriched in inflammatory genes.
(A–B) UMAP dimensionality reduction plots colored by (A) cluster or (B) group. (C) Bar plot showing the proportion of spots in each cluster by sample. (D) Number of spots in cluster 11 by group (N=11; Student’s two-tailed t-test). (E) Representative spatial feature plots of naïve and experimental autoimmune encephalomyelitis (EAE) samples showing the spatial distribution of each cluster. (F) MA plot comparing differences in gene expression between cluster 11 and all other clusters averaged across samples. Red and blue spots represent genes in cluster 11 that are significantly increased or decreased, respectively (adjusted p-value<0.05, log 2 fold change >1). (G) Bar plot of top 15 genes enriched in cluster 11 compared to other clusters. (H) Tree plot displaying gene set enrichment results using the gene ontology (GO) database. Spots in cluster 11 were compared to other spots and gene set sizes ranging from 10 to 500 were included (adjusted p-value<0.05).
Figure 3—figure supplement 1
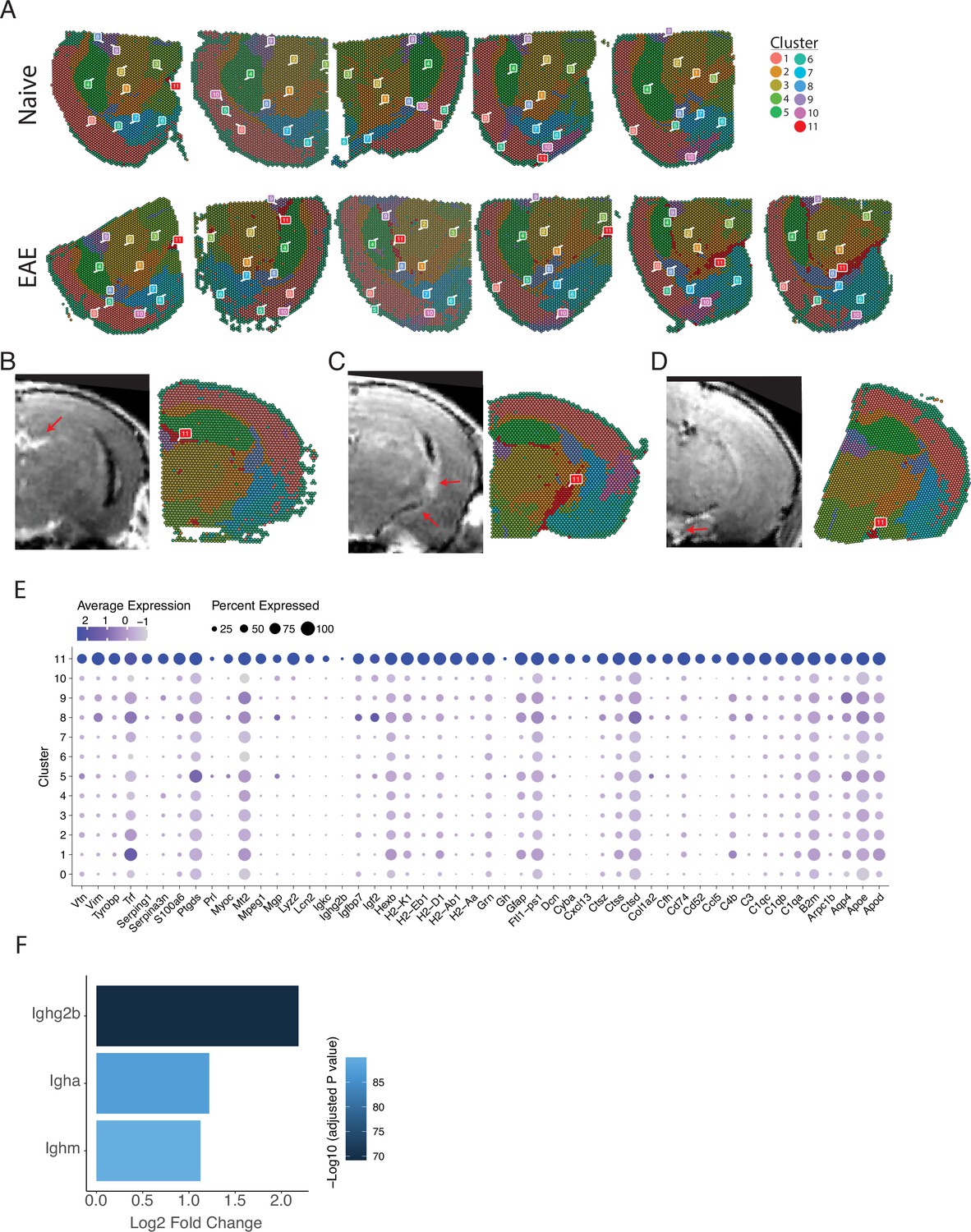
Spatial and transcriptional properties of inflammatory clusters.
(A) Representative spatial feature plots of naïve (top row) and experimental autoimmune encephalomyelitis (EAE) (bottom row) samples colored by cluster show consistent labeling of anatomic regions and the distribution of meningeal inflammation (cluster 11). (B–D) Representative images of gadolinium-enhanced magnetic resonance imaging (MRI) scans collected 10 weeks after immunizations and samples collected from the same animal. Areas of meningeal enhancement (red arrows) correlate with cluster 11. (E) Dot plot of the top 100 significantly enriched genes (sorted by adjusted p-value) in cluster 11 compared to other clusters. Dot color represents expression value and dot size represents the percent of spots within that cluster expressing the gene.
Figure 4 with 2 supplements

Subclustering of spots adjacent to meningeal immune follicles reveals a subset of active immune patterns.
(A) UMAP dimensionality reduction plots showing subclustering of cluster 1 colored by (left) group or (right) cluster. (B) UMAP dimensionality reduction plots showing subclustering of cluster 2 colored by (left) group or (right) cluster. (C) Representative spatial feature plot showing the locations of cluster 1 and 2 subclusters. (D) Distance from the center of indicated subclusters to the nearest point of cluster 11 (N=11; Student’s two-tailed t-test). (E–G) Tree plot displaying gene set enrichment results using the gene ontology (GO) database for subcluster 1_3 (E), 1_4 (F), and 2_6 (G) compared to other spots in their respective clusters. (H) Venn diagram shows overlap of significantly enriched GO gene sets between cluster 11 and subclusters 1_3, 1_4, and 2_6, with (I) 31 gene sets elevated in all. GO gene set of size ranging from 10 to 500 were included (adjusted p-value<0.05).
Figure 4—figure supplement 1
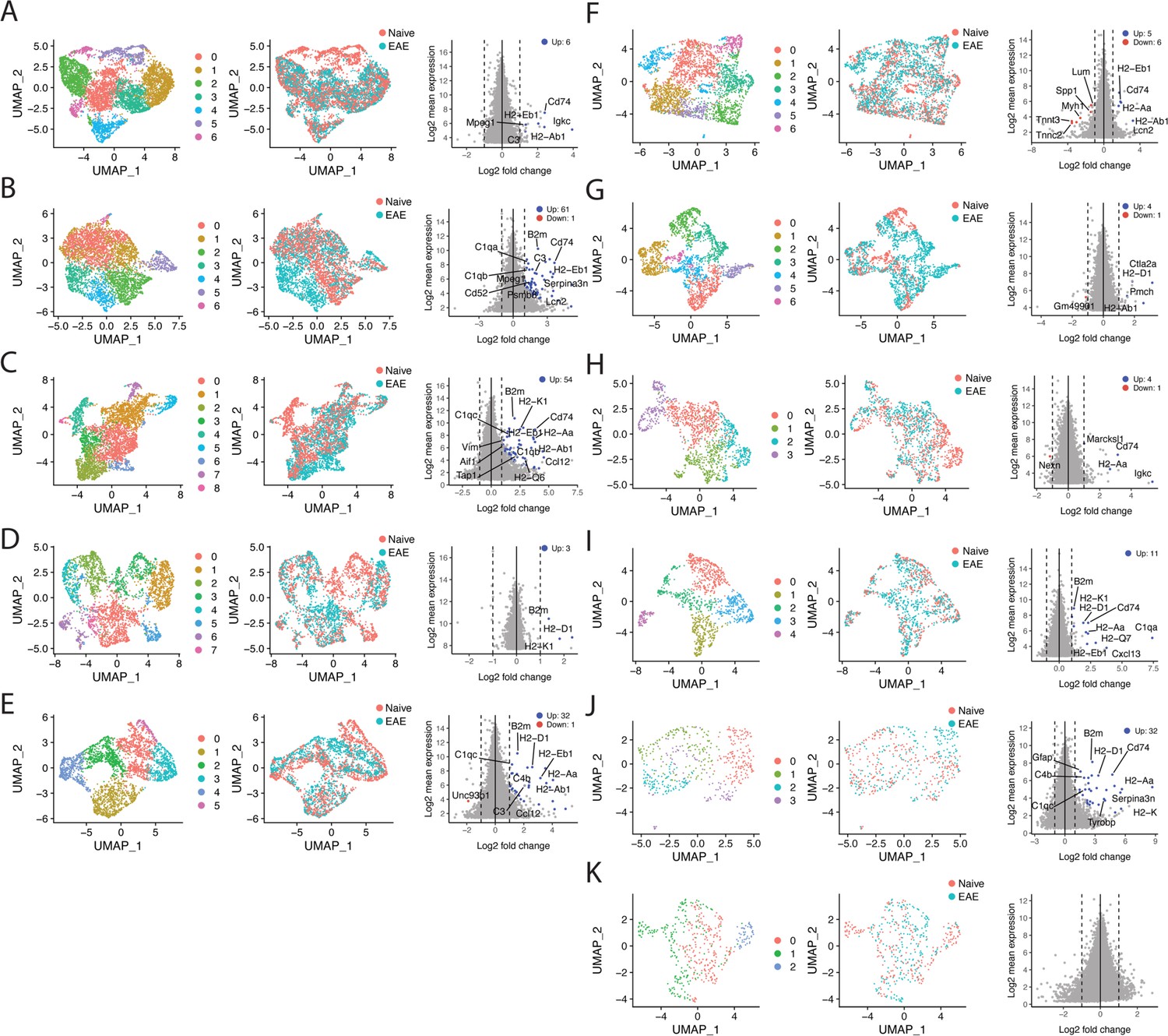
Subcluster analysis reveals experimental autoimmune encephalomyelitis (EAE)-specific subclusters and gene enrichment.
(A–K) Data from each individual cluster was extracted and unbiased subclustering performed based on gene expression; (A) cluster 0, (B) cluster 1, (C) cluster 2, (D) cluster 3, (E) cluster 4, (F) cluster 5, (G) cluster 6, (H) cluster 7, (I) cluster 8, (J) cluster 9, (K) cluster 10. UMAP plots are shown colored by subcluster (left panel) and group (middle panel). Data from each cluster was isolated, pseudobulked by sample, and groups were compared using DESeq2 for gene enrichment (right panel; adjusted p-value<0.05, log 2 fold change >1).
Figure 4—figure supplement 2
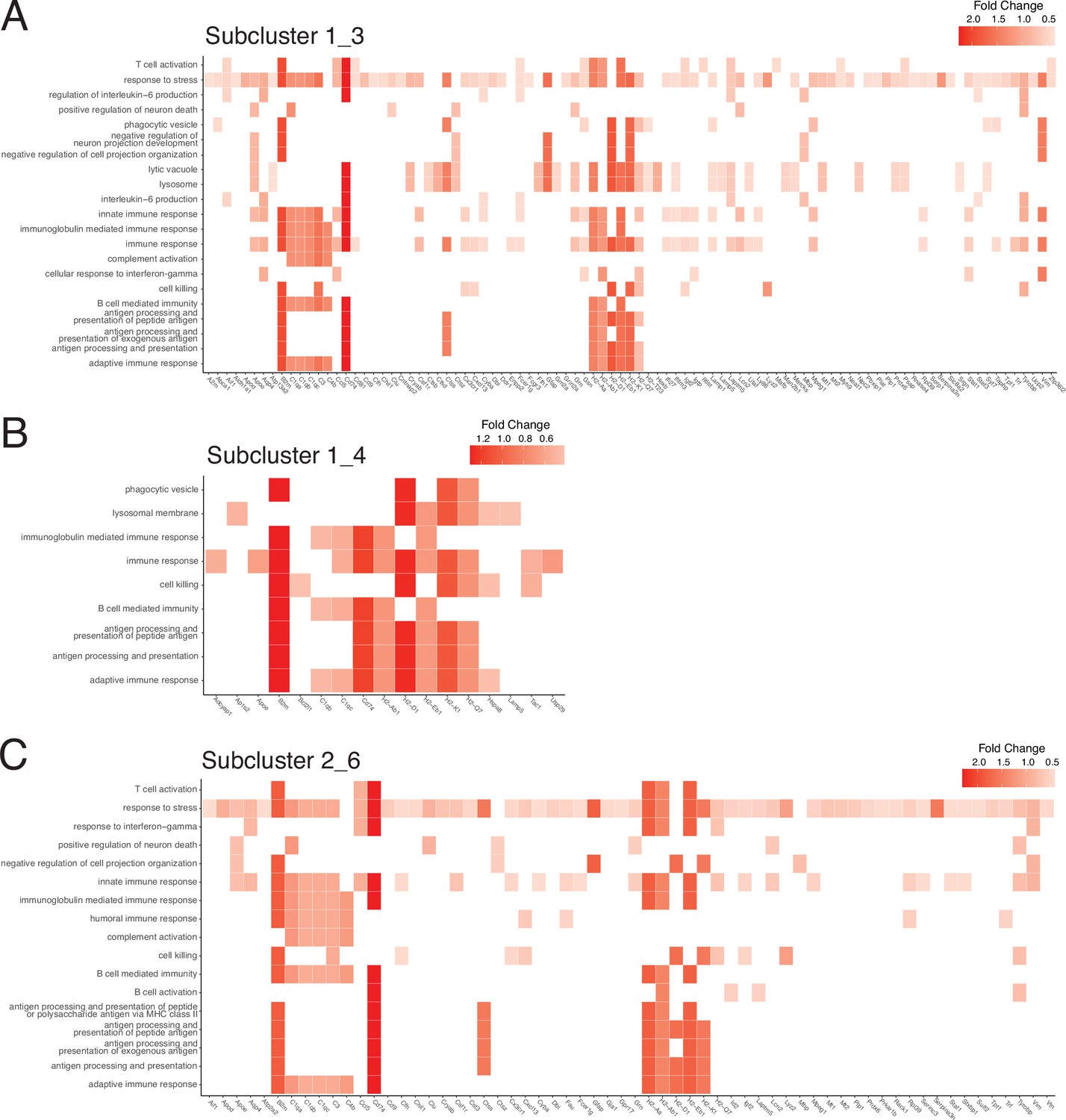
Gene contributions to enriched gene sets in subclusters 1_3, 1_4, and 2_6.
(A–C) Heatmaps of gene ontology (GO) gene sets enriched in subcluster 1_3 (A), 1_4 (B), and 2_6 (C). Individual genes contributing to gene sets are displayed on the x axis. Data from each subcluster was pseudobulked by sample and groups were compared using DESeq2 for gene enrichment (adjusted p-value<0.05, log 2 fold change >1). Heatmaps of select gene sets were generated using the enrichPlot package.
Figure 5 with 1 supplement
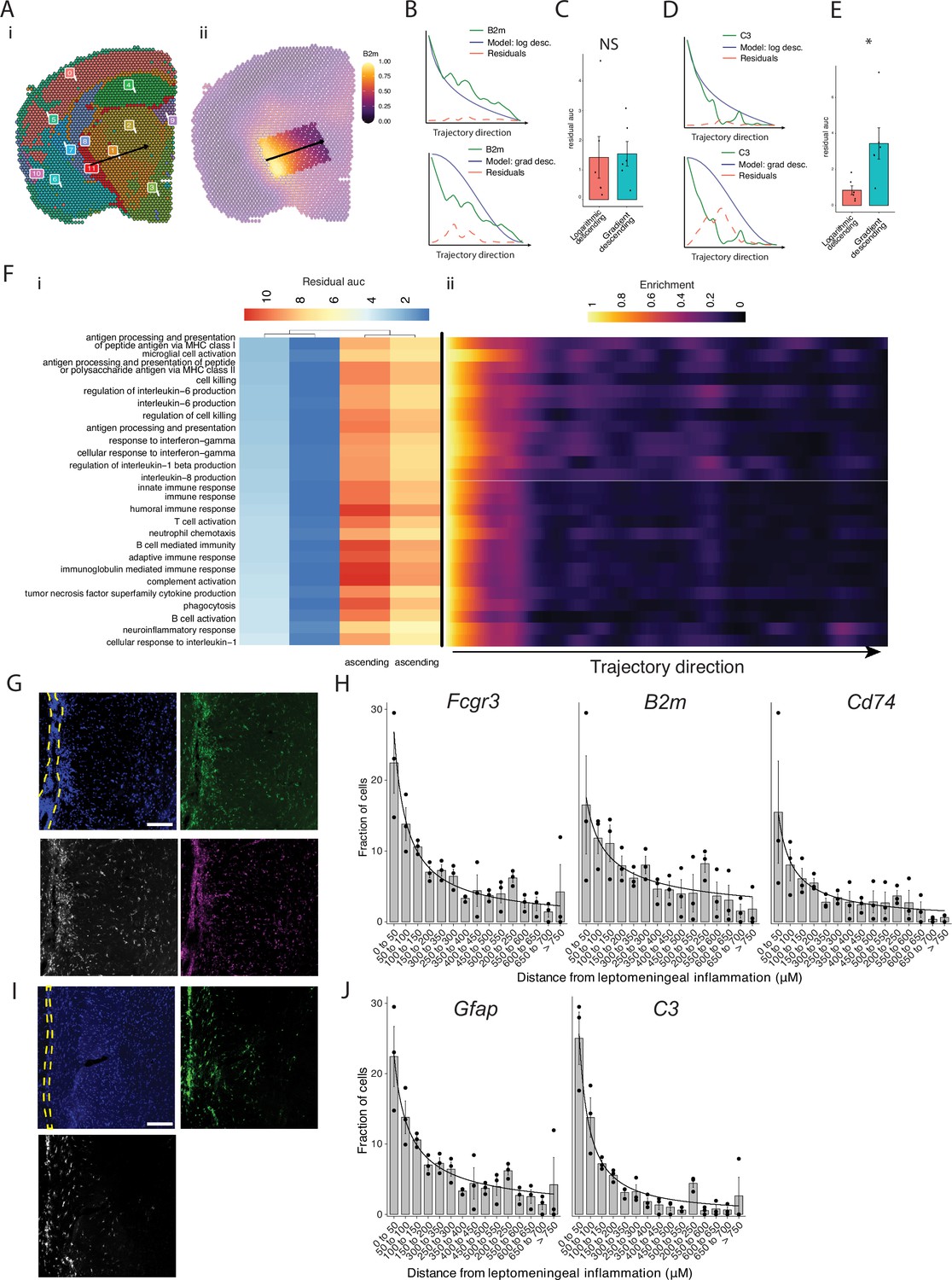
Trajectory analysis reveals gradients of gene expression originating from meningeal lymphoid follicles.
(A) Trajectories were drawn based on spatial cluster plot (i) from C11 to C2 (ii). (B) Representative plot of B2m relative expression along the trajectory length. Green line: B2m expression; black line: ideal model fit, ‘logarithmic descending’ (top) or ‘gradient descending’ (bottom); red line: residual area under the curve (AUC) representing the difference between B2m expression and the ideal model. (C) Bar plot showing residual AUC of B2m relative expression along the trajectory direction compared to ‘logarithmic descending’ or ‘gradient descending’ (Student’s two-tailed t-test). (D) Representative plot of C3 relative expression along the trajectory length. Green line: C3 expression; black line: ideal model fit, ‘logarithmic descending’ (top) or ‘gradient descending’ (bottom); red line: residual area under the curve (AUC) representing the difference between C3 expression and the ideal model. (C) Bar plot showing residual AUC of C3 relative expression along the trajectory direction compared to ‘logarithmic descending’ or ‘gradient descending’ (Student’s t-test). (F) Gene sets that were previously identified as significantly enriched in C11 were selected for trajectory analysis. Residual AUCs were calculated for ‘logarithmic descending’, ‘gradient descending’, ‘logarithmic ascending’, and ‘gradient ascending’ ideal fits and displayed on (i) a heatmap sorted by ‘gradient descending’. (ii) Representative feature plot demonstrating deeper penetration of upper gene sets (related to antigen presentation and processing, microglial activation, IL-6 production, interferon gamma) response relative to other gene sets (B cell activation, T cell activation, TNF production, complement, humoral immune response). (G, I) Representative images of RNAscope labeling for (G) Fcgr3, B2m, Cd74, and (I) Gfap, C3 in Swiss Jim Lambert (SJL) mice 11 weeks after experimental autoimmune encephalomyelitis (EAE) induction. Yellow dashed lines indicate the areas of leptomeningeal inflammation, scale bars represent 100 μM. (H, J) Bar plots representing the percent of marker-positive cells present at distances from leptomeningeal inflammation. Lines represent best fit curves from exponential regression. N=3 animals per group; bars represent mean, error bars represent standard error.
Figure 5—figure supplement 1
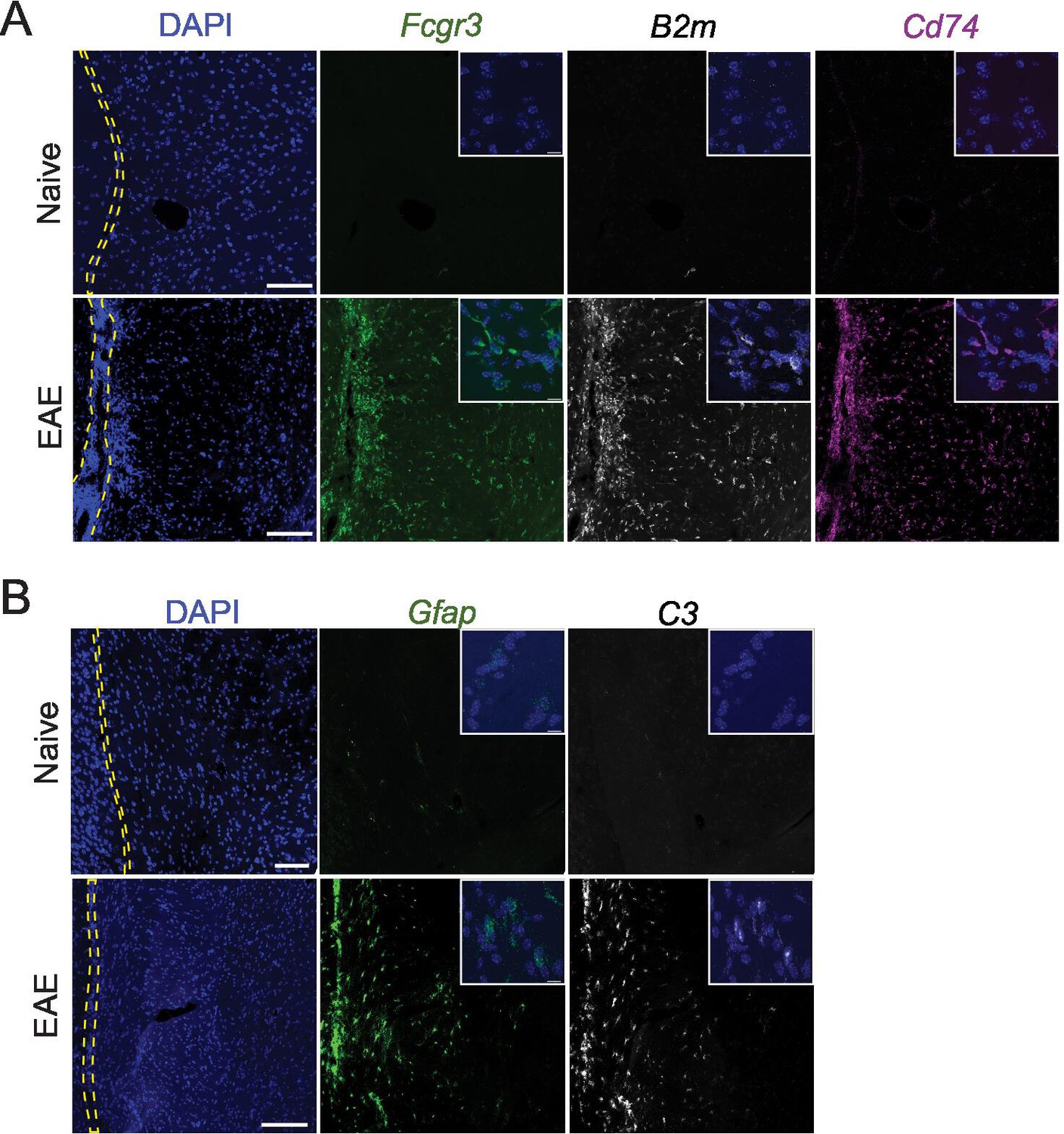
Representative images of RNAscope labeling in naïve and experimental autoimmune encephalomyelitis (EAE) tissues.
RNAscope labeling of (A) Fcgr3, B2m, Cd74, and (B) Gfap, C3. Images are representative of N=3 animals per group. Yellow dashed lines outline the subarachnoid space. Scale bars represent 100 μM (main image) and 10 μM (inset).
Additional files
-
Supplementary file 1
Results of differentially expressed gene analysis comparing experimental autoimmune encephalomyelitis (EAE) and naive brain slices.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp1-v1.csv
-
Supplementary file 2
Results of differentially expressed gene analysis comparing cluster 11 with other clusters.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp2-v1.csv
-
Supplementary file 3
Results of gene set enrichment analysis based on cluster 11 differentially expressed genes.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp3-v1.csv
-
Supplementary file 4
Results of gene set enrichment analysis based on cluster 1_3 differentially expressed genes.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp4-v1.csv
-
Supplementary file 5
Results of gene set enrichment analysis based on cluster 1_4 differentially expressed genes.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp5-v1.csv
-
Supplementary file 6
Results of gene set enrichment analysis based on cluster 2_6 differentially expressed genes.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp6-v1.csv
-
Supplementary file 7
Exponential regression analysis of RNAscope data.
- https://cdn.elifesciences.org/articles/88414/elife-88414-supp7-v1.csv
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Spatial transcriptomics of meningeal inflammation reveals inflammatory gene signatures in adjacent brain parenchyma
eLife 12:RP88414.
https://doi.org/10.7554/eLife.88414.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}