Heat Shock Factor 1 forms nuclear condensates and restructures the yeast genome before activating target genes
- Department of Biochemistry and Molecular Biology Louisiana State University Health Sciences Center, United States
eLife assessment
This is a valuable contribution to our understanding of how different cell stressors (ethanol or heat-shock) elicit unique responses at the genomic and topographical level under the regulation of yeast transcription factor Hsf1, providing solid evidence documenting the temporal coupling (or lack thereof) between Hsf1 aggregation and long-range communication among co-regulated heat-shock loci versus chromatin remodeling and transcriptional activation. A particular strength is the combination of genomic and imaging-based experimental approaches applied to genetically engineered in vivo systems.
https://doi.org/10.7554/eLife.92464.4.sa0Significance of the findings:
Valuable: Findings that have theoretical or practical implications for a subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Solid: Methods, data and analyses broadly support the claims with only minor weaknesses
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
In insects and mammals, 3D genome topology has been linked to transcriptional states yet whether this link holds for other eukaryotes is unclear. Using both ligation proximity and fluorescence microscopy assays, we show that in Saccharomyces cerevisiae, Heat Shock Response (HSR) genes dispersed across multiple chromosomes and under the control of Heat Shock Factor (Hsf1) rapidly reposition in cells exposed to acute ethanol stress and engage in concerted, Hsf1-dependent intergenic interactions. Accompanying 3D genome reconfiguration is equally rapid formation of Hsf1-containing condensates. However, in contrast to the transience of Hsf1-driven intergenic interactions that peak within 10–20 min and dissipate within 1 hr in the presence of 8.5% (v/v) ethanol, transcriptional condensates are stably maintained for hours. Moreover, under the same conditions, Pol II occupancy of HSR genes, chromatin remodeling, and RNA expression are detectable only later in the response and peak much later (>1 hr). This contrasts with the coordinate response of HSR genes to thermal stress (39°C) where Pol II occupancy, transcription, histone eviction, intergenic interactions, and formation of Hsf1 condensates are all rapid yet transient (peak within 2.5–10 min and dissipate within 1 hr). Therefore, Hsf1 forms condensates, restructures the genome and transcriptionally activates HSR genes in response to both forms of proteotoxic stress but does so with strikingly different kinetics. In cells subjected to ethanol stress, Hsf1 forms condensates and repositions target genes before transcriptionally activating them.
Introduction
Genomes of higher eukaryotes are organized into multiple hierarchical levels. Chromosomes are segregated within individual territories, and within chromosomal territories active and inactive regions are separated into topologically associated domains (TADs; Wendt and Grosveld, 2014). Within TADs, DNA loops are formed to permit the interaction between enhancers or silencers and the promoters of their target loci. Although this hierarchy suggests a static view of the genome, recent studies have suggested that the genome is quite dynamic, as multiple points of interaction form in response to developmental cues and other stimuli. This dynamic restructuring ranges from the interactions between co-regulated genes (Fanucchi et al., 2013; Papantonis et al., 2012; Park et al., 2014; Schoenfelder et al., 2010), to the convergence of enhancers dispersed across multiple chromosomes into a hub that regulates a single gene (Monahan and Lomvardas, 2015), to the reorganization of the genome that occurs in the zygote (Schulz and Harrison, 2019).
Despite its evolutionary distance, the yeast Saccharomyces cerevisiae also possesses an organized genome. Centromeres are located in a cluster at the spindle pole body, while chromosomal arms are extended with telomeres and the nucleolus located at the opposite side of the nucleus (Duan et al., 2010), resulting in a Rabl-like configuration (Taddei and Gasser, 2012). And as in higher eukaryotes, the budding yeast genome is organized into TAD-like structures (Eser et al., 2017) subdivided into smaller loop domains (Hsieh et al., 2015). Also as is the case in mammalian cells, the yeast genome is not static. Genes, including INO1, GAL1, and GAL10, have been observed to reposition from the nuclear interior to the nuclear periphery upon their activation (Brickner et al., 2019; Brickner and Walter, 2004; Cabal et al., 2006; Casolari et al., 2004; Dieppois et al., 2006; Green et al., 2012). In response to methionine starvation, several Met4-regulated genes have been observed to cluster (Du et al., 2017; Lee et al., 2024).
More dramatic still are Heat Shock Response (HSR) genes under the regulation of Heat Shock Factor 1 (Hsf1). Upon exposure to acute thermal stress (heat shock [HS]), HSR genes dispersed across multiple chromosomes transcriptionally activate and engage in novel cis- and trans-intergenic interactions between one another – principally although not exclusively involving coding regions – that culminate in their coalescence into intranuclear foci (Chowdhary et al., 2017; Chowdhary et al., 2019). These stress-induced foci, comprised of Hsf1 and components of the transcriptional machinery, form quickly, rearrange rapidly and dissolve suddenly (Chowdhary et al., 2022). As such, Hsf1 clusters exhibit properties of biomolecular condensates, defined as self-organized membrane-free compartments enriched in specific macromolecules that may or may not be phase-separated (Banani et al., 2017). Hsf1 condensate formation elicited by heat shock, like that of HSR gene interactions, parallels the kinetics of induction and attenuation of Hsf1-dependent genes (Chowdhary et al., 2017; Chowdhary et al., 2022).
The Hsf1-driven heat shock response is a fundamental, evolutionarily conserved transcriptional program characterized by the gene-specific transcription factor (TF) Hsf1, its DNA recognition element (heat shock element [HSE]) and a core set of target genes encoding molecular chaperones and co-chaperones (reviewed in Verghese et al., 2012). In absence of proteotoxic stress, yeast Hsf1 is bound by Hsp70 and its co-chaperone Sis1 in the nucleoplasm (Feder et al., 2021; Krakowiak et al., 2018; Peffer et al., 2019; Zheng et al., 2016). Upon encountering stress, Hsp70 is titrated by unfolded proteins, particularly orphan ribosomal proteins located in the nucleolus and nascent polypeptides in the cytosol (Albert et al., 2019; Ali et al., 2023; Tye et al., 2019; Tye and Churchman, 2021). This results in the release of Hsf1 which then trimerizes and binds to HSEs located upstream of ~50 genes whose transactivation is dependent on this factor (Pincus et al., 2018). Once proteostasis is reestablished, excess Hsp70 binds Hsf1, inactivating it, thereby closing the negative feedback loop that regulates Hsf1 transcriptional activity (Krakowiak et al., 2018; Zheng et al., 2016). Note that in yeast, a fraction of Hsf1 is constitutively trimeric and it is this species that binds high-affinity HSEs even under non-stressful conditions (Giardina and Lis, 1995; Pincus et al., 2018).
In addition to thermal stress, Hsf1 can be activated by chemical stressors such as ethanol. Ethanol is a metabolite of glucose breakdown that budding yeast cells secrete into their surroundings. Ethanol production helps yeast outcompete other microbes in the environment (Liti, 2015; Piskur et al., 2006; Rozpędowska et al., 2011). Furthermore, once glucose is depleted, ethanol serves as an alternative carbon source (Piskur et al., 2006). Given this strategic use of ethanol, it is of fundamental importance for yeast to have a mechanism in place to respond to the stress that ethanol elicits. Similar to thermal stress, exposure to ethanol causes a large number of cellular perturbations including disruption of the plasma membrane (Piper et al., 1994); disruption of the H+ ATPase and intracellular acidification (Rosa and Sá-Correia, 1991; Rosa and Sá-Correia, 1996; Triandafillou et al., 2020); production of reactive oxygen species (Bandas and Zakharov, 1980; Davidson et al., 1996); depolymerization of the actin cytoskeleton (Homoto and Izawa, 2018; Tan et al., 2017); cell cycle arrest (Johnston and Singer, 1980; Kubota et al., 2004); disruption of mRNP transport to the daughter cell and formation of stress granules (Grousl et al., 2009; Kato et al., 2011); global inhibition of transcription and translation (Bresson et al., 2020; Gasch et al., 2000); and formation of protein aggregates (Piper, 1995; Plesset et al., 1982; Stanley et al., 2010). The cell counteracts many of these perturbations through the enhanced production of molecular chaperones (Desroches Altamirano et al., 2024; Verghese et al., 2012).
Here, we investigate activation of the HSR in yeast exposed to 8.5% ethanol (ethanol stress [ES]) and compare it to the response induced by exposure to 39°C (HS). We find that like HS, exposure to ES induces transcription of Hsf1-regulated HSR genes and induces concerted intergenic interactions between them. In contrast to HS, however, Hsf1 condensate formation and HSR intergenic interactions cells peak well before transcription. The delay in transcriptional induction may be linked to a widespread increase in nucleosome occupancy that occurs upon exposure of cells to ethanol. At longer times of ethanol exposure, HSR gene transcript accumulation continues to increase while HSR gene coalescence has already dissipated. Likewise, ES-induced Hsf1 condensates are present for a prolonged period, in contrast to HS-induced condensates that begin to dissipate shortly after they appear. Collectively, our data indicate that different stimuli drive distinct transcription, chromatin, topologic and condensation phenomena, yet all are dependent on Hsf1.
Results
Thermal stress and ethanol stress elicit distinct proteotoxic responses in yeast
Saccharomyces cerevisiae in the wild metabolizes glucose and other sugars into ethanol, which the yeast secretes into the environment to suppress microbial competition. Therefore, it is likely that yeast has evolved mechanisms to contend with ethanol toxicity. Indeed, a common laboratory strain (W303) retains viability when cultivated in the presence of 8.5% ethanol, although its ability to proliferate is diminished (Figure 1A and B). As assessed by the presence of Hsp104-containing foci (a measure of protein aggregation; Liu et al., 2010), the rate of cytosolic protein aggregation is similar in cells exposed to either 8.5% ethanol or 39°C thermal stress (Figure 1D and E; see Figure 1C for experimental design). However, it is notable that the number and volume of Hsp104 foci, and by extension protein aggregation, is substantially higher in cells subjected to ethanol stress.
Figure 1
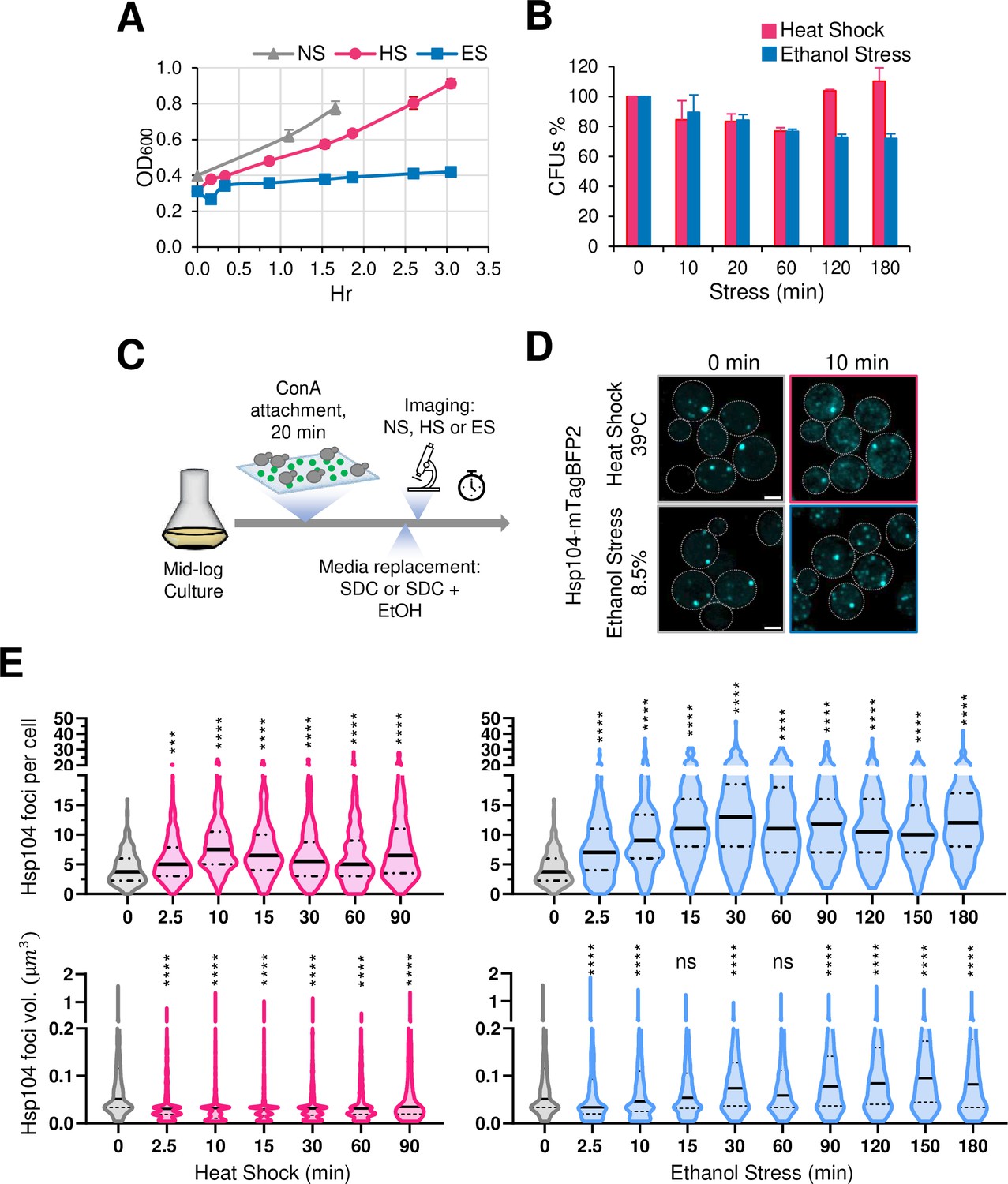
Thermal and chemical stresses used in this study elicit distinct proteotoxic responses.
(A) Growth curve of strain W303-1B grown in liquid culture (YPDA). Mid-log phase cultures were diluted to OD600=0.4 and shifted to different conditions: no stress (NS, 25°C), heat shock (HS, 39°C), or ethanol stress (ES, 8.5% v/v, 25°C). OD600 was monitored over time. Means and SD are shown. N=2. (B) Viability assay of W303-1B cells following exposure to heat shock (25° to 39°C upshift for the indicated time) or ethanol stress (8.5% v/v ethanol for the indicated time at 25°C). An aliquot was taken from each condition at the indicated stress timepoints and diluted in rich media. Cells were spread on YPDA plates and grown at 30°C for 3 days. Colony forming units (CFUs) were determined using ImageJ/FIJI. Plotted are percentages of CFUs of stressed cells normalized to those of the 0 min control. Graphs depict means + SD. N=2. (C) Experimental strategy for imaging Hsp104 foci. Cells were attached to a concanavalin A (ConA)-coated surface, followed by heat shock or ethanol stress treatment (see Materials and Methods). Synthetic complete media (SDC) was supplemented with ethanol to a final concentration of 8.5% for ES samples. Scale bar: 2 µm. (D) Both heat shock and ethanol stress induce formation of Hsp104 foci. DPY1561 haploid cells were attached to a VAHEAT substrate using Concanavalin A and subjected to an instantaneous heat shock (25° to 39°C) or to ethanol stress (25°C, 8.5% v/v). The 0 min control was kept at 25°C without stress. Hsp104-mTagBFP2 foci were visualized by confocal microscopy. Shown are maximal projections of 11 z-planes, taken with an interplanar distance of 0.5 µm. Scale bar: 2 µm. (E) Cells subjected to the above treatments were assayed for Hsp104 puncta number and volume. Violin plots summarizing this analysis are depicted. An average of 200 cells per timepoint per condition was quantified using Imaris image analysis software (v.10.0.1). For this analysis, we made the assumption that the diffuse Hsp104 clusters seen in HS cells are comparable to the compact Hsp104 foci in ES cells. N=2. Significance was determined by Mann Whitney test, stress vs. no stress (0 min). ***, p<0.001; ****, p<0.0001; ns, not significant.
-
Figure 1—source data 1
Spreadsheet tabulates the number of Hsp104 foci per cell and the volume of individual Hsp104 foci (Figure 1E).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig1-data1-v1.xlsx
We next investigated the subcellular localization of the Hsp70 co-chaperone, Sis1, in cells exposed to ethanol versus thermal stress. Previous work has shown that Sis1 is diffusely localized within the nucleus under control conditions where it promotes binding of Hsp70 to Hsf1, thereby repressing the HSR (Feder et al., 2021). In response to acute HS, Sis1 relocates to nucleolus where it forms a perinucleolar ring, and to the cytoplasm where it forms cytosolic clusters that colocalize with Hsp104 and spatially associate with the endoplasmic reticulum (Feder et al., 2021). Consistent with these previous observations, we observed rapid relocalization of Sis1 in response to a 39°C HS. It formed a ring-like structure within 2.5 min consistent with a perinuclear location. Such relocalization lasted at least 60 min and spatially separated Sis1 from Hsf1 (Figure 2A and B). A similar result was obtained when cells were exposed to 42°C. In response to ES, Sis1 remained largely co-localized with Hsf1 within the nucleoplasm although some enrichment at the nuclear periphery was evident. This was the case whether cells were exposed to 5% or 8.5% ethanol (Figure 2A and B). In addition, a 60 min exposure to 8.5% ethanol resulted in the formation of prominent cytosolic Sis1 puncta that were less evident in the HS sample (Figure 2A). These observations suggest that these two stressful treatments elicit a qualitatively distinct response. Experiments described below provide further support for this possibility.
Figure 2
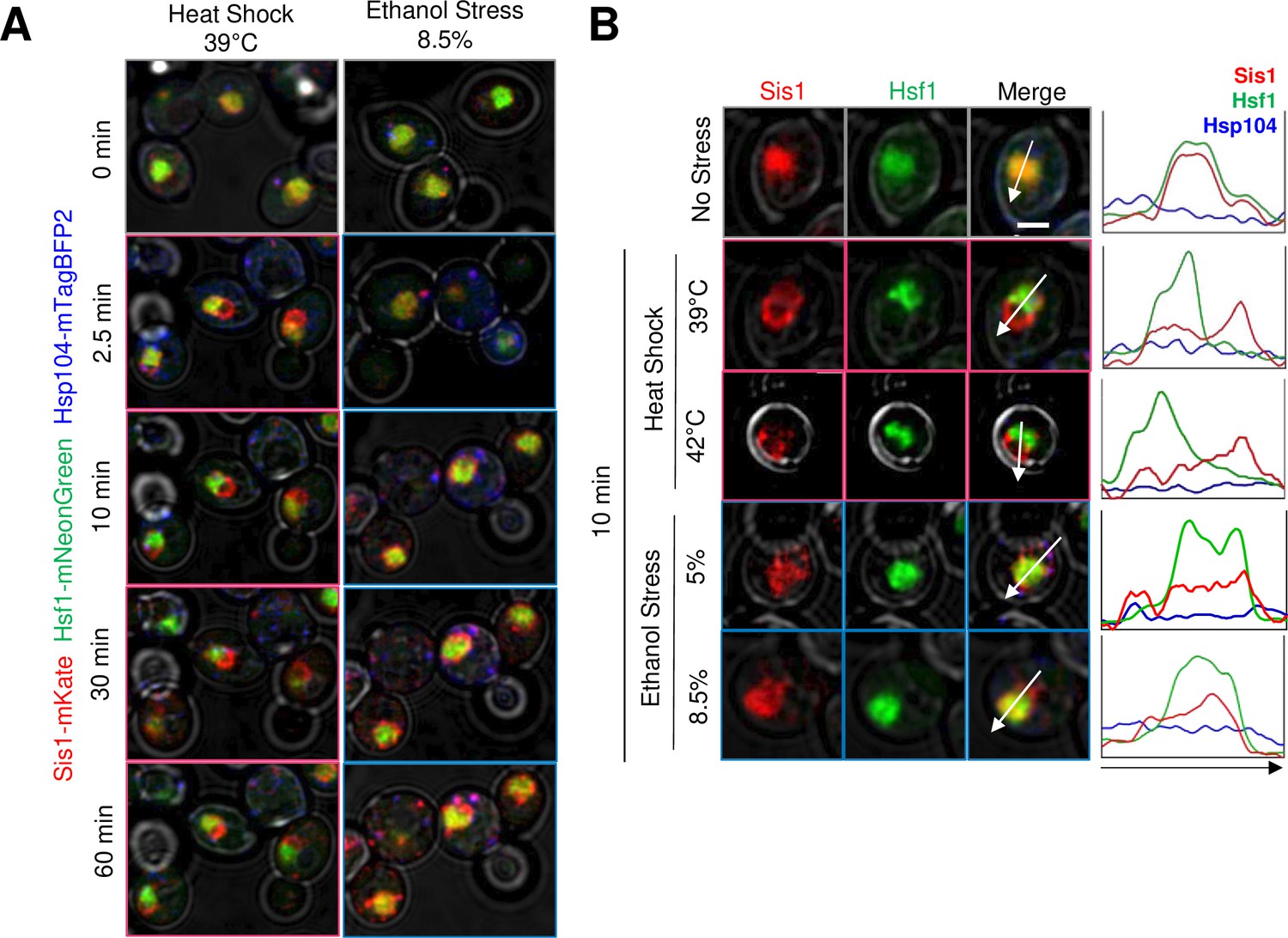
Heat shock and ethanol stress elicit distinct patterns of Sis1 subcellular relocalization.
(A) Live cell confocal microscopy of the diploid strain LRY033 expressing Sis1-mKate, Hsf1-mNeonGreen, and Hsp104-mTagBFP2. Cells were treated as in Figure 1D. 11 z-planes were captured with an interplanar distance of 0.5 µm. Shown is a representative plane for each timepoint. (B) Subcellular localization analysis of Sis1, Hsf1, and Hsp104 in cells subjected to no stress (25°C), heat shock (at 39° or 42°C), or ethanol stress (at 5% or 8.5% v/v [25°C]) for 10 min. Cells from strain LRY033 were treated as described in Figure 1D. A representative plane is shown for each condition. Line profiles are plotted for each channel on the right. Arrows were drawn to bisect the nucleus. Scale bar: 2 µm.
Ethanol stress induces transcriptional activation of Hsf1-dependent genes but with delayed kinetics and reduced expression compared to thermal stress
To gain further insight into the mechanism by which S. cerevisiae contends with ethanol-induced proteotoxicity, we assessed the kinetics of transcriptional activation of HSR genes in cells exposed to ethanol stress. Cells were cultivated to early log phase in rich YPD medium, then ethanol was added to a final concentration of 8.5% and cell aliquots were removed at 0-, 10-, 20-, and 60 min. Transcription was terminated through addition of sodium azide (see Materials and methods). A parallel culture was exposed to an instantaneous 30° to 39°C heat shock and cells were removed at the corresponding time points. Transcription was terminated as above.
While cells exposed to heat shock displayed a rapid and substantial increase in HSR mRNA levels (typically >10 fold increase within 10 min of thermal upshift), those exposed to ethanol stress only weakly induced the same cohort of genes (Figure 3A). However, while HS induced a transient increase in RNA expression, ES induced a sustained increase that was evident at all Hsf1-dependent genes tested (see also Figure 3—figure supplements 1 and 2). A corresponding immunoblot analysis of two Hsf1 targets, Hsp104 and Btn2, strengthens the notion that HS induces a rapid yet transient response, whereas ES induces a delayed, yet far more sustained increase in gene expression (Figure 3—figure supplement 3). In the case of HSP12 and HSP26, whose transcription is under the dual regulation of Msn2 and Hsf1, exposure to heat shock resulted in a high level of induction as previously observed (Chowdhary et al., 2019) yet exposure to 8.5% ethanol failed to cause detectable activation during the first 20 min (Figure 3B; Figure 3—figure supplement 1). Nonetheless, as described below, both HSP12 and HSP26 respond to acute ethanol stress but do so through their inducible and dramatic 3D genomic repositioning.
Figure 3 with 3 supplements see all
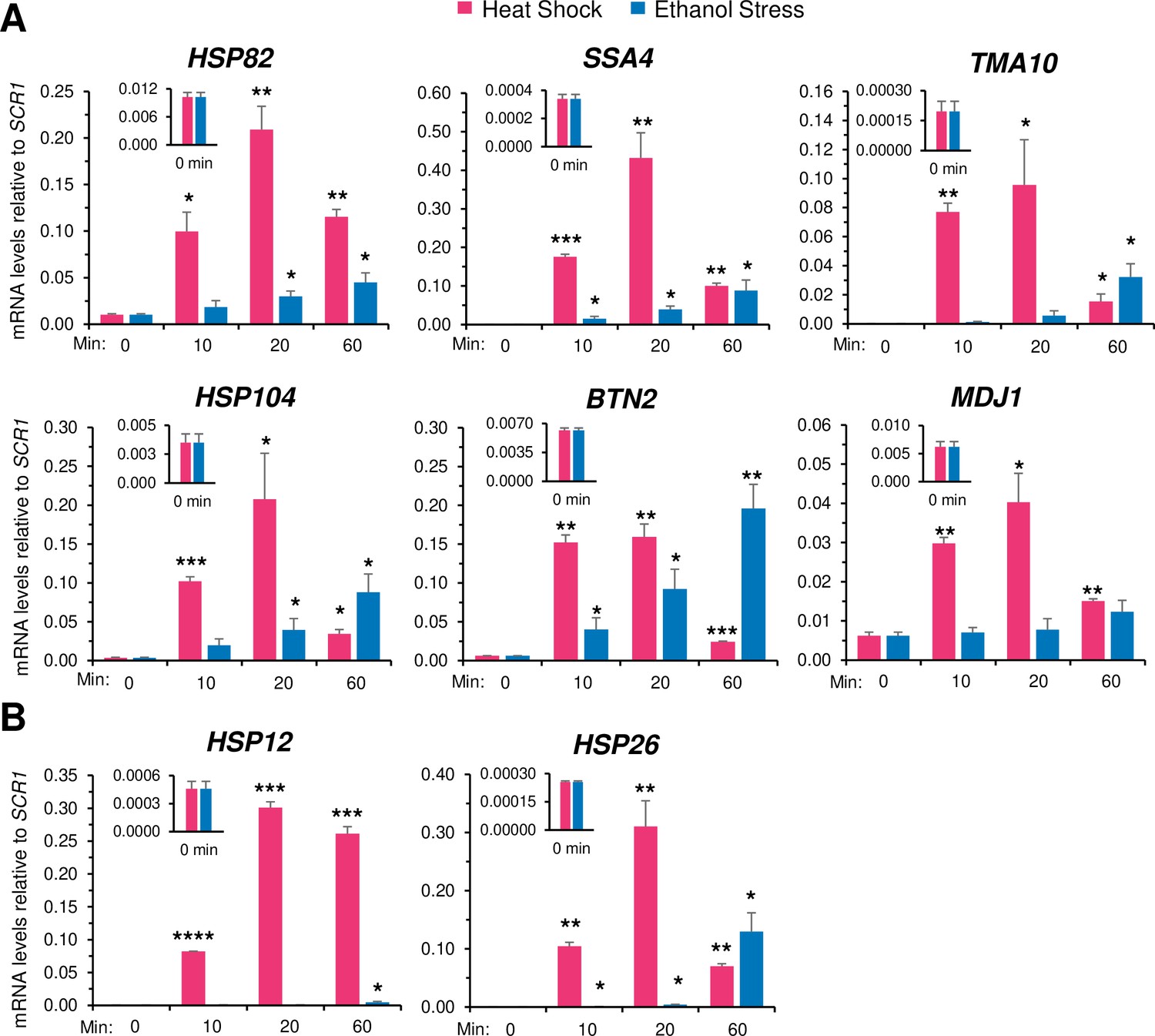
Ethanol stress transcriptionally induces Heat Shock Response (HSR) genes but with markedly slower kinetics than thermal stress.
(A) RNA abundance of Hsf1-dependent HSR genes was determined by Reverse Transcription-qPCR in strain W303-1B. Heat shock was performed at 39°C; ethanol stress was done using 8.5% (v/v) ethanol at 25°C. Insets display transcript abundance using a zoomed-in scale. Depicted are means + SD. N=2, qPCR = 4. Statistical analysis: T-test, one-tailed, no stress vs. stress conditions. *, p<0.05; **, p<0.01, ***; p<0.001. (B) As in (A), but the Hsf1-, Msn2-dual regulated genes HSP12 and HSP26 were evaluated. *, p<0.05; ***, p<0.001; ****, p<0.0001.
-
Figure 3—source data 1
Spreadsheet contains the RT-qPCR data plotted for HSR RNA analysis (Figure 3).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig3-data1-v1.xlsx
Pol II recruitment and histone eviction are delayed in ethanol-stressed cells and this correlates with a transient, widespread increase in nucleosome density
The delayed transcriptional response of HSR genes in ES- versus HS-treated cells prompted us to investigate occupancy of Hsf1, RNA Pol II and histones at these genes over a time course. A possible explanation for the delay in activation in cells exposed to ethanol stress is less rapid (or reduced) Hsf1 binding to the genes’ upstream regulatory regions. To explore this possibility, we exposed cells to either thermal or chemical stress and processed them for chromatin immunoprecipitation (ChIP) analysis. As previously observed (Kim and Gross, 2013; Pincus et al., 2018; Sekinger and Gross, 2001), occupancy of Hsf1 at its target loci increases many-fold following a brief heat shock (Figure 4B, left, dark red; see Figure 4A for location of primers). Hsf1 occupancy typically declines after 60 min of continuous thermal stress and in the case of TMA10, dissociation begins much sooner. In response to ethanol stress, Hsf1 occupancy steadily increased, in most cases reaching maximal levels by 20 min and plateauing thereafter (Figure 4B, left, black). These results suggest that ethanol stress induces binding of Hsf1 to its cognate HSEs to a degree similar to heat shock, yet such binding is more gradual. Moreover, given the above, Hsf1’s binding to these sites fails to elicit a corresponding transcriptional response.
Figure 4 with 2 supplements see all
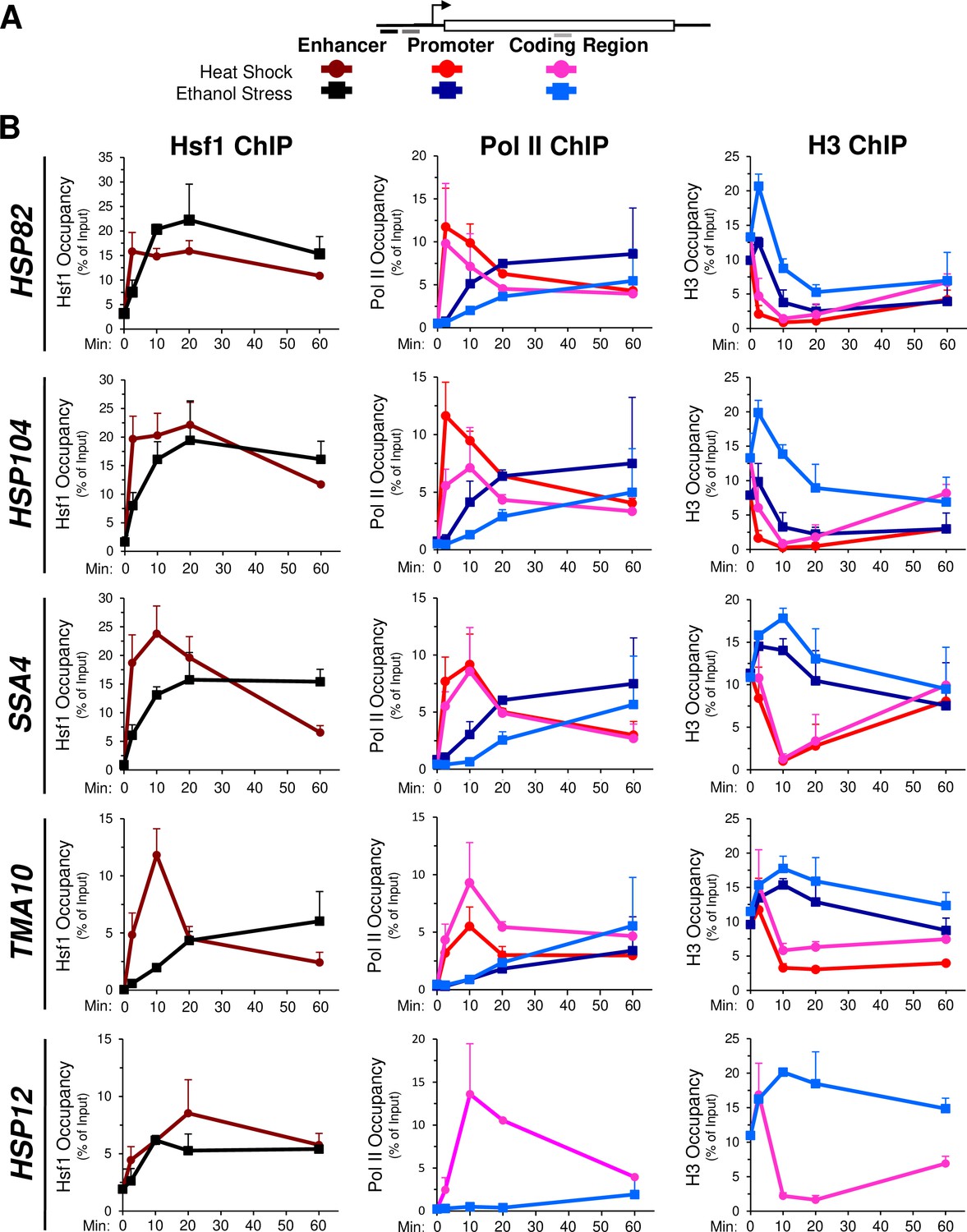
Hsf1 and Pol II recruitment to HSR genes is delayed in ethanol stressed cells, while histone occupancy transiently increases.
(A) Map of a representative HSR gene depicting locations of primers used for chromatin immunoprecipitation (ChIP) analysis. Heat shock: shades of red and pink. Ethanol stress: shades of blue. (B) ChIP analysis of Hsf1, Pol II (Rpb1) and histone H3 occupancy to the enhancer (UAS), promoter and coding regions of the indicated genes. Mid-log cultures of strain BY4741 were subjected to the indicated times of heat shock (39°C) or ethanol stress (8.5% v/v, 25°C). Time points evaluated for all three factors: 0-, 2.5-, 10-, 20-, and 60 min. Antibodies raised against full-length Hsf1, CTD of Rbp1 or the globular domain of histone H3 were used (see Materials and methods). ChIP signals were normalized to input. Shown are means + SD. N=2, qPCR = 4.
-
Figure 4—source data 1
Spreadsheet tabulates Hsf1, Pol II, and histone H3 occupancy.
Chromatin immunoprecipitation (ChIP) data for HSR genes in cells exposed to heat shock or ethanol stress (Figure 4B).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig4-data1-v1.xlsx
In light of this disconnect between Hsf1 binding and HSR mRNA production, we evaluated abundance of the large subunit of Pol II at representative genes over the same time course. In response to heat shock, Rpb1 is rapidly recruited to the promoters and coding regions of each HSR gene, peaking within 2.5 min and then gradually declining over the next ~60 min (Figure 4B, middle, red and pink traces). By contrast, its occupancy is noticeably delayed in cells exposed to ethanol stress (blue traces), consistent with reduced transcript levels (Figure 4—figure supplement 1B). The increase in HSR mRNA in heat-shocked cells parallels, yet consistently lags, the abundance of Pol II within HSR gene coding regions (Figure 4—figure supplement 1A). This delay in reaching peak accumulation may reflect contributions beyond RNA synthesis, such as transient enhanced stability of HSR transcripts during the acute phase of heat shock.
To obtain further insight into the chromatin landscape present during the two stresses, we assayed histone H3 abundance as a measure of nucleosome density. In response to heat shock, nucleosomes are rapidly displaced over the promoter and coding regions of the strongly expressed HSR genes. Following this initial phase (~20 min), nucleosomes reassemble over these genes and in certain cases return to their original density by 60 min (Figure 4B, right, red and pink traces). This dramatic and dynamic remodeling has been previously observed (Kremer and Gross, 2009; Zhang et al., 2014; Zhao et al., 2005). In contrast, ethanol stress elicits a transient increase in H3 abundance over all five genes (Figure 4B, right, blue traces). This apparent increase in chromatin compaction is not restricted to HSR genes; a variety of unrelated loci, including a stress-responsive, Msn2-regulated gene (PGM2), two constitutively expressed genes (TUB1, ACT1), two genes assembled into SIR-dependent heterochromatin (HMLα1, YFR057w) and a non-transcribed region (ARS504) also exhibit a transient increase in nucleosome density in ethanol-exposed cells (Figure 4—figure supplement 2A).
In agreement with this idea, chromatin volume (as assessed by the signal arising from an H2A-mCherry protein fusion) decreases upon exposure of cells to ethanol stress (and much more transiently to heat stress; Figure 4—figure supplement 2[B, C]). These data suggest that the increase in nucleosome density antagonizes Pol II recruitment and its subsequent release into the coding regions of HSR genes in ES cells. Once Pol II has been stably recruited, elongation and concomitant histone eviction ensues (Figure 4B, light blue). Altogether, our ChIP data indicate that Hsf1 binds to its target enhancers less readily in ethanol-stressed than in thermally stressed cells. This impediment to Hsf1 occupancy is magnified by a corresponding, and more severe, hindrance to Pol II recruitment resulting in a pronounced delay in HSR gene transcription.
Acute ethanol stress induces rapid and profound 3D genomic repositioning of HSR loci
An intriguing feature of HSR genes is the fact that they coalesce into discrete intranuclear foci in response to heat shock. Such interactions have been documented using both molecular (Chromosome Conformation Capture [3C]) and imaging (fluorescence microscopy) approaches (Chowdhary et al., 2017; Chowdhary et al., 2019; Chowdhary et al., 2022; Rubio and Gross, 2023). Moreover, such physical interactions specifically involve Hsf1 targets irrespective of their location in the genome. Other loci, including adjacent, transcriptionally active genes, show little or no tendency to interact with Hsf1-dependent genes. Such cis- and trans-interactions principally involve gene coding regions and are highly dynamic, typically peaking at 2.5 min and dissipating by 30- to 60 min. The kinetics of HS-induced coalescence often, although not always, correlate with kinetics of transcriptional induction; they also parallel the formation of Hsf1 condensates (Chowdhary et al., 2017; Chowdhary et al., 2019; Chowdhary et al., 2022) as discussed further below.
Given these previous observations, we wished to know if ethanol stress induced similar 3D genome restructuring. It seemed unlikely that such topological changes would occur during the initial phase of ES since only weak Pol II occupancy and low HSR transcript levels are observed (Figures 3 and 4). However, as shown in Figure 5, exposure to 8.5% ethanol triggered frequent intergenic interactions between Hsf1 targets during the first 10 min as revealed by Taq I-3C, a highly sensitive, quantitative version of 3C (Chowdhary et al., 2020) (see Figure 5—figure supplement 1 for location of 3C primers). Both intra- and interchromosomal interactions can be detected. Moreover, the interaction frequencies following this exposure in most cases equaled, and in some instances exceeded, those detected in cells heat-shocked for 2.5 min (Figure 5A and B), when peak 3C interactions occur in thermally stressed cells (Chowdhary et al., 2017). A detailed kinetic analysis revealed that intergenic interactions elicited by ethanol stress, similar to those induced by thermal stress, are highly dynamic: detectable within 2.5 min, peak shortly thereafter (within 10–20 min) and largely attenuate by 60 min (Figures 5 and 6A).
Figure 5 with 2 supplements see all
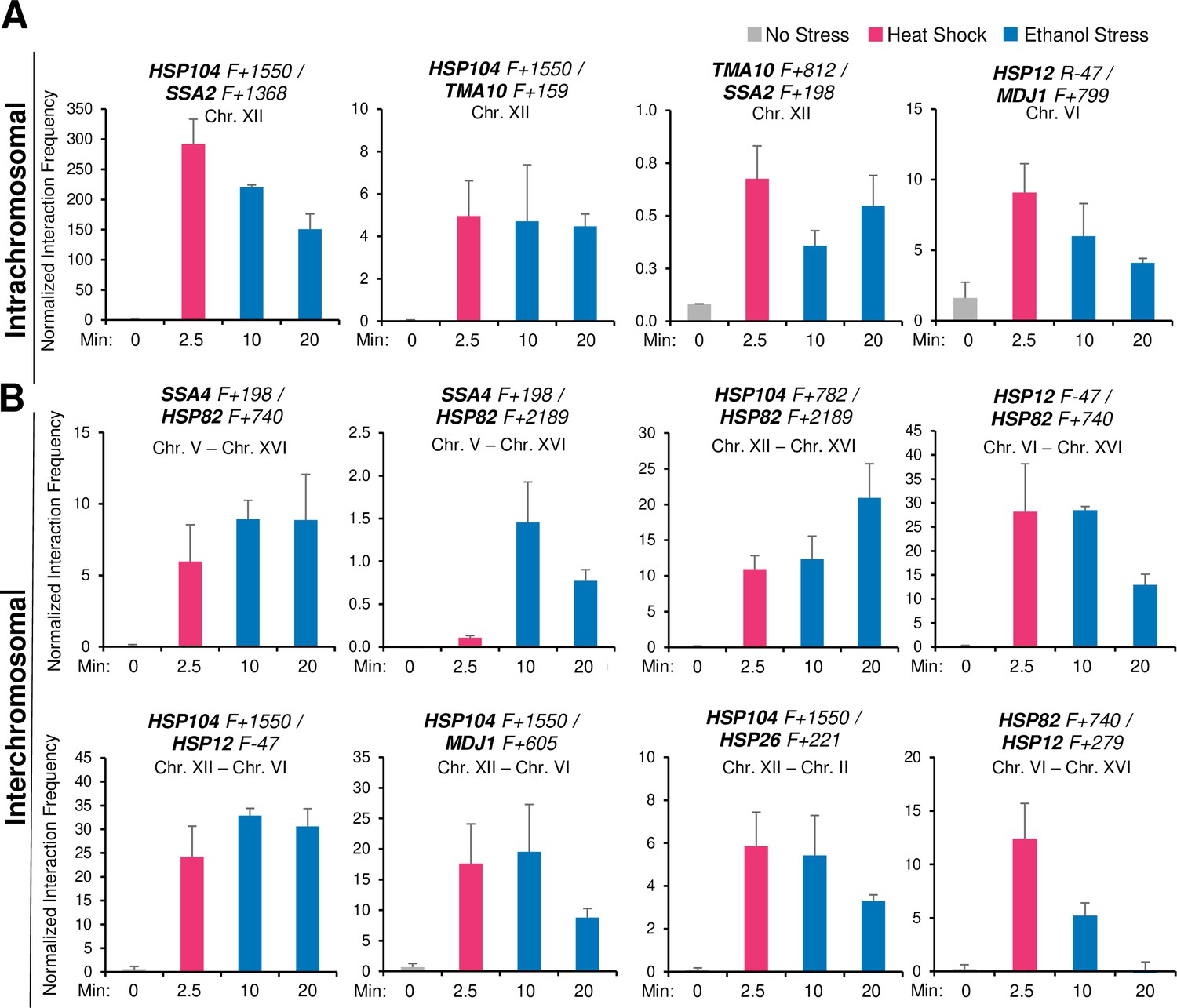
Ethanol stress induces intergenic interactions between HSR genes that are comparable to those induced by acute thermal stress.
(A) Intrachromosomal (cis) interactions between HSR genes were analyzed by Taq I-3C. W303-1B cells were instantaneously shifted from 30° to 39°C for 2.5 min (HS) or exposed to 8.5% v/v ethanol at 30°C for 10 or 20 min (ES). No stress samples were kept at 30°C. Location of Taq I coordinates are provided in Figure 5—figure supplement 1. F (forward) primers are positioned near the indicated Taq I restriction site. 3C signals were normalized to the 3C signal derived from using a naked genomic DNA template. Graphs depict means + SD; N=2; qPCR=4. (B) Interchromosomal (trans) interactions between HSR genes were detected as in (A).
-
Figure 5—source data 1
Spreadsheet tabulates 3C data of cells under no stress, 2.5 min heat shock, and 10- or 20 min ethanol stress.Intrachromosomal and interchromosomal interactions are analyzed (Figure 5A and B).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig5-data1-v1.xlsx
Figure 6
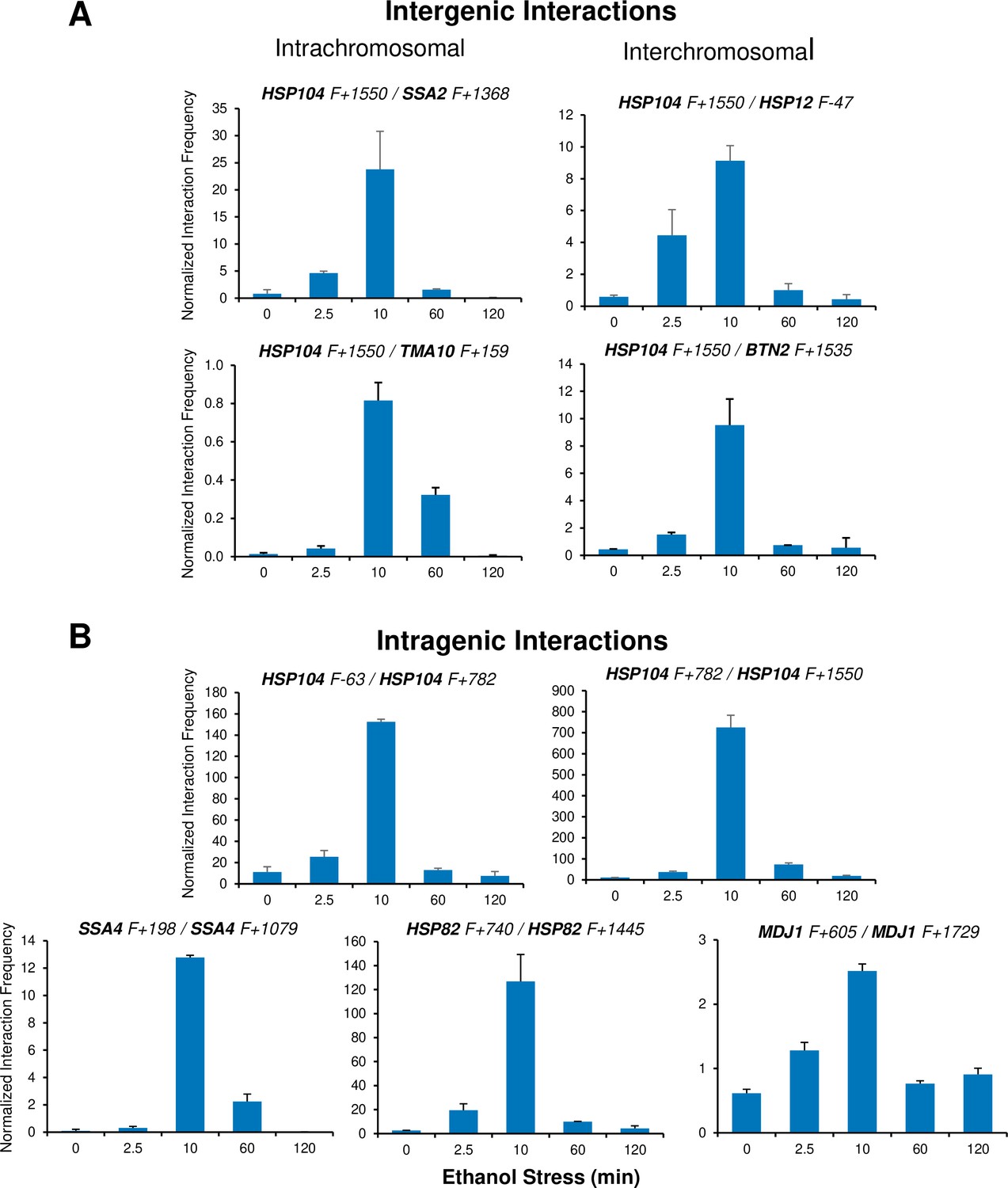
Ethanol-induced HSR gene interactions are detectable by 2.5 min but typically dissipate within 60 min.
(A) Taq I-3C analysis of intergenic interactions occurring during ethanol stress was conducted as described in Figure 5. All samples were kept at 25°C. Plotted are means + SD. N=2, qPCR=4. (B) As in (A), but for intragenic interactions.
-
Figure 6—source data 1
Spreadsheet tabulates 3C data of cells subjected to chronic ethanol stress (Figure 6).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig6-data1-v1.xlsx
It has been previously suggested that a functional link exists between gene looping and transcriptional activation (Ansari and Hampsey, 2005; O’Sullivan et al., 2004). Indeed, gene loops and other intragenic interactions (Chowdhary et al., 2017) are readily detected within HSR genes in cells exposed to ethanol. However, as is the case with intergenic interactions, these topological changes are kinetically uncoupled from both transcription and Pol II occupancy. They are detected quite early (within 2.5 min), peak soon thereafter (at 10 min) and attenuate by 60 min (Figure 6B and Figure 5—figure supplement 2). Taken together, our 3C and expression analyses indicate that 3D genomic repositioning and intragenic looping of HSR genes precedes the maxima of transcript levels and Pol II occupancy. Moreover, for certain loci (e.g. HSP12 and HSP26), they argue that neither transcription nor Pol II recruitment is required to drive 3D topological changes in these genes. Further evidence in support of this is provided below.
Live cell imaging reveals that HSR genes coalesce to a similar degree under ethanol stress and heat stress conditions
To provide an orthogonal line of evidence for HSR gene interaction, we employed fluorescence microscopy to image live cells bearing LacO-tagged HSP104 and TetO-tagged TMA10 loci in cells expressing GFP-LacI and TetR-mCherry fusion proteins. Both genes are located on Chromosome XII, on opposite arms, and are physically separated by the nucleolus (100–200 rDNA repeats) that lies between them (Duan et al., 2010) (schematically depicted in Figure 7—figure supplement 1A). In the absence of stress, fluorescence signals representing these two genes are typically well-separated (Figure 7—figure supplement 1B, 0 min). Upon heat shock, they rapidly converge, usually within 2.5 min. Upon exposure to ethanol, gene convergence is also observed, albeit less rapidly (Figure 7—figure supplement 1B; see also below). Despite the slight delay, these results demonstrate that HSP104 and TMA10 coalesce in ethanol stressed cells and do so with similar frequency as in thermally stressed cells (Figure 7—figure supplement 1C), consistent with the 3C analysis described above.
Having confirmed the physical interaction of Hsf1-dependent genes under ethanol stress, we assessed the transcriptional status of coalesced genes. Our RT-qPCR analysis indicated that the increase in HSR mRNA levels in ES-induced cells is delayed compared to HS-induced cells (Figure 3). To obtain insight into HSR gene transcription kinetics in single cells, we integrated a stem loop array (24xMS2) upstream of HSP104, allowing production of a chimeric transcript visualized upon binding of the MCP-mCherry fusion protein (Haim et al., 2007). This strain also harbored LacO-tagged HSP104 and HSP12 genes and expressed GFP-LacI (schematically illustrated in Figure 7A). We were unable to detect an MCP-mCherry focus adjacent to HSP104 under no stress conditions (Figure 7B; 0 min), consistent with very low HSP104 basal transcript levels (Figure 3A). Heat shock induced rapid coalescence between HSP104 and HSP12, as well as transcription from HSP104. These phenomena were detectable by 2.5 min as a merged signal of the chimeric transcript and the two GFP-labeled genes (Figure 7B). This visualization method allowed us to quantify the percentage of the population that is actively engaged in transcription, revealing that during heat shock, transcription, and coalescence are positively correlated (Figure 7C and F).
Figure 7 with 1 supplement see all
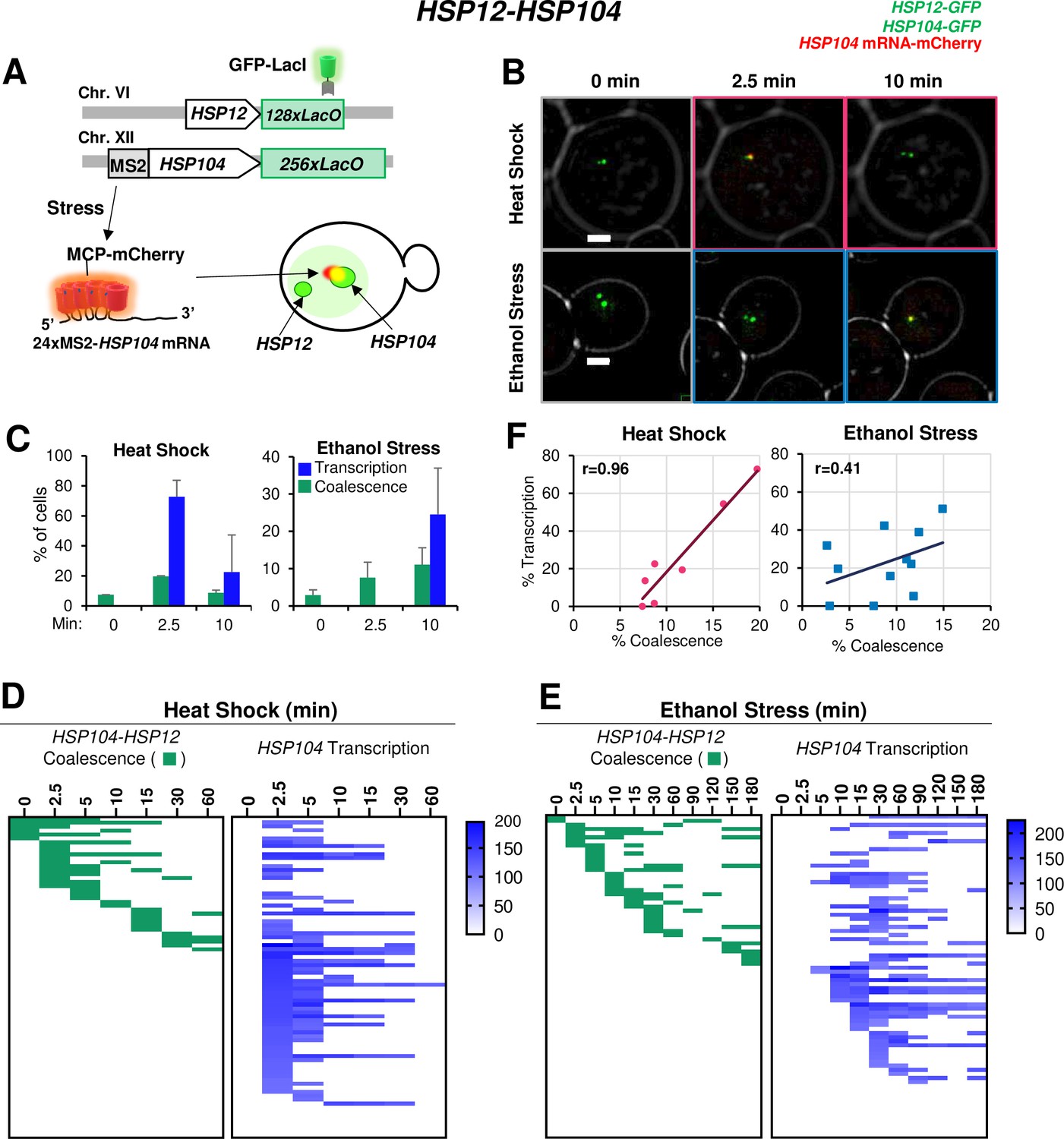
HSR gene transcription and coalescence are strongly correlated in heat-shocked but not ethanol-stressed cells.
(A) HSP12 and HSP104 are flanked by LacO arrays in the heterozygous diploid strain VPY705. In addition, HSP104 has a 24xMS2 loop array integrated within its 5’-UTR. MCP-mCherry binds to the nascent chimeric HSP104 transcript and is visualized as a red dot adjacent to the gene which appears as a green dot. (B) Live cell confocal fluorescence microscopy of strain VPY705 heat-shocked at 39°C using a VAHEAT device or exposed to 8.5% ethanol at 25°C for the indicated times. An Olympus spinning disk confocal microscope system was used for imaging. Scale bar: 2 µm. (C) Quantification of VPY705 cells treated as above and scored for the coalescence of HSP104-HSP12 and the presence of chimeric MS2x24-HSP104 mRNA. Cells were scored positive for coalescence only when a single green dot could be visualized in the nucleus across the 11 z-planes. Transcription was scored as positive only when a red dot above background could be seen near the large green dot (HSP104). Approximately 40 cells were scored per timepoint, per condition. Graphs represent means + SD. N=2. (D) Single cell analysis of HSP12-HSP104 coalescence (green) and HSP104 transcription (blue) at discrete timepoints over a heat shock time course. Each row in the transcription analysis corresponds to the same cell in the coalescence analysis. Blue gradient represents the intensity of mCherry signal (HSP104 transcript) in each cell, as quantified by ImageJ/Fiji (v. 1.54f). (E) As in (D) but for ethanol stress. (F) Pearson correlation coefficient analysis showing the correlation (r) between percent of cells positive for transcription and percent of cells positive for coalescence under HS and ES conditions (derived from Figure 7—figure supplement 1D). Each plotted value corresponds to a different stress timepoint.
-
Figure 7—source data 1
Spreadsheet tabulates the percentage of cells with HSP104-HSP12 coalescence and HSP104 mRNA foci upon ES or HS treatment (Figure 7C and F).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig7-data1-v1.xlsx
A detailed live cell analysis supports the strong spatiotemporal correlation between HSR gene coalescence and transcription in heat-shocked cells (Figure 7D). In contrast, under ethanol stress, HSP104 RNA was not detected in most cases until 10 min even though HSP12 and HSP104 coalesced as early as 2.5 min (Figure 7E; Figure 7—figure supplement 1D). Indeed, such an analysis suggests temporal uncoupling between HSR gene coalescence and HSR gene transcription in ethanol stressed cells (Figure 7F). Underscoring the disconnect between 3D genome repositioning and transcription is the fact that in ethanol stressed cells, enhanced HSP12 mRNA levels were undetectable until 60 min (Figure 3B and Figure 3—figure supplement 1). Collectively, our RT-qPCR, 3C and imaging data argue that ethanol stress induces striking topological changes in HSR genes within the first 10–20 min, yet these are accompanied by a minimal increase in transcript levels. This provides a sharp contrast to heat-shocked cells where there exists a strong temporal correlation between HSR gene transcription and HSR gene repositioning (Figure 7F).
Ethanol stress induces formation of long-lived Hsf1 condensates
Recently, TF condensates have been proposed as a mechanism for transcriptional regulation (Boija et al., 2018; Cho et al., 2018; Hnisz et al., 2017; Nair et al., 2019; Sabari et al., 2018). Heat shock-activated Hsf1 forms small nuclear condensates (diameter of ≤300 nm) that localize at HSR gene loci in both human (Zhang et al., 2022) and budding yeast cells (Chowdhary et al., 2022), and their presence positively correlates with HSR gene transcriptional activity. The tendency of Hsf1 to form condensates may be linked to its extensive intrinsically disordered structure (Figure 8A), a feature proposed to be critical for the biomolecular condensation of proteins (reviewed in Alberti et al., 2019; Banani et al., 2017). Additionally, nuclear stress bodies comprised of HSF1 bound to satellite III DNA repeats in human cells have been described (Jolly et al., 1997; Jolly et al., 2002), although formation of these large foci (diameter of several µm) is independent of the sites of active Heat Shock Response genes and their presence may repress HSR gene activation (Gaglia et al., 2020).
Figure 8 with 1 supplement see all
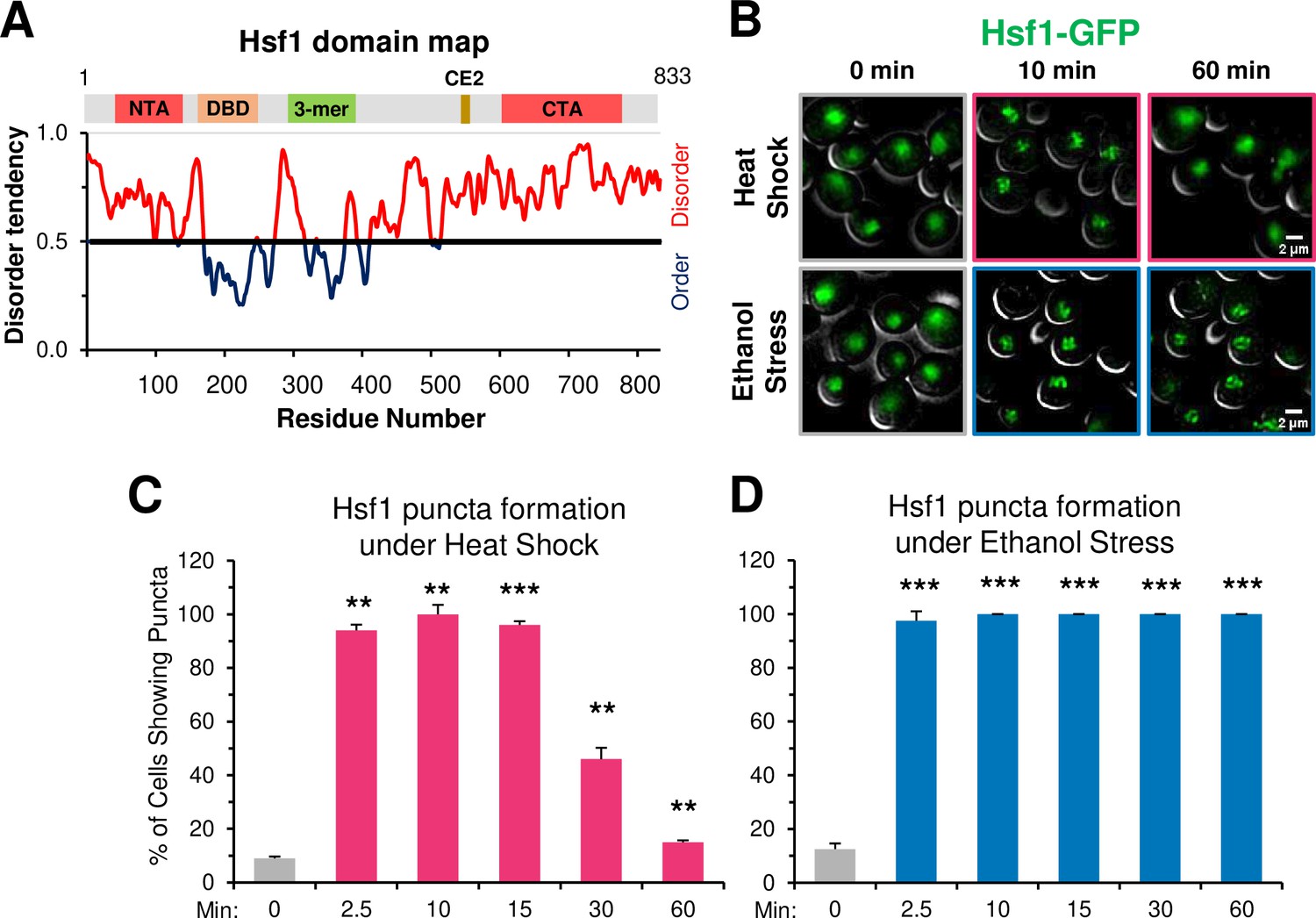
Ethanol stress induces rapid formation of long-lasting Hsf1 condensates.
(A) Hsf1 is a transcription factor bearing N- and C-terminal domains with high disorder tendency (determined by IUPRED2). NTA, N-terminal activator; DBD, DNA binding domain; 3-mer, trimerization domain; CE2, conserved element 2 (Hsp70 binding site); CTA, C-terminal activator. (B) Hsf1-GFP condensates form in response to ethanol stress. Diploid cells expressing Hsf1-GFP (ASK741) were grown in synthetic complete medium supplemented with adenine (SDC +Ade) and mounted onto ConA-coated coverslips. Live cell widefield microscopy was performed on cells exposed to either heat shock (38°C) or ethanol stress (8.5% v/v) or left untreated (25°C). A representative plane is shown for each condition out of 11 z-planes imaged (interplanar distance of 0.5 µm). Scale bar: 2 µm. (C) ASK741 cells were subjected to a 38°C heat shock for the indicated times and scored for the presence of Hsf1 condensates. A cell was scored as positive if it contained at least one clearly defined puncta. Approximately 200 cells were evaluated per timepoint. A one-tailed t-test was used to assess significance (stress versus no stress condition). N=2. **, p<0.01; ***, p<0.001.(D) ASK741 cells were exposed to 8.5% v/v ethanol for the indicated times and the presence of Hsf1 condensates were scored from a total of 200 cells per timepoint. Significance was determined as in (C).
-
Figure 8—source data 1
Spreadsheet tabulates the percentage of cells containing Hsf1-GFP puncta under heat shock or ethanol stress (Figure 8C and D).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig8-data1-v1.xlsx
Given the temporal link between Hsf1 condensation and transcription established in heat-shocked yeast cells, we anticipated that the appearance of Hsf1 condensates in ethanol-stressed cells would be delayed relative to what is seen in heat-shocked cells. However, we found that ethanol stress induced formation of Hsf1-GFP condensates as rapidly as did heat shock. These were visible in virtually all ES cells as early as 2.5 min, paralleling their rapid appearance in HS cells (Figure 8B–D). However, in contrast to the rapid dissolution of condensates in HS cells, those formed in cells exposed to 8.5% ethanol showed no evidence of dissipating even after 60 min of continuous exposure (Figure 8B and D). An independent analysis of Hsf1 tagged with a monomeric GFP, mNeonGreen, gave virtually identical results:>90% of cells exhibited Hsf1 puncta as early as 2.5 min and such puncta were stably maintained for 150 min (Figure 8—figure supplement 1). Moreover, in an independent analysis they remained visible at 5.5 hr (data not shown). Therefore, in 8.5% ethanol-stressed cells, formation of Hsf1 condensates is uncoupled from HSR gene transcription and their maintenance is uncoupled from HSR gene repositioning. An important implication is that although condensates may initiate or promote HSR gene repositioning, they cannot maintain the 3D restructured state of the genome.
Does the enhanced stability of ES-induced puncta arise from the nature of the stress or its intensity? To test this, we examined the behavior of yeast cells exposed to a lower concentration of ethanol. As shown in Figure 8—figure supplement 1, Hsf1 puncta formed in >95% of 5% ethanol-treated cells within 2.5 min, resembling what was seen in cells exposed to either 8.5% ethanol or 39°C HS. Yet such puncta dissipated in a majority of 5% ES cells within 30 min, resembling the case with HS but strongly contrasting to what was seen with 8.5% ethanol. Therefore, although 5% and 8.5% ES elicit a similar pattern of Sis1 subcellular relocalization, one that is distinct from that caused by either a 39° or 42°C HS (Figure 2), it is the greater intensity of 8.5% ethanol stress, not its intrinsic nature, that most closely correlates with the stability of Hsf1 condensates.
Hsf1 and Pol II are required for HSR gene interactions in response to both heat shock and ethanol stress
The above analyses reveal several unexpected differences in the way yeast responds to ethanol stress versus heat stress. Given these differences, we asked whether either Hsf1 or RNA Pol II are required for the repositioning of HSR genes in response to ethanol stress; both have been shown to be necessary for 3D genome restructuring in response to heat shock (Chowdhary et al., 2019; Chowdhary et al., 2022). To do so, we used the auxin-induced degradation system to conditionally degrade Hsf1 and the Rpb1 subunit of RNA Pol II in appropriately engineered strains. As schematically summarized in Figure 9A, cells expressing degron-tagged Hsf1 or Rpb1 were pre-treated with 1 mM indole-3-acetic acid (IAA) for 30–40 min, at which time each protein was >90% degraded (Figure 9—figure supplement 1A). Although minimal short-term growth defects were detectable, loss of Rpb1 and Hsf1 resulted in loss of cell viability at all temperatures (Figure 9—figure supplement 1B, C).
Figure 9 with 1 supplement see all
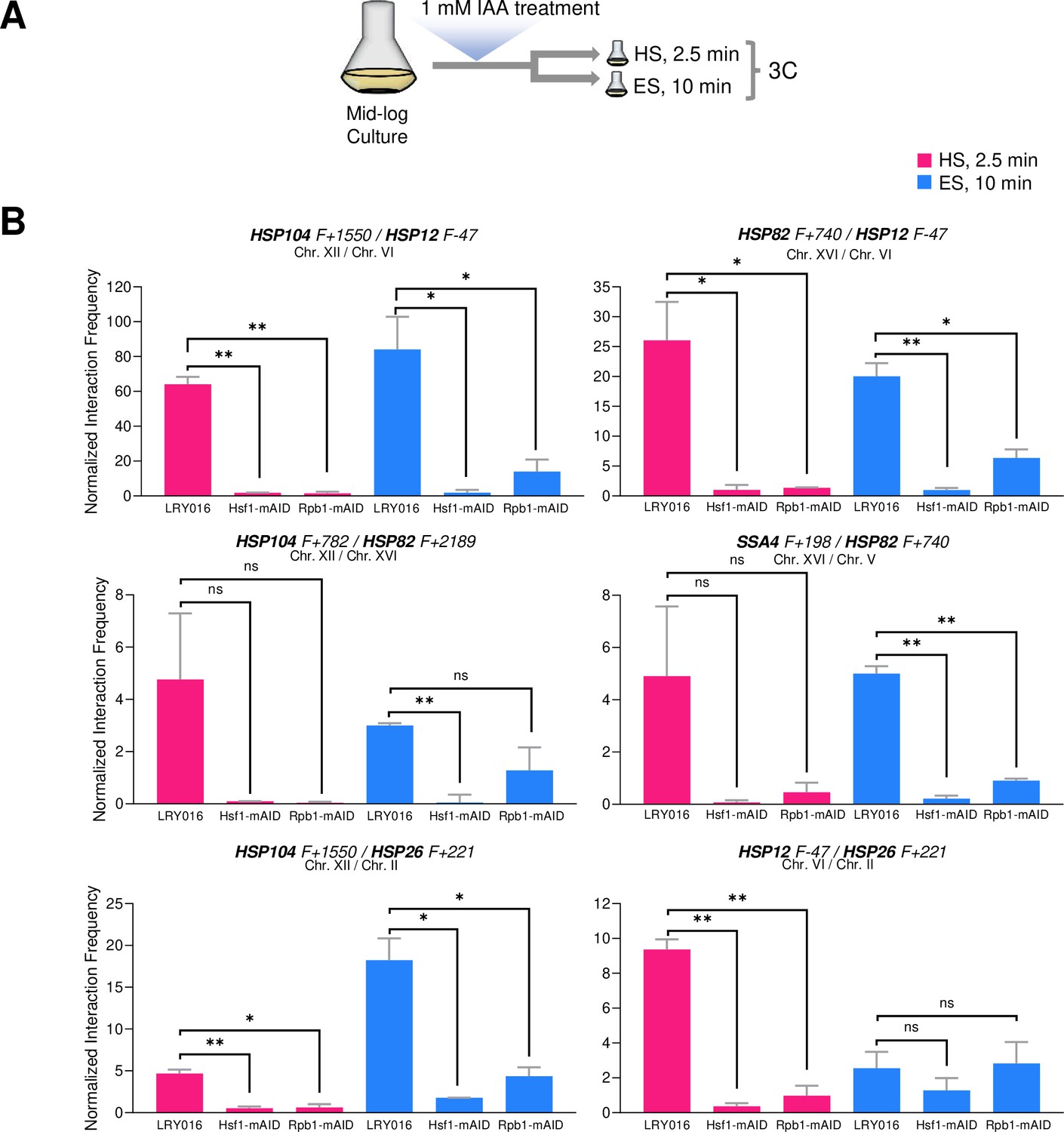
Hsf1 and Pol II are critically required for HSR gene interactions in response to either heat shock or ethanol stress.
(A) Experimental strategy. Degron-tagged cells were treated with 1 mM IAA at 25°C for 30–40 min prior to exposure to either heat shock (HS, 39°C for 2.5 min) or ethanol stress (ES, 8.5% v/v ethanol for 10 min) followed by HCHO crosslinking and 3C analysis. (B) Strains LRY016 (W303-1B; OsTIR1), LRY100 (LRY016; Hsf1-mAID), and LRY102 (LRY016; Rpb1-mAID) were subjected to the above protocol and physical interactions between the indicated chromosomal loci were detected by Taq I-3C as in Figure 5. Representative interchromosomal interactions are shown. Graphs represent means + SD. Statistical significance between the indicated interaction frequencies was determined using a one-tailed t-test. *, p<0.05; **, p<0.01; ns, not significant. A no stress sample, maintained at 30°C for 10 min following IAA treatment and then crosslinked, was handled in parallel. No signal above background was detected for any pairwise test.
-
Figure 9—source data 1
Spreadsheet tabulates 3C analysis of cells pretreated with 1 mM auxin followed by 2.5 min HS or 10 min ES (Figure 9B).
- https://cdn.elifesciences.org/articles/92464/elife-92464-fig9-data1-v1.xlsx
Consistent with previous observations (Chowdhary et al., 2019), intergenic interactions between HSR genes, located on the same or different chromosomes, were nearly obviated in cells conditionally depleted of either Hsf1 or Rpb1 and then heat shocked (Figure 9B and data not shown). While a similar strong dependence on Hsf1 is observed in 8.5% ethanol-treated cells, residual HSR-HSR gene interactions are retained in Rpb1-depleted cells for certain loci. Particularly noteworthy is the inducible interaction between HSP12 and HSP26, genes co-regulated by Msn2 and Hsf1. Neither is detectably activated following a 10 min exposure to ethanol (Figure 3B), yet the genes engage in an intergenic interaction that is unaffected by prior degradation of Rpb1 (Figure 9B). Together with the kinetic uncoupling of HSR gene repositioning with Pol II recruitment/transcription and the relative permanence of Hsf1 condensates described above, these observations raise the possibility that 8.5% (v/v) ethanol-induced Hsf1 condensates are compositionally different from those formed in response to heat shock and drive HSR gene repositioning and transcription in a mechanistically distinct way (see Figure 10 for model).
Figure 10
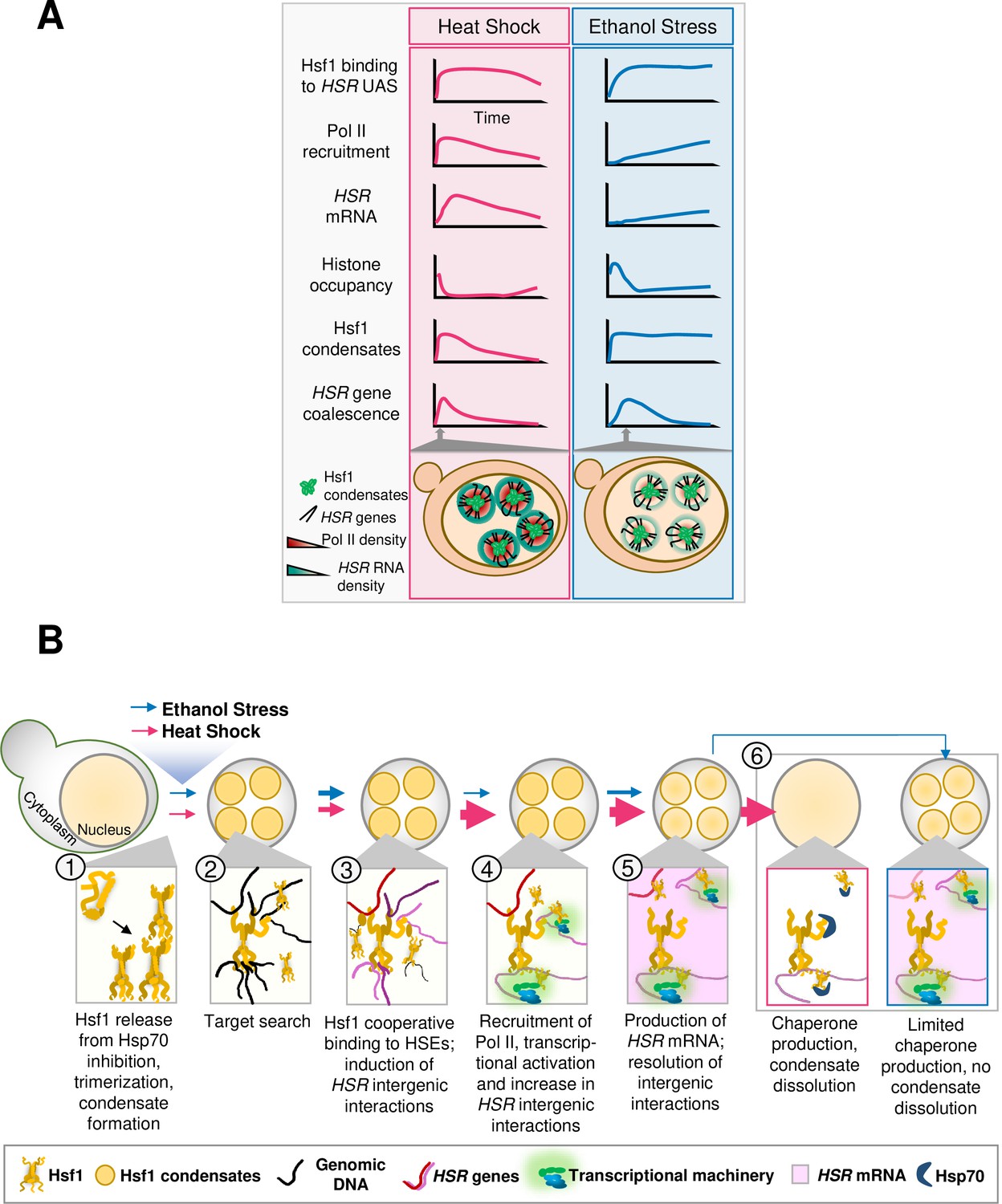
Central findings of this study.
(A) Kinetics of the yeast HSR to thermal and chemical stresses. (B) Hsf1 forms condensates, restructures the genome and transcriptionally activates HSR genes in a distinct fashion in response to thermal vs. chemical stress. Heat Shock (pink arrows): S. cerevisiae cells exposed to heat shock (30° to 39°C upshift) undergo protein misfolding (proteotoxicity), leading to chaperone titration and activation of Hsf1 accompanied by formation of Hsf1 condensates (1). Rapid target search is hypothesized to occur via multivalent interactions between Hsf1’s activation domains and chromatin-bound proteins (Brodsky et al., 2020) (2). Subsequently, Hsf1 cooperatively binds HSEs and induces the 3D reorganization of Heat Shock Responsive (HSR) genes (3). Simultaneously, RNA Pol II is recruited to HSR genes and transcription is induced (4). Productive transcription (pink shading) ensues, followed by dissolution of HSR gene interactions (5). Rapid export of HSR mRNAs (Zander et al., 2016) facilitates production of chaperone and cytoprotective proteins, which aid in restoration of proteostasis and disassembly of Hsf1 condensates (Chowdhary et al., 2022) (6). Thickness of arrows (for both HS and ES) symbolizes rapidity and/or magnitude of the subsequent step. Ethanol Stress (blue arrows): Exposure to 8.5% (v/v) ethanol induces proteotoxicity, titration of chaperones, and subsequent activation of Hsf1, triggering formation of condensates (1, 2). Condensates may aid Hsf1 target search by facilitating the multivalent interactions described above (2). Hsf1 then binds HSEs, inducing HSR gene repositioning (3). Subsequently, Pol II is recruited and transcription is induced (4). Ethanol stress-induced interactions dissipate well before maximal HSR gene transcriptional induction is achieved (5). The weak transcriptional HSR gene output, coupled with suppressed HSR mRNA export under ES (Izawa et al., 2008), results in a low level of chaperone synthesis (6) that fails to resolve proteotoxicity. This likely contributes to the persistence of Hsf1 condensates.
Discussion
Ethanol stress induces HSR gene transcription, HSR gene coalescence and formation of Hsf1 condensates
Here we have shown that exposure of budding yeast to a high, but sub-lethal, concentration of ethanol strongly stimulates the binding of Hsf1 to the upstream regulatory regions of HSR genes. Unexpectedly, such binding – which is evident as early as 2.5 min – does not lead to concurrent recruitment of Pol II and transcription of HSR genes. Instead, Pol II recruitment and transcription are delayed, typically for 10 min or longer. As exposure to 8.5% ethanol causes a global yet transient increase in H3 occupancy – which we interpret as an increase in the compaction of chromatin (see below) – the increase in nucleosome density may present a barrier to both Pol II recruitment and elongation. In addition, another feature of heat shocked-induced Hsf1 activation, repositioning of HSR genes within the 3D genome, is observed in cells exposed to ethanol. However, unlike transcription, this phenomenon occurs rapidly and is transient, resembling what is observed in heat shocked cells. The lack of temporal linkage between HSR gene transcription and HSR intergenic interactions is consistent with the idea that HSR gene coalescence and transcription are distinct phenomena and that Hsf1 can instigate long-range changes in 3D genome structure independently of inducing transcription. Thus, Hsf1 represents an example of a gene-specific TF that has functions independent of regulating transcription (discussed further below).
It has recently been demonstrated that in response to heat shock, inducible transcriptional condensates drive 3D genome reorganization in budding yeast. This conclusion arose from several features, including the tight temporal linkage between Hsf1 condensation and HSR intergenic interactions and the similar sensitivity of these two phenomena to the aliphatic alcohol, 1,6-hexanediol (Chowdhary et al., 2022). Similar to previous observations of heat-shocked cells (and confirmed here), we have found that in cells exposed to 8.5% ethanol, Hsf1 forms discrete puncta within 2.5 min. However, Hsf1-containing condensates formed in response to 8.5% ES are stable – persisting for hours – while such assemblies dissipate within 30 min in cells exposed to HS. In addition, while Hsf1 condensates formed equally rapidly in cells exposed to 5% and 8.5% ethanol, those formed in the presence of 5% ethanol proved to be transient, resembling their counterparts in HS cells. Relevant to this observation is the finding that Hsf1 activation in yeast exposed to 5% ethanol is dependent on the misfolding of nascent proteins, while in cells exposed to 8% ethanol, Hsf1 activation is dependent on the misfolding of both nascent and mature proteins (Tye and Churchman, 2021). Therefore, the persistence of Hsf1 condensates in 8.5% ethanol may derive from widespread unfolded mature proteins that are more difficult to refold than short nascent polypeptides attached to ribosomes.
Additional features might account for the way in which these two proteotoxic stresses elicit the HSR in budding yeast. For example, while ethanol denatures proteins similar to thermal stress (Furutani and Izawa, 2022; Kato et al., 2011), it impacts the cell at other levels. Actively growing yeast preferentially consume sugars and even in the presence of oxygen can produce ethanol (Dashko et al., 2014). Once glucose is depleted, cells start consuming ethanol as an alternative carbon source. This change in carbon source, termed the diauxic shift, is marked by a reduction in growth rate due to the less efficient generation of energy as well as accumulation of waste products and depletion of nutrients (Herman, 2002). Consistent with activation of the HSR, this physiological adjustment is accompanied by increased chaperone synthesis (Piper, 1995; Piper et al., 1994) as well as an increase in the activity of alcohol metabolism-related enzymes. Such an adaptation likely takes time. In contrast to the gradual increase in ethanol that occurs under natural conditions, here we have imposed an instantaneous ethanol shock. The addition of ethanol elicits the heat shock transcriptional response and synthesis of chaperones. The cell growth arrest seen here (Figure 1A) may arise as part of the adaptive program deployed upon encountering ethanol stress, such as a transition into quiescence. However, we fail to see activation of Msn2 targets, a signature of quiescence (Breeden and Tsukiyama, 2022), early in the exposure to ES at a time when Hsf1 itself is activated (Figure 3B). While other possibilities exist, our results suggest that the activation of programs involved in proteome protection are likely the first line of defense against sudden ethanol stress. Salient differences by which yeast cells respond to heat versus ethanol stress are summarized in Table 1; a graphical summary highlighting such differences is presented in Figure 10A. A model of the Hsf1 transcriptional response suggested by the data presented here and elsewhere is provided in Figure 10B.
Table 1
Kinetics of select nuclear phenomena in response to heat shock and ethanol stress.
| Parameter | Heat Shock (39°C) | Ethanol Stress (8.5% v/v) |
|---|---|---|
| Hsf1 binding to HSEs | Rapid, peak at 2.5 min | Slight delay, peak at 10 min |
| Pol II recruitment to HSR genes | Rapid, peak at 2.5 min | Severely delayed, detection starts at 10 min |
| Histone H3 occupancy at HSR genes | Rapid depletion | Transient increase, followed by gradual depletion |
| Genome compaction* | Transient | Sustained |
| HSR gene transcription | Rapid | Delayed |
| HSR gene coalescence | Rapid, transient | Slightly delayed, transient |
| Hsf1 condensates | Rapidly induced, transient, well defined | Rapidly induced, stable, poorly defined |
-
*
As inferred from enhanced H3 ChIP signal at non-Hsf1 regulated loci.
Exposure to ethanol transiently induces the global compaction of chromatin
An important observation is that exposure of cells to 8.5% ethanol leads to a widespread increase in H3 ChIP signal that we interpret as increased compaction of chromatin. Although this effect is temporary, both euchromatic and heterochromatic regions are impacted, and this is consistent with measurements of total chromatin volume that reveal a decrease lasting nearly 60 min. A similar outcome was recently reported for both yeast and mammalian cells exposed to aliphatic di-alcohols (Itoh et al., 2021; Meduri et al., 2022). It is probable that the effect of ethanol exposure is nearly instantaneous since, as discussed above, ethanol is known to denature proteins (Kato et al., 2019), likely by dehydration (disruption of biomolecules’ hydration shell). While such denaturation may contribute to chromatin compaction, the effect is reversible, possibly due to refolding / renaturation mediated by Hsp70 and other molecular chaperones whose intracellular concentration increases during ethanol exposure. As mentioned above, this temporary increased density of chromatin could suppress Pol II recruitment and subsequent elongation. Also not ruled out is a direct effect of 8.5% ethanol on one or more components of the Pol II machinery. Possible advantages of chromatin compaction could be to downregulate global transcription, as well as to limit chromatin damage by reactive oxygen species or other potentially damaging molecules present in the cell during ethanol stress (Bradley et al., 2021; Costa et al., 1997; Davidson et al., 1996; Davidson and Schiestl, 2001; Shen et al., 2020; Voordeckers et al., 2020).
A novel function for a TF that is uncoupled from regulating the transcription of its target gene
A key finding is that in response to ethanol stress, HSR genes reposition and Hsf1 condensates form well before transcription of Hsf1-dependent genes peaks, and in certain cases, is even detected. This suggests that Hsf1 has a function distinct from transactivating its target genes: it drives HSR gene repositioning that culminates in their physical coalescence. Likewise, deletion of the N-terminal IDR (NTA, amino acids 2–146; see Figure 8A) was observed to have little effect on HSR gene transcription during an acute heat shock yet formation of Hsf1 condensates and intergenic 3C interactions were suppressed (Chowdhary et al., 2022). Furthermore, as discussed above, recruitment of Hsf1 to its target HSEs is delayed in response to ES versus HS. Nevertheless, formation of Hsf1 condensates occurs with similar kinetics under both conditions, well prior to the peak of Hsf1 DNA binding in ES cells. This suggests the possibility that two types of Hsf1 clusters exist: one that is DNA-bound (detected by ChIP) and the other that hovers over the HSR genes (detected by imaging). It is possible that this cloud-forming Hsf1 cluster aids in target searching, by analogy with Gal4 that can also form clusters without binding DNA (Meeussen et al., 2023).
Conclusion
While Hsf1 is known to induce interactions between transcriptionally active HSR genes in response to heat shock, here we have demonstrated a similar role for this transcription factor in response to ethanol stress. Despite minimal HSR gene transcription during the initial 10 min exposure to ethanol, HSR genes engage in robust physical interactions, rivaling those seen for 2.5 min HS. This interaction correlates with an increase in definition of Hsf1 condensates yet is not accompanied by concurrent recruitment of Pol II to promoters, perhaps due to the compaction of chromatin in ethanol-stressed cells. Hsf1 therefore forms condensates and drives 3D repositioning of its target genes without appreciably activating these genes. Furthermore, Hsf1 does this through formation of condensates that may be materially different than those that form in response to HS. Our results thus argue that Hsf1 condensate formation, while sufficient for TF DNA binding and 3D genome restructuring, is not sufficient to drive transcription. Additional factors and/or activities – such as the opening of chromatin – are necessary. Further research into the biophysical properties, molecular regulation, and functional consequences of HSF1 condensates will deepen our understanding of how cells respond to both thermal and chemical stress and maintain cellular homeostasis.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (S. cerevisiae) | BY4741 | Research Genetics | ||
| Cell line (S. cerevisiae) | W303-1B | Rodney Rothstein | ||
| Recombinant DNA reagent | pFA6a-link-ymNeonGreen-SpHis5 plasmid | Addgene | Cat# 125704 RRID:Addgene_125704 | PMID:30783202 |
| Recombinant DNA reagent | pGZ154 plasmid | C.K. Govind, University of Oakland | ||
| Recombinant DNA reagent | pHyg-AID*–9myc | Addgene | Cat# 99518 RRID:Addgene_99518 | PMID:23836714 |
| Chemical compound, drug | Indole-3-acetic acid (IAA) | Sigma-Aldrich | Cat# I3750 | |
| Chemical compound, drug | Sodium azide | Sigma-Aldrich | Cat# S2002 | |
| Chemical compound, drug | Ethanol | Decon Labs | Cat# 04-355-223 | |
| Chemical compound, drug | Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat# P7626 | |
| chemical compound, drug | Phenol:Chloroform:Isoamyl Alcohol mixture | Sigma-Aldrich | Cat# 77617 | |
| Chemical compound, drug | Formaldehyde | Fisher Scientific | Cat# 14-650-250 | |
| Chemical compound, drug | Glycine | Bio-Rad | Cat# 1610724 | |
| chemical compound, drug | Protein A-Sepharose beads | GE Healthcare (Cytiva) | Cat# 17096303 | |
| Commercial assay or kit | High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 | |
| Commercial assay or kit | iTaq Universal SYBR Green Supermix | BioRad Laboratories | Cat# 1725125 | |
| Antibody | Anti-Btn2 (rabbit polyclonal) | Bernd Bukau, University of Heidelberg | WB (1:5000) | |
| Antibody | Anti-cMyc (mouse monoclonal) | Santa Cruz Biotechnology | Cat# sc-40 RRID:AB_627268 | WB (1:1000) |
| Antibody | Anti-Histone H3 (rabbit polyclonal) | Abcam | Cat# ab1719 | WB (1:1000) ChIP (1 µL/rxn) |
| Antibody | Anti-Hsf1 (rabbit polyclonal) | PMID:8943356 | ChIP (1.5 µL/rxn); Gross Lab | |
| Antibody | Anti-Hsp104 (rabbit polyclonal) | Enzo Life Sciences | Cat# ADI-SPA-1040-F RRID:AB_11181448 | WB (1:1000) |
| Antibody | Anti-Pgk1 (mouse monoclonal) | ThermoFisher Scientific | Cat# 459250 RRID:AB_2532235 | WB (1:10,000) |
| Antibody | Anti-Rpb1 (rabbit polyclonal) | PMID:16199876 | ChIP (1.5 µL/rxn); Gross Lab | |
| Antibody | Anti-Mouse, Horseradish peroxidase conjugated (goat) | Santa Cruz Biotechnology | Cat# sc-2005 RRID:AB_631736 | WB (1:5000) |
| Peptide, recombinant protein | Concanavalin A | Sigma-Aldrich | Cat# C2010 | |
| Peptide, recombinant protein | TaqI restriction enzyme | New England Biolabs | Cat# R0149L | |
| Commercial assay or kit | Quick Ligation Kit | New England Biolabs | Cat# M2200L | |
| Software, algorithm | FIJI/ImageJ | https://imagej.net/software/fiji/ | ||
| Software, algorithm | Imaris | https://imaris.oxinst.com/ | ||
| Software, algorithm | IUPRED2 | https://iupred2a.elte.hu/ | ||
| Other | VAHEAT device | Interherence GmbH | Temperature controller for live cell imaging |
Yeast strain construction
Request a detailed protocolThe HSF1-mNeonGreen (HSF1-mNG) diploid strain LRY033 expressing a yeast-optimized version of mNeonGreen, derived from Branchiostoma lanceolatum (Shaner et al., 2013), and co-expressing Sis1-mKate and Hsp104-BFP (Blue Fluorescent Protein; mTagBFP2) was created as follows. First, DPY1561 (Feder et al., 2021) was crossed to W303-1B to create strain LRY031. This diploid was sporulated and a strain homozygous for HSF1 and retention of one allele each of SIS-mKate and HSP104-mTagBFP2 was obtained after back crossing. The resultant diploid was named LRY032. LRY032 was transformed with a PCR amplicon containing 50 bp of homology sequences flanking the HSF1 stop codon, targeting the mNG tag to HSF1 at its C-terminus flanked by the HIS3 selectable marker. The plasmid template for this amplification was pFA6a-link-ymNeonGreen-SpHis5 (Botman et al., 2019). LRY033 is heterozygous for HSF1-mNG, SIS1-mKate and HSP104-mTagBFP2.
Other strains were created as follows. LRY037 was constructed using the HSF1-targeted mNeonGreen amplicon to transform strain W303-1B. LRY040 was constructed by transforming LRY037 with an amplicon containing RPB3-mCherry::hphMX6, obtained using genomic DNA from strain SCY004 (Chowdhary et al., 2022) as template. LRY100 and LRY102 were constructed using LRY016 (Rubio and Gross, 2023) as recipient of the mini-degron tag amplified from pHyg-AID*–9Myc (Morawska and Ulrich, 2013), targeted to the C-terminus of HSF1 and RPB1, respectively.
A complete list of strains as well as plasmids and primers used in strain construction are listed in Supplementary file 1a, b, and c.
Yeast culture and treatment conditions
Request a detailed protocolCells were grown at 30°C in YPDA (1% w/v yeast extract, 2% w/v peptone, 2% w/v dextrose and 20 mg/L adenine) to mid-log density (OD600=0.6–0.8). For ethanol stress, the cell culture was mixed with an equal volume of YPDA containing 17% v/v ethanol (yielding a final concentration of 8.5%) and incubated at 25°C for different lengths of time as indicated in the figures. For heat shock, the mid-log culture was mixed with an equal volume of 55°C YPDA medium to achieve an instantaneous temperature upshift to 39°C, and the culture was maintained at 39°C for the indicated times. The no stress samples were diluted with an equivalent volume of YPDA and maintained at 25°C. Samples were kept at their respective temperatures using a water bath with constant shaking.
Cell viability and growth assays
Cell viability assay
Request a detailed protocolCells were grown at 30°C in YPDA to OD600=0.6 and then diluted to OD600=0.4 using an equivalent volume of medium as described above for ethanol stress, heat shock or the no stress control (YPDA at 25°C). Cells were kept under these conditions for 3 hr; during this time aliquots were taken at different timepoints and diluted 1:26,000 for plating onto YPDA. Plates were incubated at 30°C for 3 days then scanned. Colonies were quantified using ImageJ/Fiji (v. 1.53t) (Schindelin et al., 2012) - ‘Analyze Particles’ option. The number of colony-forming units (CFUs) obtained in stress samples were normalized to the no stress samples and expressed as a percentage of the number of CFUs obtained in the no stress sample.
Growth assay
Request a detailed protocolCells were grown in liquid culture and subjected to the same treatments as described above. OD600 readings of each sample were taken at intervals over 3 hr (see Figure 1). The average OD600 from two samples was plotted versus time.
Spot dilution
Request a detailed protocolSpot dilution was conducted as described previously (Rubio and Gross, 2023).
Auxin-induced degradation
Request a detailed protocolCells expressing the F-box protein osTIR1 in combination with a degron-tagged protein were grown in YPDA medium to mid-log phase and indole-3-acetic acid (IAA) was then added to a final concentration of 1 mM. IAA stocks (10 mg/mL [57 mM]) were prepared fresh in 95% ethanol and filter-sterilized before use. For immunoblot analysis, cells were treated for varying times up to 1 hr prior to metabolic arrest achieved through addition of sodium azide to a final concentration of 20 mM, followed by cell harvesting. 0 min control samples were treated with vehicle alone (1.7% v/v ethanol). For growth curve analysis, samples were kept at 30°C with constant shaking and aliquots were removed at various timepoints to monitor OD600. For 3C analysis, cells were similarly grown in YPDA to OD600=0.6, then treated with 1 mM IAA for either 30 min (LRY016 and LRY100) or 40 min (LRY102) prior to cell cross-linking and subsequent cell harvesting.
Reverse transcription-qPCR (RT-qPCR)
Request a detailed protocolRT-qPCR was conducted as previously described using 25 mL cell culture aliquots (Rubio and Gross, 2023). Transcription was terminated via addition of sodium azide to a final concentration of 20 mM (Lee and Garrard, 1991). PCR primers used are listed in Supplementary file 1d.
Immunoblot analysis
Request a detailed protocolImmunoblotting was performed as previously reported using 20 mL cell culture aliquots (Rubio and Gross, 2023).
Chromatin immunoprecipitation
Request a detailed protocolChromatin immunoprecipitation was performed as previously described (Chowdhary et al., 2019) with modifications. Briefly, cells from a 50 mL mid-log culture were exposed to 8.5% v/v ethanol for 0, 2.5, 10, 20 or 60 min and then fixed using 3.01% formaldehyde (HCHO), resulting in a net concentration of 1%. Glycine was then added to 0.363 M to quench excess formaldehyde. We note that HCHO is consumed in a reaction with ethanol. Since 8.5% ethanol equals 1.46 M, acetal formation at 1:2 stoichiometry consumes 0.730 M (2.01%) of HCHO, leaving an effective concentration of 0.363 M (1%). The reaction resulting in formation of an acetal is illustrated in Figure 11.
Figure 11
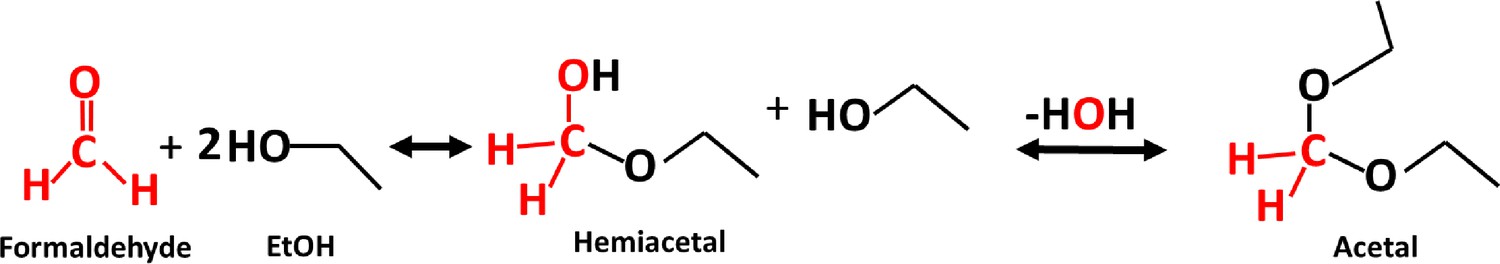
Acetal formation reaction (McMurry, 2012).
For heat shock, cells from a 50 mL mid-log culture were fixed with 1% (0.363 M) formaldehyde following 39°C upshift for the times indicated (Figure 4; Figure 4—figure supplements 1 and 2). Glycine was then added at a final concentration of 0.363 M to quench unreacted formaldehyde. Chromatin lysates, prepared as previously described (Chowdhary et al., 2019), were incubated using 20% of the lysate with one of the following antibodies: 1.5 µL of anti-Hsf1 (Erkine et al., 1996), 1.5 µL anti-Rpb1 antiserum (Zhao et al., 2005), or 1 µL of H3 antibody (Abcam, ab1791). Incubation of lysate with antibody was done for 16 hr at 4°C. Chromatin fragments bound to antibody were captured on Protein A-Sepharose beads (GE Healthcare) for 16 hr at 4°C. Wash, elution, and DNA purification were conducted as described (Chowdhary et al., 2019). The ChIP DNA template was quantified by qPCR (7900HT Fast Real Time PCR System, Applied Biosystems). A standard curve was generated using genomic DNA and ChIP DNA quantities were deduced by interpolation. The qPCR signal for each primer combination was normalized relative to the corresponding signal arising from the input DNA. Primers used in ChIP analysis are listed in Supplementary file 1e.
Taq I chromosome conformation capture (Taq I-3C)
View detailed protocolTaqI-3C was performed as previously described (Rubio and Gross, 2023). A master cell culture was grown at 30°C in YPDA from OD600=0.15 to a final OD600=0.8. Aliquots of 50 mL were used for each condition. Heat shock and ethanol stress were conducted as described above. Primers for analysis of 3C templates are listed Supplementary file 1f.
Fluorescence microscopy
Widefield fluorescence microscopy
Request a detailed protocolFor Figure 7—figure supplement 1B, C and Figure 8, cells were grown at 30°C in Synthetic Complete Dextrose (SDC) medium supplemented with 0.1 mg/mL adenine to early log phase. From this culture, 90 µL were removed and cells were immobilized onto a concanavalin A (ConA, Sigma Aldrich, 100 μg/mL in ddH2O)-coated coverslip for 20 min. For ethanol stress, the medium was then removed and replaced by either SDC or SDC +8.5% v/v ethanol. The coverslip was mounted onto a concave microscope slide to reduce media evaporation (2-Well Concavity Slide, Electron Microscopy Sciences). Images were acquired using an AX70 Olympus epifluorescence microscope across 11 z-planes with 0.5 µm of interplanar distance. Filter set 89021 (Chroma Technology) and a Photometrics Prime 95B camera were used to image GFP and mCherry. SlideBook Software version 6.0.15 (Intelligent Imaging Innovations) was used for image capturing and z-axis stepping motor operation (Ludl Electronic products).
For heat shock, no stress (NS) control images were taken using an Olympus Ach 100/1.25-NA objective coupled to a heating device (Bioptechs objective heater system). Heat-shocked sample imaging was done by rotating the objective away from the coverslip, switching the heating system on, and allowing it to reach 38°C before returning the objective to the coverslip for an instantaneous heat shock. The same field of view on the coverslip was imaged for both NS and HS timepoints. For ethanol stress, an Olympus UPlanFl 100/1.4-NA objective was used for image acquisition. Analysis of the images after acquisition was done using ImageJ (v. 1.52).
In the HSP104-TMA10 coalescence analysis, cells in which the fluorophore-tagged genes had undergone replication (two green or two red fluorescent spots, indicative of late S/G2 phase) were excluded from the analysis. The locations of the two tagged loci were analyzed over 11 different z-planes, encompassing the whole nucleus. Cells were deemed coalescence positive if they displayed colocalized, non-resolvable fluorophore signals with a distance of <0.4 µm between centroids.
Spinning disk confocal microscopy
Request a detailed protocolFor all other imaging, cells were grown as described above and image acquisition was done using an UPlan Apo 100 x/1.50 NA objective in an Olympus Yokogawa CSU W1 Spinning Disk Confocal System coupled to sCMOS cameras (Hamamatsu Fusion) controlled by cellSens Dimension software. A 50 µm pinhole disk was used for imaging in combination with 10% of laser power employed for excitation using 405 nm, 488 nm, and 561 nm lasers. Z-stacks were captured as for widefield fluorescence. For heat shock, cells were attached to a VAHEAT substrate (Interherence GmbH, Germany) (Icha et al., 2022) using ConA as above. The substrate was mounted onto the VAHEAT holder and control images were captured before heat was applied. Samples were instantaneously heated to 39° or 42°C. The substrate reservoir was covered with a coverslip to prevent media evaporation. Imaging was done over multiple timepoints as indicated in the figures. For ethanol stress, cells were mounted onto a coverslip as above, petroleum jelly was used to hold the coverslip against the concave slide and to reduce media evaporation. The slide was inverted and placed on the stage for visualization at room temperature.
Image reconstruction and analysis were done using FIJI/ImageJ (v. 1.53t) (Schindelin et al., 2012). Hsp104-BFP (LRY033) and Hsf1-mNeonGreen (LRY040) foci count were performed using the ‘Cells’ feature in Imaris (v.10.0.0). The background fluorescence was used to delimit the region of interest (ROI). This ROI was then used to quantify the number of foci per cell. For Hsp104, the ROI (whole cell) was delimited to volumes between 20 and 140 μ (Uchida et al., 2011). Foci size was measured to an average of 0.4 μ in diameter, which was used to train Imaris to recognize foci of distinct sizes (‘Different Spot Sizes [Region Growing]’ option). A similar approach was taken to identify the nucleus (as the cell) and the Hsf1 foci (as nucleus) using the ‘Cells’ feature in Imaris. Nuclei with volume of 10 μ ±3.7 μ were used for the analysis (Larson et al., 2011). Data obtained from these analyses were plotted using GraphPad Prism 8. Plot profile for signal arising from Hsf1-mNeonGreen, Sis1-mKate, and Hsp104-BFP was done using FIJI/ImageJ (v. 1.53t).
For analysis of transcription, 24xMS2-HSP104 mRNA was visualized using the signal arising from MCP-mCherry binding to the chimeric transcript. Cells were interrogated for transcription by assaying the presence of an mCherry focus adjacent to HSP104-LacO256 bound by LacI-GFP. The brightest voxel next to HSP104 was used as a proxy for the intensity of transcription and plotted for each cell over the different timepoints. Intensity was measured using FIJI/ImageJ (v. 1.53t) and plotted employing GraphPad Prism 8.
Statistical tests
Request a detailed protocolThe statistical significance of the differences in mean values for a variety of assays was determined as specified in the figure legends using GraphPad Prism 8.
Materials availability statement
Request a detailed protocolYeast strains, plasmids and antibodies are available upon request. All data generated in this study are presented in the manuscript, source data, and supplementary files.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Adaptive preservation of orphan ribosomal proteins in chaperone-dispersed condensatesNature Cell Biology 25:1691–1703.https://doi.org/10.1038/s41556-023-01253-2
-
A role for the CPF 3’-end processing machinery in RNAP II-dependent gene loopingGenes & Development 19:2969–2978.https://doi.org/10.1101/gad.1362305
-
Biomolecular condensates: organizers of cellular biochemistryNature Reviews. Molecular Cell Biology 18:285–298.https://doi.org/10.1038/nrm.2017.7
-
Acute ethanol stress induces sumoylation of conserved chromatin structural proteins in Saccharomyces cerevisiaeMolecular Biology of the Cell 32:1121–1133.https://doi.org/10.1091/mbc.E20-11-0715
-
Quiescence in Saccharomyces cerevisiaeAnnual Review of Genetics 56:253–278.https://doi.org/10.1146/annurev-genet-080320-023632
-
Why, when, and how did yeast evolve alcoholic fermentation?FEMS Yeast Research 14:826–832.https://doi.org/10.1111/1567-1364.12161
-
Mitochondrial respiratory electron carriers are involved in oxidative stress during heat stress in Saccharomyces cerevisiaeMolecular and Cellular Biology 21:8483–8489.https://doi.org/10.1128/MCB.21.24.8483-8489.2001
-
Cotranscriptional recruitment to the mRNA export receptor Mex67p contributes to nuclear pore anchoring of activated genesMolecular and Cellular Biology 26:7858–7870.https://doi.org/10.1128/MCB.00870-06
-
Subcellular localization of the J-protein Sis1 regulates the heat shock responseThe Journal of Cell Biology 220:e202005165.https://doi.org/10.1083/jcb.202005165
-
Adaptability of wine yeast to ethanol-induced protein denaturationFEMS Yeast Research 22:1–8.https://doi.org/10.1093/femsyr/foac059
-
HSF1 phase transition mediates stress adaptation and cell fate decisionsNature Cell Biology 22:151–158.https://doi.org/10.1038/s41556-019-0458-3
-
Genomic expression programs in the response of yeast cells to environmental changesMolecular Biology of the Cell 11:4241–4257.https://doi.org/10.1091/mbc.11.12.4241
-
Dynamic protein-DNA architecture of a yeast heat shock promoterMolecular and Cellular Biology 15:2737–2744.https://doi.org/10.1128/MCB.15.5.2737
-
A negative feedback loop at the nuclear periphery regulates GAL gene expressionMolecular Biology of the Cell 23:1367–1375.https://doi.org/10.1091/mbc.E11-06-0547
-
Stationary phase in yeastCurrent Opinion in Microbiology 5:602–607.https://doi.org/10.1016/s1369-5274(02)00377-6
-
1,6-hexanediol rapidly immobilizes and condenses chromatin in living human cellsLife Science Alliance 4:e202001005.https://doi.org/10.26508/lsa.202001005
-
Ribosomal precursor RNA metabolism and cell division in the yeast Saccharomyces cerevisiaeMolecular & General Genetics 178:357–360.https://doi.org/10.1007/BF00270484
-
HSF1 transcription factor concentrates in nuclear foci during heat shock: relationship with transcription sitesJournal of Cell Science 110 ( Pt 23):2935–2941.https://doi.org/10.1242/jcs.110.23.2935
-
In vivo binding of active heat shock transcription factor 1 to human chromosome 9 heterochromatin during stressThe Journal of Cell Biology 156:775–781.https://doi.org/10.1083/jcb.200109018
-
Mediator recruitment to heat shock genes requires dual Hsf1 activation domains and mediator tail subunits Med15 and Med16The Journal of Biological Chemistry 288:12197–12213.https://doi.org/10.1074/jbc.M112.449553
-
SAGA and Rpd3 chromatin modification complexes dynamically regulate heat shock gene structure and expressionThe Journal of Biological Chemistry 284:32914–32931.https://doi.org/10.1074/jbc.M109.058610
-
Effect of ethanol on cell growth of budding yeast: genes that are important for cell growth in the presence of ethanolBioscience, Biotechnology, and Biochemistry 68:968–972.https://doi.org/10.1271/bbb.68.968
-
Phase-separation antagonists potently inhibit transcription and broadly increase nucleosome densityThe Journal of Biological Chemistry 298:102365.https://doi.org/10.1016/j.jbc.2022.102365
-
Transcription factor clusters enable target search but do not contribute to target gene activationNucleic Acids Research 51:5449–5468.https://doi.org/10.1093/nar/gkad227
-
Monoallelic expression of olfactory receptorsAnnual Review of Cell and Developmental Biology 31:721–740.https://doi.org/10.1146/annurev-cellbio-100814-125308
-
Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assemblyNature Structural & Molecular Biology 26:193–203.https://doi.org/10.1038/s41594-019-0190-5
-
Gene loops juxtapose promoters and terminators in yeastNature Genetics 36:1014–1018.https://doi.org/10.1038/ng1411
-
Regulation of the Hsf1-dependent transcriptome via conserved bipartite contacts with Hsp70 promotes survival in yeastJournal of Biological Chemistry 294:12191–12202.https://doi.org/10.1074/jbc.RA119.008822
-
Genetic and epigenetic determinants establish a continuum of Hsf1 occupancy and activity across the yeast genomeMolecular Biology of the Cell 29:3168–3182.https://doi.org/10.1091/mbc.E18-06-0353
-
How did Saccharomyces evolve to become a good brewer?Trends in Genetics 22:183–186.https://doi.org/10.1016/j.tig.2006.02.002
-
Induction of heat shock proteins and thermotolerance by ethanol in Saccharomyces cerevisiaeBiochemical and Biophysical Research Communications 108:1340–1345.https://doi.org/10.1016/0006-291x(82)92147-7
-
In vivo activation by ethanol of plasma membrane ATPase of Saccharomyces cerevisiaeApplied and Environmental Microbiology 57:830–835.https://doi.org/10.1128/aem.57.3.830-835.1991
-
Fiji: an open-source platform for biological-image analysisNature Methods 9:676–682.https://doi.org/10.1038/nmeth.2019
-
Mechanisms regulating zygotic genome activationNature Reviews. Genetics 20:221–234.https://doi.org/10.1038/s41576-018-0087-x
-
The ethanol stress response and ethanol tolerance of Saccharomyces cerevisiaeJournal of Applied Microbiology 109:13–24.https://doi.org/10.1111/j.1365-2672.2009.04657.x
-
Hsf1 activation by proteotoxic stress requires concurrent protein synthesisMolecular Biology of the Cell 32:1800–1806.https://doi.org/10.1091/mbc.E21-01-0014
-
Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model systemMicrobiology and Molecular Biology Reviews 76:115–158.https://doi.org/10.1128/MMBR.05018-11
-
Ethanol exposure increases mutation rate through error-prone polymerasesNature Communications 11:3664.https://doi.org/10.1038/s41467-020-17447-3
-
Transcription in the context of the 3D nucleusCurrent Opinion in Genetics & Development 25:62–67.https://doi.org/10.1016/j.gde.2013.11.020
Article and author information
Author details
Funding
National Institutes of Health (R01 GM138988)
- David S Gross
National Institutes of Health (R15 GM128065)
- David S Gross
Ike Muslow Predoctoral Fellowship
- Linda S Rubio
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Drs. Kelly Tatchell, Lucy C Robinson, Rini Ravindran, Eric First, Surabhi Chowdhary and David Pincus for helpful discussions and technical advice; Drs. Vickky Pandit and Amoldeep Kainth for strain construction; Paula Polk for help with quantitative PCR; Drs. Axel Mogk and Bernd Bukau, University of Heidelberg, for the Btn2 antibody; Drs. Chabbi Govind, Oakland University, and Helle D Ulrich, Institute of Molecular Biology gGmbH, for OsTIR1 and mAID plasmids; and Drs. Donna and Jason Brickner, Kelly Tatchell, Surabhi Chowdhary, Amoldeep Kainth and David Pincus for generously providing yeast strains. This work was supported by NIH grants R15 GM128065 and R01 GM138988 awarded to D S G and an Ike Muslow predoctoral fellowship awarded to L S R.
Version history
- Sent for peer review:
- Preprint posted:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Reviewed Preprint version 3:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.92464. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Rubio et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,208
- views
-
- 75
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Heat Shock Factor 1 forms nuclear condensates and restructures the yeast genome before activating target genes
eLife 12:RP92464.
https://doi.org/10.7554/eLife.92464.4
Further reading
-
- Chromosomes and Gene Expression
- Evolutionary Biology
Gene regulation is essential for life and controlled by regulatory DNA. Mutations can modify the activity of regulatory DNA, and also create new regulatory DNA, a process called regulatory emergence. Non-regulatory and regulatory DNA contain motifs to which transcription factors may bind. In prokaryotes, gene expression requires a stretch of DNA called a promoter, which contains two motifs called –10 and –35 boxes. However, these motifs may occur in both promoters and non-promoter DNA in multiple copies. They have been implicated in some studies to improve promoter activity, and in others to repress it. Here, we ask whether the presence of such motifs in different genetic sequences influences promoter evolution and emergence. To understand whether and how promoter motifs influence promoter emergence and evolution, we start from 50 ‘promoter islands’, DNA sequences enriched with –10 and –35 boxes. We mutagenize these starting ‘parent’ sequences, and measure gene expression driven by 240,000 of the resulting mutants. We find that the probability that mutations create an active promoter varies more than 200-fold, and is not correlated with the number of promoter motifs. For parent sequences without promoter activity, mutations created over 1500 new –10 and –35 boxes at unique positions in the library, but only ~0.3% of these resulted in de-novo promoter activity. Only ~13% of all –10 and –35 boxes contribute to de-novo promoter activity. For parent sequences with promoter activity, mutations created new –10 and –35 boxes in 11 specific positions that partially overlap with preexisting ones to modulate expression. We also find that –10 and –35 boxes do not repress promoter activity. Overall, our work demonstrates how promoter motifs influence promoter emergence and evolution. It has implications for predicting and understanding regulatory evolution, de novo genes, and phenotypic evolution.
-
- Chromosomes and Gene Expression
- Developmental Biology
The male-specific lethal complex (MSL), which consists of five proteins and two non-coding roX RNAs, is involved in the transcriptional enhancement of X-linked genes to compensate for the sex chromosome monosomy in Drosophila XY males compared with XX females. The MSL1 and MSL2 proteins form the heterotetrameric core of the MSL complex and are critical for the specific recruitment of the complex to the high-affinity ‘entry’ sites (HAS) on the X chromosome. In this study, we demonstrated that the N-terminal region of MSL1 is critical for stability and functions of MSL1. Amino acid deletions and substitutions in the N-terminal region of MSL1 strongly affect both the interaction with roX2 RNA and the MSL complex binding to HAS on the X chromosome. In particular, substitution of the conserved N-terminal amino-acids 3–7 in MSL1 (MSL1GS) affects male viability similar to the inactivation of genes encoding roX RNAs. In addition, MSL1GS binds to promoters such as MSL1WT but does not co-bind with MSL2 and MSL3 to X chromosomal HAS. However, overexpression of MSL2 partially restores the dosage compensation. Thus, the interaction of MSL1 with roX RNA is critical for the efficient assembly of the MSL complex on HAS of the male X chromosome.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}