Novel risk loci for COVID-19 hospitalization among admixed American populations
- ERN-ITHACA-European Reference Network, Spain
- Pediatric Neurology Unit, Department of Pediatrics, Navarra Health Service Hospital, Spain
- CIBERER, ISCIII, Spain
- Centro Singular de Investigación en Medicina Molecular y Enfermedades Crónicas (CIMUS), Universidade de Santiago de Compostela, Spain
- Universidade Federal do Rio Grande do Norte, Departamento de Analises Clinicas e Toxicologicas, Brazil
- Genomics Division, Instituto Tecnológico y de Energías Renovables, Spain
- Fundación Pública Galega de Medicina Xenómica, Sistema Galego de Saúde (SERGAS), Spain
- Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz IDIPAZ, Spain
- Unit of Infectious Diseases, Hospital Universitario 12 de Octubre, Instituto de Investigación Sanitaria Hospital 12 de Octubre (imas12), Spain
- Spanish Network for Research in Infectious Diseases (REIPI RD16/0016/0002), Instituto de Salud Carlos III, Spain
- CIBERINFEC, ISCIII, Spain
- Hospital General Santa Bárbara de Soria, Spain
- Navarra Health Service, NavarraBioMed Research Group, Spain
- Hospital Universitario Virgen Macarena, Neumología, Spain
- Department of Genetics & Genomics, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital - Universidad Autónoma de Madrid (IIS-FJD, UAM), Spain
- Spanish National Cancer Research Centre, Human Genotyping-CEGEN Unit, Spain
- Department of Child and Adolescent Psychiatry, Institute of Psychiatry and Mental Health, Hospital General Universitario Gregorio Marañón (IiSGM), Spain
- School of Medicine, Universidad Complutense, Spain
- Biocruces Bizkai HRI, Spain
- Cruces University Hospital, Osakidetza, Spain
- Centre for Biomedical Network Research on Mental Health (CIBERSAM), Instituto de Salud Carlos III, Spain
- Fundació Docència I Recerca Mutua Terrassa, Spain
- Spanish National Cancer Research Centre, CNIO Biobank, Spain
- Hospital General de Occidente, Mexico
- Centro Universitario de Tonalá, Universidad de Guadalajara, Mexico
- Centro de Investigación Multidisciplinario en Salud, Universidad de Guadalajara, Mexico
- Universidad Católica San Antonio de Murcia (UCAM), Spain
- Instituto Murciano de Investigación Biosanitaria (IMIB-Arrixaca), Spain
- Hospital Universitario de Salamanca-IBSAL, Servicio de Medicina Interna-Unidad de Enfermedades Infecciosas, Spain
- Escola Tecnica de Saúde, Laboratorio de Vigilancia Molecular Aplicada, Brazil
- Federal University of Pernambuco, Genetics Postgraduate Program, Brazil
- Hospital Universitario Mutua Terrassa, Spain
- Instituto de Investigación Sanitaria de Santiago (IDIS), Xenética Cardiovascular, Spain
- CIBERCV, ISCIII, Spain
- Cardiovascular Genetics Center, Institut d’Investigació Biomèdica Girona (IDIBGI), Spain
- Medical Science Department, School of Medicine, University of Girona, Spain
- Hospital Josep Trueta, Cardiology Service, Spain
- Institute of Biomedicine of Seville (IBiS), Consejo Superior de Investigaciones Científicas (CSIC)- University of Seville- Virgen del Rocio University Hospital, Spain
- Departamento de Medicina, Hospital Universitario Virgen del Rocío, Universidad de Sevilla, Spain
- CIBERESP, ISCIII, Spain
- Hospital Universitario de Salamanca-IBSAL, Servicio de Medicina Interna, Spain
- Universidad de Salamanca, Spain
- Osakidetza, Cruces University Hospital, Spain
- Centre for Biomedical Network Research on Diabetes and Metabolic Associated Diseases (CIBERDEM), Instituto de Salud Carlos III, Spain
- University of Pais Vasco, UPV/EHU, Spain
- Oncology and Genetics Unit, Instituto de Investigacion Sanitaria Galicia Sur, Xerencia de Xestion Integrada de Vigo-Servizo Galego de Saúde, Spain
- Hospital Universitario Río Hortega, Spain
- Servicio de Medicina intensiva, Complejo Hospitalario Universitario de A Coruña (CHUAC), Sistema Galego de Saúde (SERGAS), Spain
- Tecnológico de Monterrey, Mexico
- Department of Microgravity and Translational Regenerative Medicine, Otto von Guericke University, Germany
- Hospital Universitario Mostoles, Unidad de Genética, Spain
- Instituto Aragonés de Ciencias de la Salud (IACS), Spain
- Instituto Investigación Sanitaria Aragón (IIS-Aragon), Spain
- Preventive Medicine Department, Instituto de Investigacion Sanitaria Galicia Sur, Xerencia de Xestion Integrada de Vigo-Servizo Galego de Saúde, Spain
- Unidad Diagnóstico Molecular, Fundación Rioja Salud, Spain
- Hospital Universitario de Salamanca-IBSAL, Servicio de Cardiología, Spain
- IDIVAL, Spain
- Hospital U M Valdecilla, Spain
- Universidad de Cantabria, Spain
- Universidad Nacional de Asunción, Facultad de Politécnica, United States
- Urgencias Hospitalarias, Complejo Hospitalario Universitario de A Coruña (CHUAC), Sistema Galego de Saúde (SERGAS), Spain
- Unidad de Infección Viral e Inmunidad, Centro Nacional de Microbiología (CNM), Instituto de Salud Carlos III (ISCIII), Spain
- Grupo de Investigación en Interacciones Gen-Ambiente y Salud (GIIGAS) - Instituto de Biomedicina (IBIOMED), Universidad de León, Spain
- IDIS, Republic of Korea
- Hospital Universitario de Getafe, Servicio de Genética, Spain
- Ministerio de Salud Ciudad de Buenos Aires, Argentina
- Hospital Universitario Virgen de las Nieves, Servicio de Análisis Clínicos e Inmunología, Spain
- IIS La Fe, Plataforma de Farmacogenética, Spain
- Universidad de Valencia, Departamento de Farmacología, Spain
- Data Analysis Department, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital - Universidad Autónoma de Madrid (IIS-FJD, UAM), Spain
- Universidad de los Andes, Facultad de Ciencias, Colombia
- SIGEN Alianza Universidad de los Andes - Fundación Santa Fe de Bogotá, Colombia
- Hospital General de Segovia, Medicina Intensiva, Spain
- Facultad de Farmacia, Universidad San Pablo-CEU, CEU Universities, Urbanización Montepríncipe, Spain
- Hospital Universitario 12 de Octubre, Department of Immunology, Spain
- Instituto de Investigación Sanitaria Hospital 12 de Octubre (imas12), Transplant Immunology and Immunodeficiencies Group, Spain
- Fundación Santa Fe de Bogota, Departamento Patologia y Laboratorios, Colombia
- Unidad de Genética y Genómica Islas Baleares, Spain
- Hospital Universitario Son Espases, Unidad de Diagnóstico Molecular y Genética Clínica, Spain
- Genomics of Complex Diseases Unit, Research Institute of Hospital de la Santa Creu i Sant Pau, IIB Sant Pau, Spain
- Universidade de Brasília, Faculdade de Medicina, Brazil
- Programa de Pós-Graduação em Ciências Médicas (UnB), Brazil
- Programa de Pós-Graduação em Ciencias da Saude (UnB), Brazil
- Hospital El Bierzo, Unidad Cuidados Intensivos, Spain
- Hospital Universitario Mostoles, Medicina Interna, Spain
- Universidad Francisco de Vitoria, Spain
- Departamento de Genética e Morfologia, Instituto de Ciências Biológicas, Universidade de Brasília, Brazil
- Programa de Pós-Graduação em Biologia Animal (UnB), Brazil
- Programa de Pós-Graduação Profissional em Ensino de Biologia (UnB), Brazil
- Universidad Complutense de Madrid, Department of Immunology, Ophthalmology and ENT, Spain
- Universidade Federal do Pará, Núcleo de Pesquisas em Oncologia, Brazil
- Infectious Diseases, Microbiota and Metabolism Unit, CSIC Associated Unit, Center for Biomedical Research of La Rioja (CIBIR), Spain
- Inditex, A Coruña, Spain
- GENYCA, Spain
- Instituto Mexicano del Seguro Social (IMSS), Centro Médico Nacional Siglo XXI, Unidad de Investigación Médica en Enfermedades Infecciosas y Parasitarias, Mexico
- Instituto Mexicano del Seguro Social (IMSS), Centro Médico Nacional La Raza, Hospital de Infectología, Mexico
- Clinica Comfamiliar Risaralda, Colombia
- Bellvitge Biomedical Research Institute (IDIBELL), Neurometabolic Diseases Laboratory, L’Hospitalet de Llobregat, Spain
- Catalan Institution of Research and Advanced Studies (ICREA), Spain
- Hospital Ophir Loyola, Departamento de Ensino e Pesquisa, Brazil
- Unidad de Cuidados Intensivos, Hospital Clínico Universitario de Santiago (CHUS), Sistema Galego de Saúde (SERGAS), Spain
- Department of Preventive Medicine and Public Health, School of Medicine, Universidad Autónoma de Madrid, Spain
- IdiPaz (Instituto de Investigación Sanitaria Hospital Universitario La Paz), Spain
- IMDEA-Food Institute, CEI UAM+CSIC, Spain
- Complejo Asistencial Universitario de León, Spain
- Instituto de Investigación Biosanitaria de Granada (ibs GRANADA), Spain
- Universidad de Granada, Departamento Bioquímica, Biología Molecular e Inmunología III, Spain
- Hospital Infanta Elena, Allergy Unit, Valdemoro, Spain
- Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital - Universidad Autónoma de Madrid (IIS-FJD, UAM), Spain
- Faculty of Medicine, Universidad Francisco de Vitoria, Spain
- Hospital Universitario Infanta Leonor, Spain
- Complutense University of Madrid, Spain
- Gregorio Marañón Health Research Institute (IiSGM), Spain
- Haemostasis and Thrombosis Unit, Hospital de la Santa Creu i Sant Pau, IIB Sant Pau, Spain
- Hospital Clinico Universitario de Valladolid, Servicio de Anestesiologia y Reanimación, Spain
- Universidad de Valladolid, Departamento de Cirugía, Spain
- Hospital Clinico Universitario de Valladolid, Servicio de Hematologia y Hemoterapia, Spain
- Hospital de Niños Ricardo Gutierrez, Argentina
- Fundación Universitaria de Ciencias de la Salud, Colombia
- Spanish National Cancer Research Centre, Familial Cancer Clinical Unit, Spain
- University Hospital of Burgos, Spain
- Universidad Simón Bolívar, Facultad de Ciencias de la Salud, Colombia
- Centro para el Desarrollo de la Investigación Científica, Paraguay
- Centre for Biomedical Network Research on Neurodegenerative Diseases (CIBERNED), Instituto de Salud Carlos III, Spain
- Research Center and Memory clinic, ACE Alzheimer Center Barcelona, Universitat Internacional de Catalunya, Spain
- CIEN Foundation/Queen Sofia Foundation Alzheimer Center, Spain
- Hospital Universitario de Valme, Unidad Clínica de Enfermedades Infecciosas y Microbiología, Spain
- Sección Genética Médica - Servicio de Pediatría, Hospital Clínico Universitario Virgen de la Arrixaca, Servicio Murciano de Salud, Spain
- Departamento Cirugía, Pediatría, Obstetricia y Ginecología, Facultad de Medicina, Universidad de Murcia (UMU), Spain
- Grupo Clínico Vinculado, Centre for Biomedical Network Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Spain
- Department of Anthropology, University of Toronto at Mississauga, Canada
- Tecnologico de Monterrey, Escuela de Medicina y Ciencias de la Salud, Mexico
- Research Unit, Hospital Universitario Nuestra Señora de Candelaria, Instituto de Investigación Sanitaria de Canarias, Spain
- Department of Clinical Sciences, University Fernando Pessoa Canarias, Spain
- Centre for Biomedical Network Research on Respiratory Diseases (CIBERES), Instituto de Salud Carlos III, Spain
Figures
Figure 1 with 1 supplement
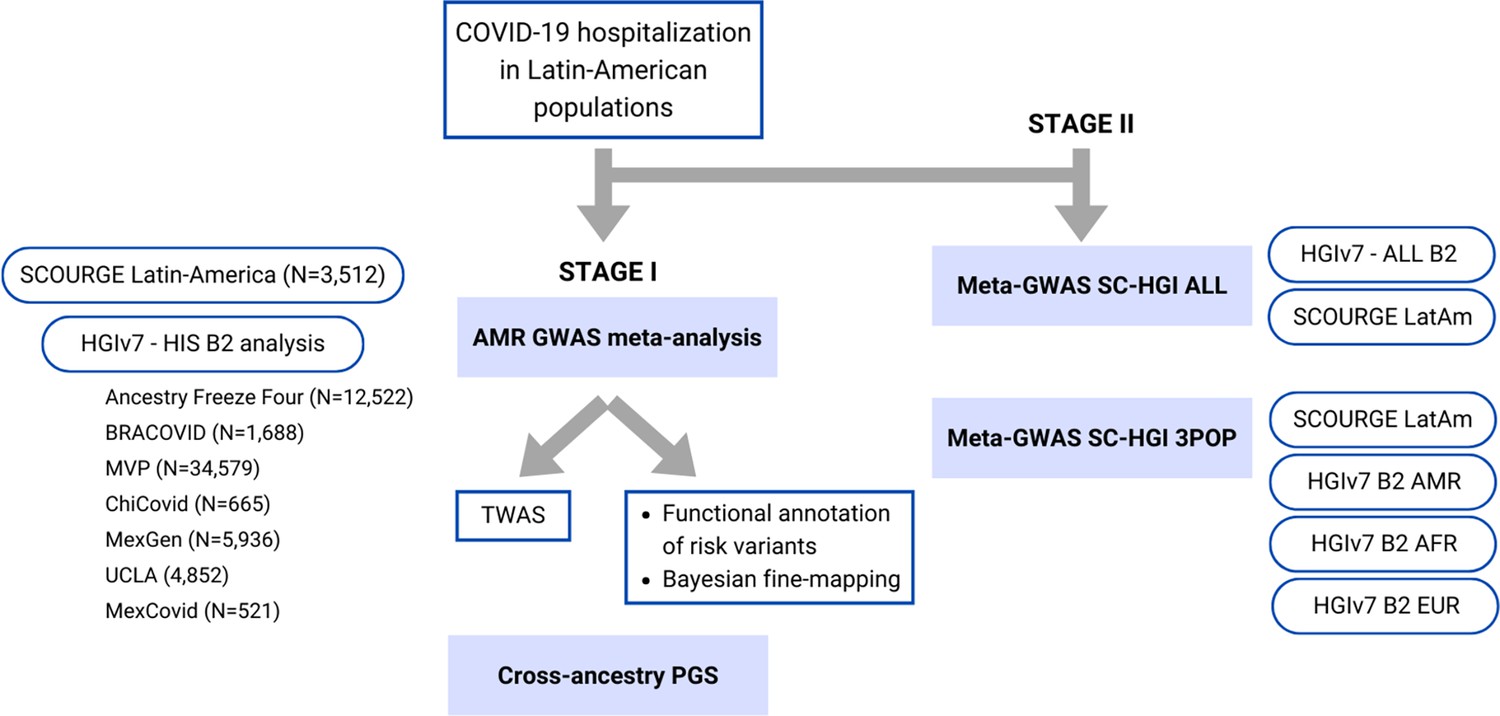
Flow chart of this study.
Stage I of the study involved a meta-analysis of the Latin American genome-wide association studies (GWAS) from SCOURGE and the COVID-19 Host Genetics Initiative. The resulting meta-analysis was leveraged to prioritize genes by using a transcriptome-wide association study (TWAS), Bayesian fine-mapping and functional annotations, and to assess the generalizability of polygenic risk score (PGS) cross-population models in Latin Americans. Stage II involved two additional cross-population GWAS meta-analyses to further investigate the replicability of findings.
Figure 1—figure supplement 1
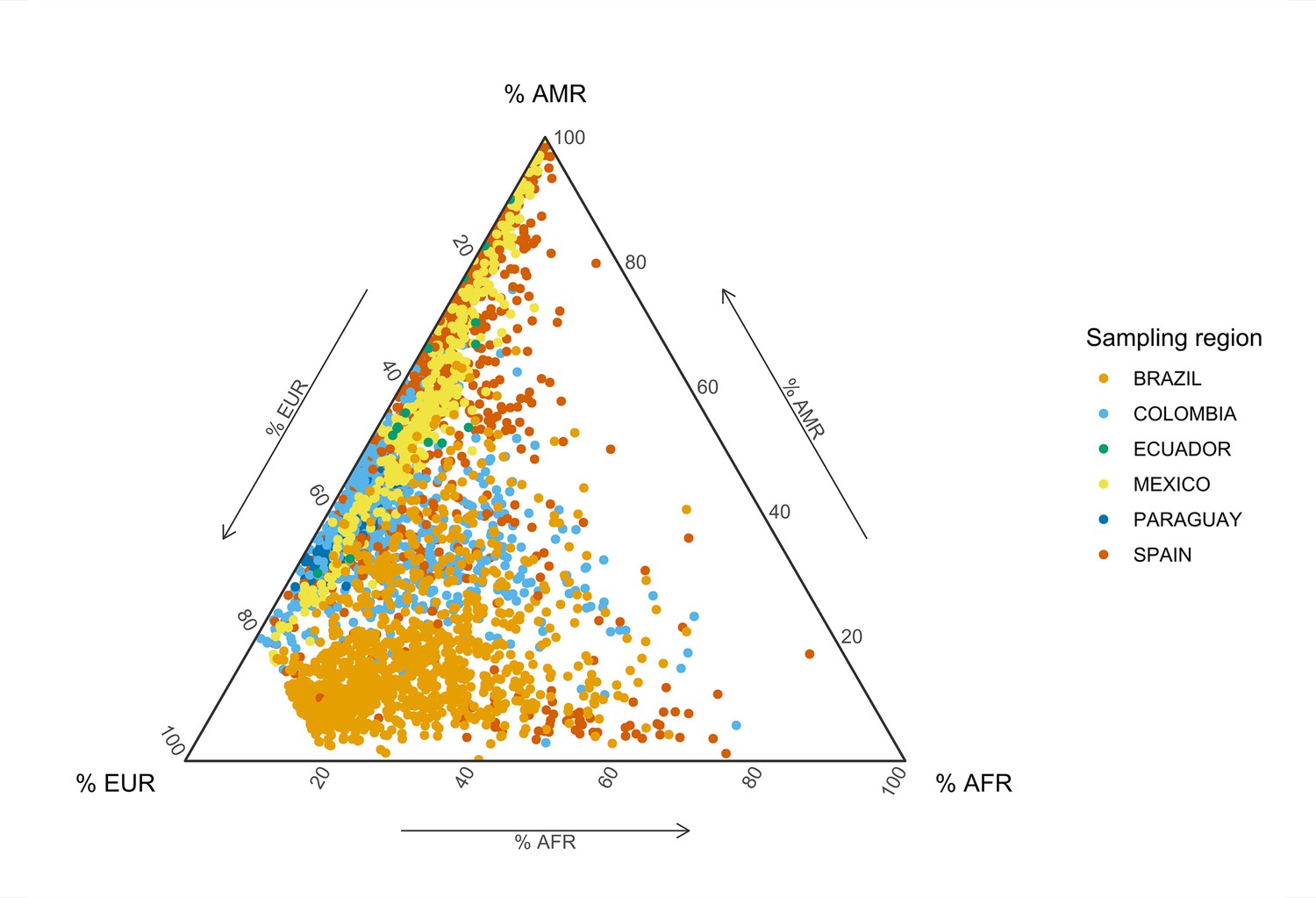
Global genetic inferred ancestry (GIA) composition in the SCOURGE Latin American cohort.
European (EUR), African (AFR), and Native American (AMR) GIA was derived with ADMIXTURE from a reference panel composed of Aymaran, Mayan, Nahuan, and Quechuan individuals of Native American genetic ancestry and randomly selected samples from the EUR and AFR 1KGP populations. The colors represent the different geographical sampling regions from which the admixed American individuals from SCOURGE were recruited.
Figure 2 with 1 supplement
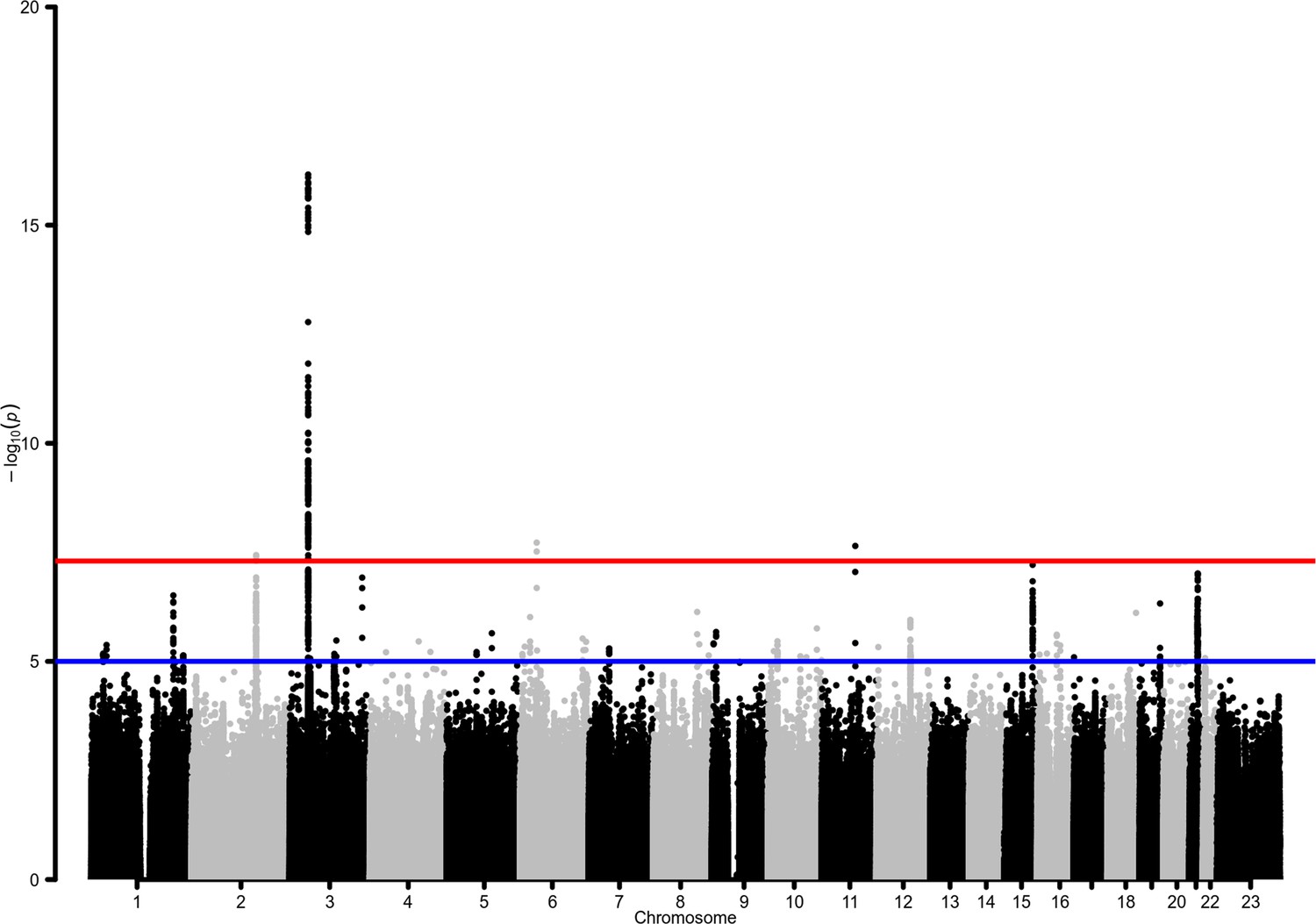
Manhattan plot for the admixed AMR genome-wide association studies (GWAS) meta-analysis.
Probability thresholds at p=5 × 10–8 and p=5 × 10–5 are indicated by the horizontal lines. Genome-wide significant associations with COVID-19 hospitalizations were found on chromosome 2 (within BAZ2B), chromosome 3 (within LZTFL1), chromosome 6 (within FOXP4), and chromosome 11 (within DDIAS).
Figure 2—figure supplement 1
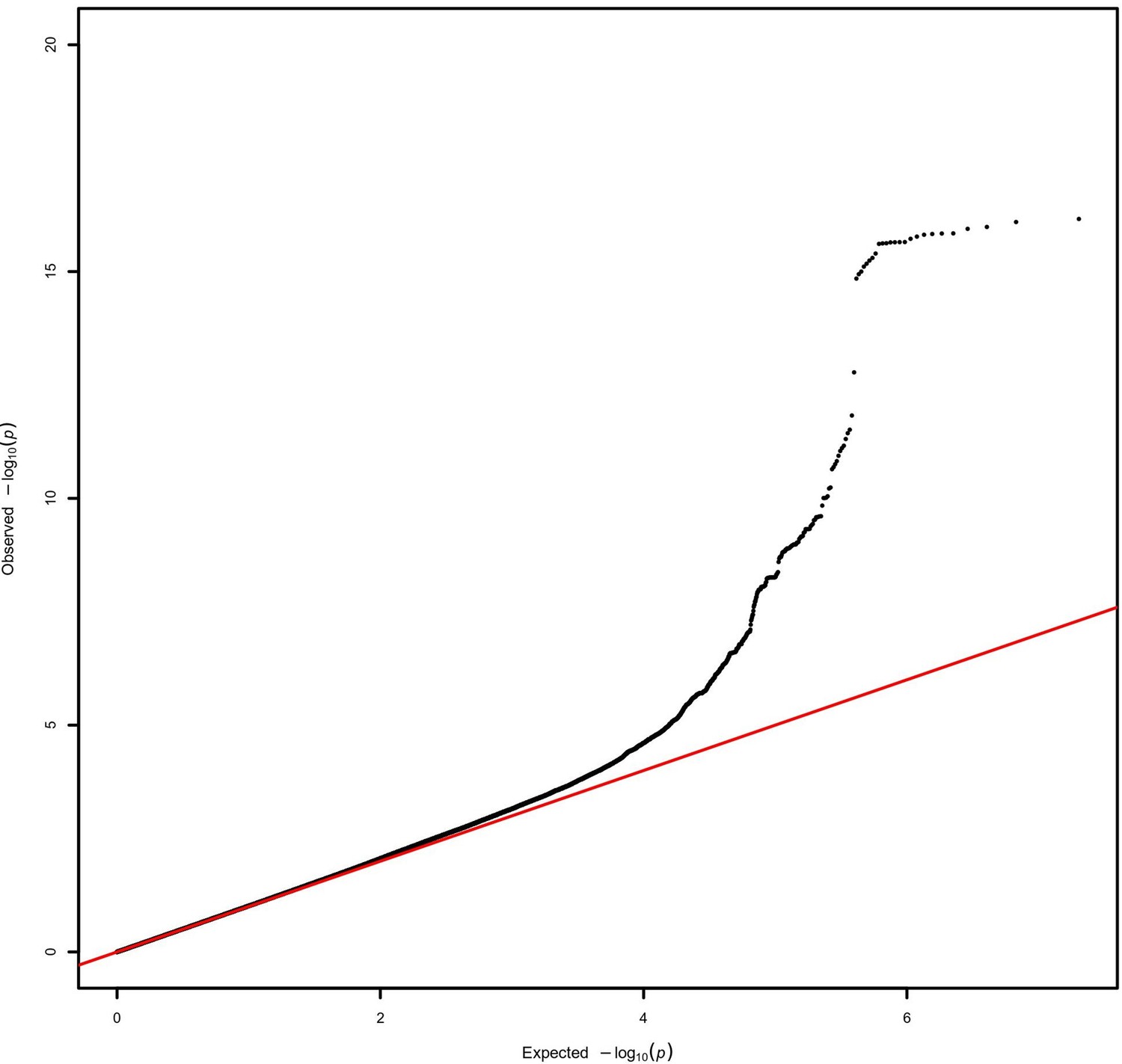
Quantile–quantile plot for the AMR genome-wide association studies (GWAS) meta-analysis.
A lambda inflation factor of 1.015 was obtained.
Figure 3 with 1 supplement
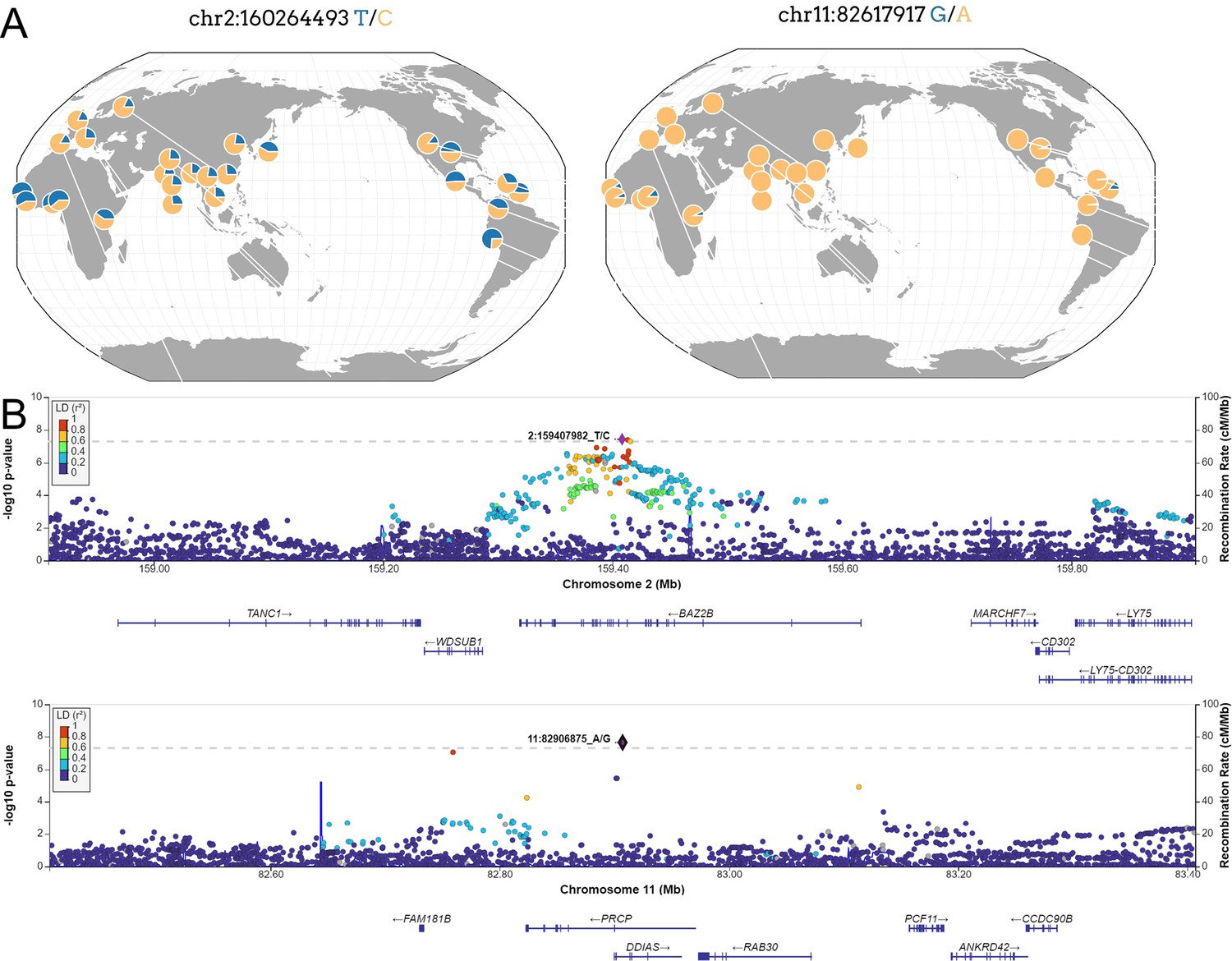
New loci associated with COVID-19 hospitalization in Admixed american populations.
(A) Regional association plots for rs1003835 at chromosome 2 and rs77599934 at chromosome 11. (B) Allele frequency distribution across the 1000 Genomes Project populations for the lead variants rs1003835 and rs77599934. Retrieved from The Geography of Genetic Variants Web or GGV.
Figure 3—figure supplement 1
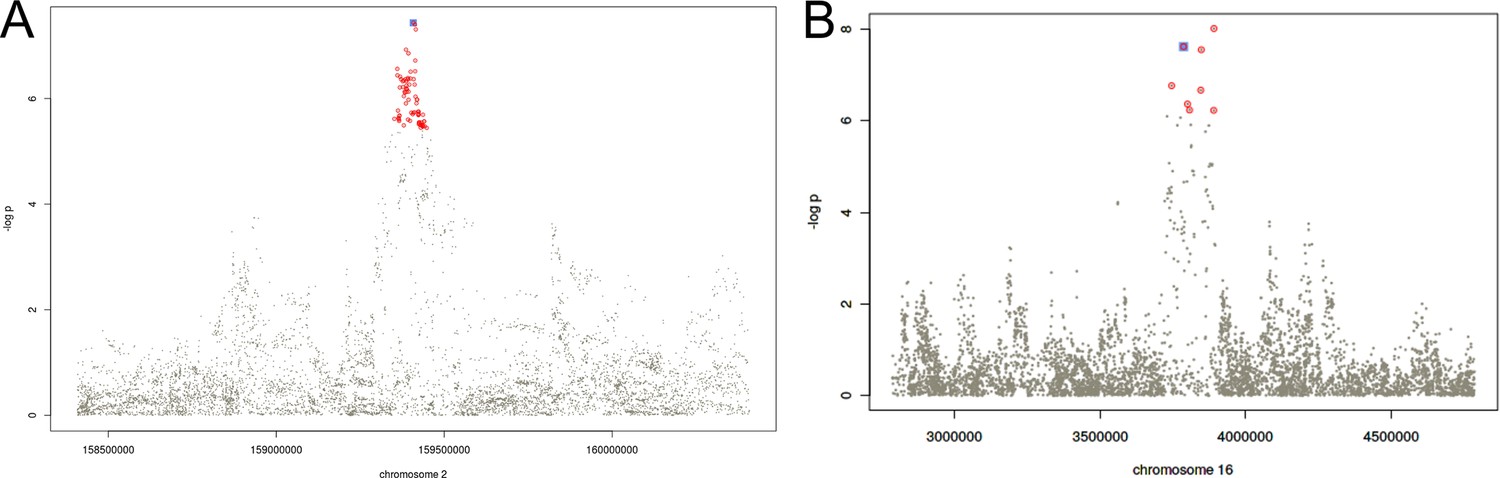
Regional association plots for the fine mapped loci in chromosomes 2 (A) and 16 (B).
Colored in red, the variants allocated to the credible set at the 95% confidence according to the Bayesian fine mapping. In blue, the sentinel variant.
Figure 4 with 1 supplement
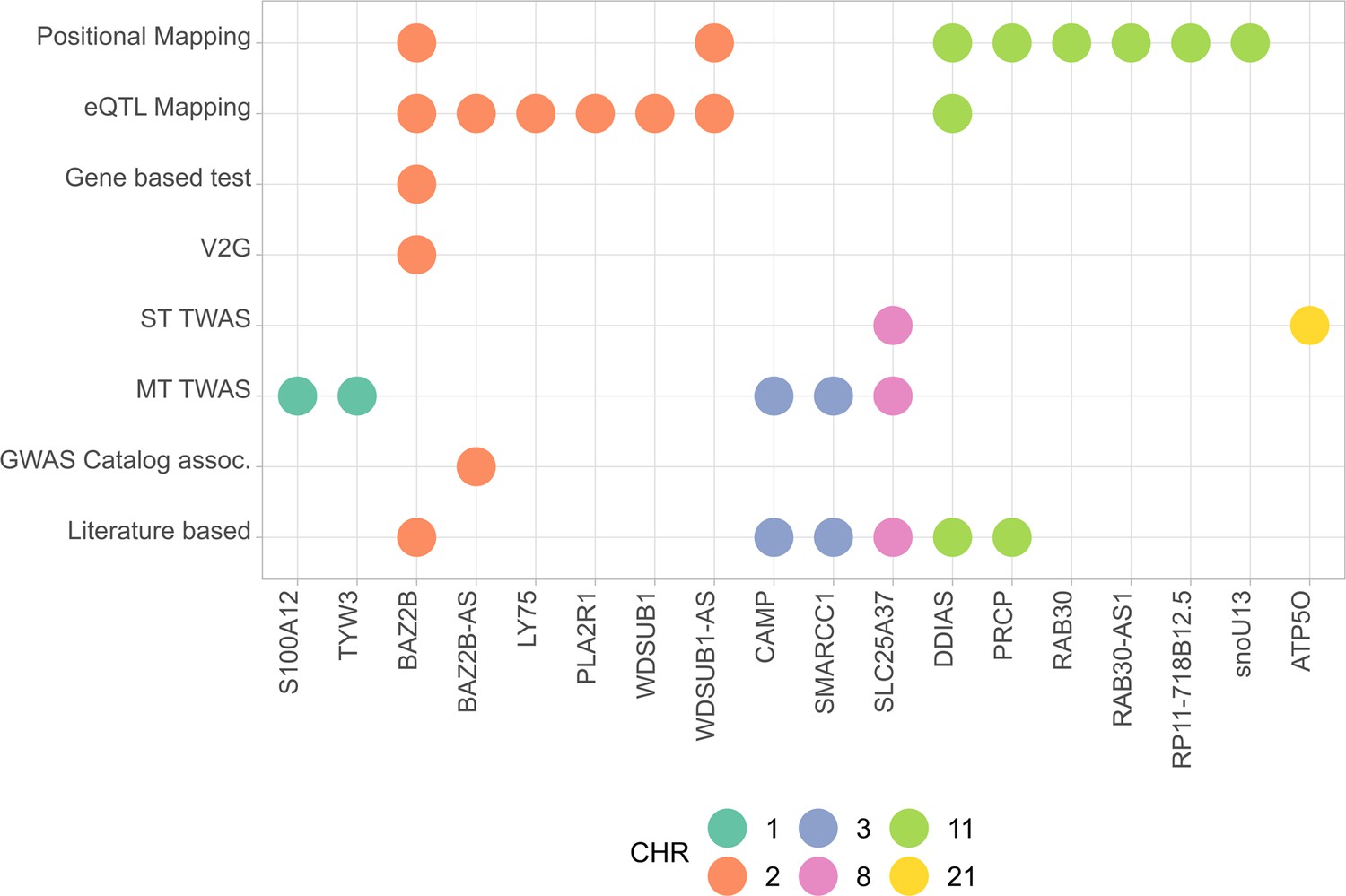
Summary of the results from gene prioritization strategies used for genetic associations in AMR populations.
Genome-wide association studies (GWAS) catalog association for BAZ2B-AS was with FEV/FCV ratio. Literature-based evidence is further explored in ‘Discussion’.
Figure 4—figure supplement 1
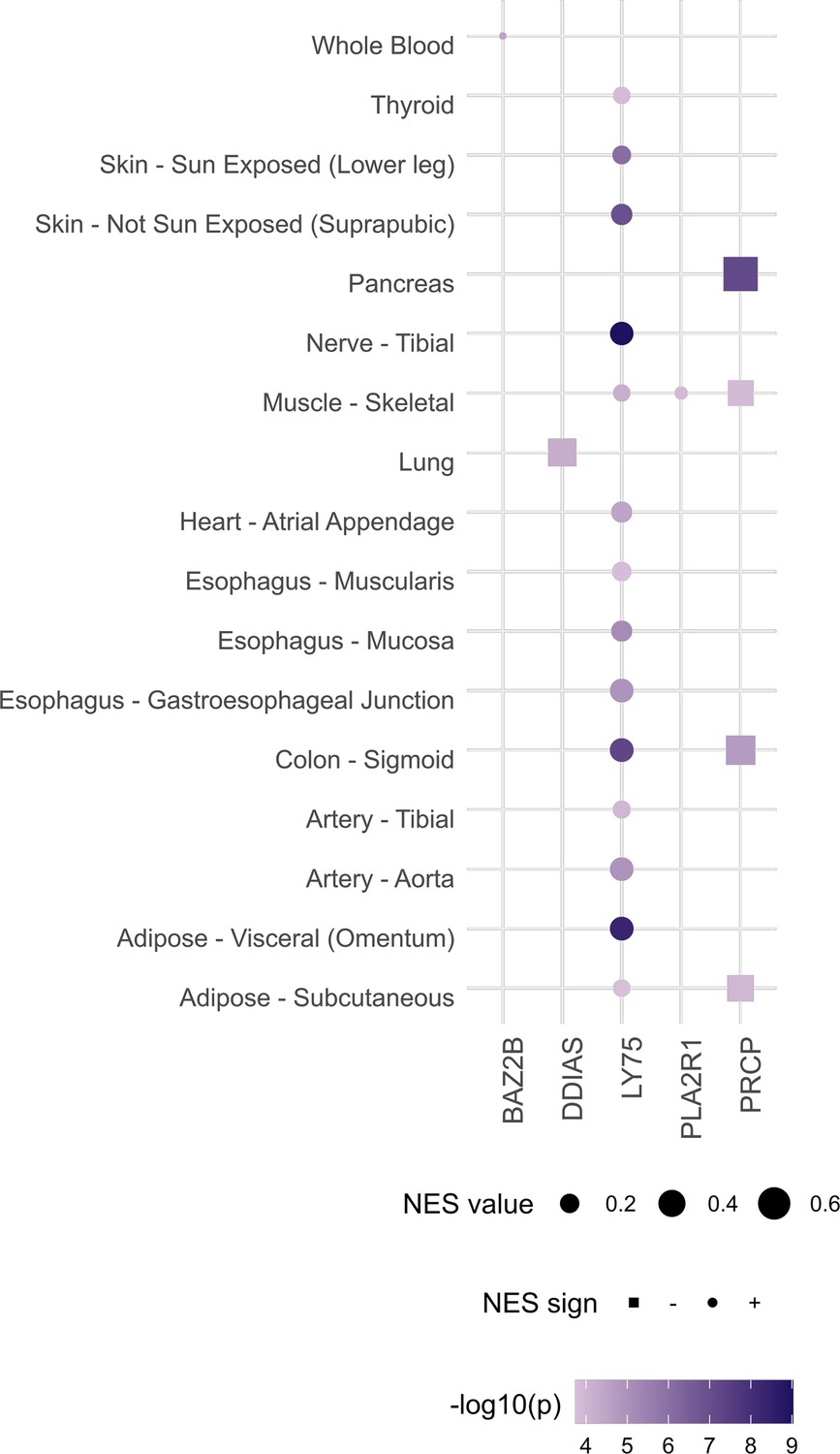
Gene‒tissue pairs for which either rs1003835 or rs60606421 are significant expression quantitative trait loci (eQTL) at false discovery rate (FDR) < 0.05 (data retrieved from https://gtexportal.org/home/snp/).
rs1003835 (chromosome 2) maps to BAZ2B, LY75, and PLA2R1 genes. As for the lead variant of chromosome 11, rs77599934, since it was not an eQTL, we used an LD proxy variant (rs60606421). DDIAS and PRCP genes map closely to this variant. NES and p-values correspond to the normalized effect size (and direction) of eQTL-gene associations and the p-value for the tissue, respectively.
Figure 5
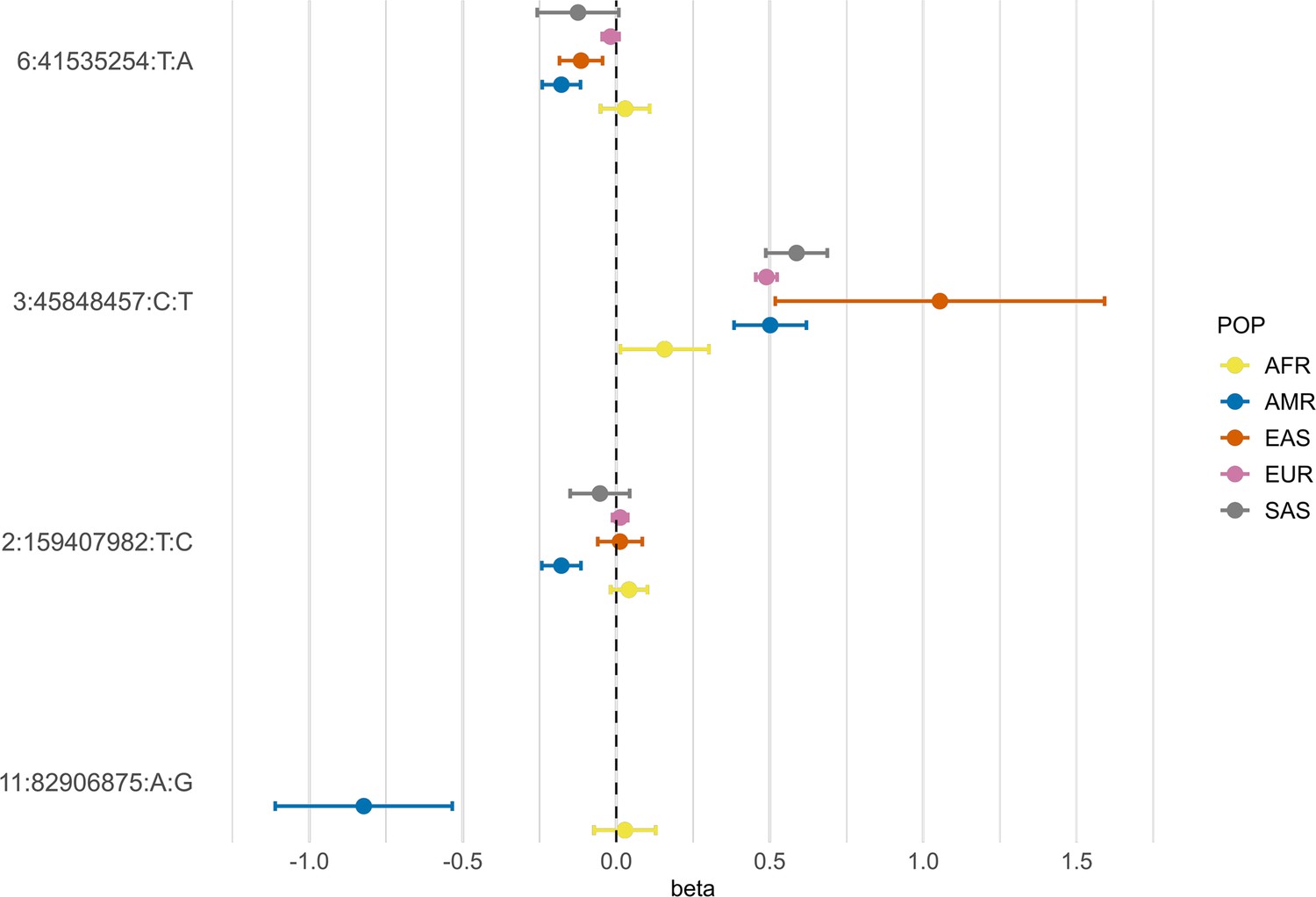
Forest plot showing effect sizes and the corresponding confidence intervals for the sentinel variants identified in the AMR meta-analysis across populations.
All beta values with their corresponding CIs were retrieved from the B2 population-specific meta-analysis from the HGI v7 release, except for AMR, for which the beta value and IC from the HGIAMR-SCOURGE meta-analysis are represented.
Figure 6
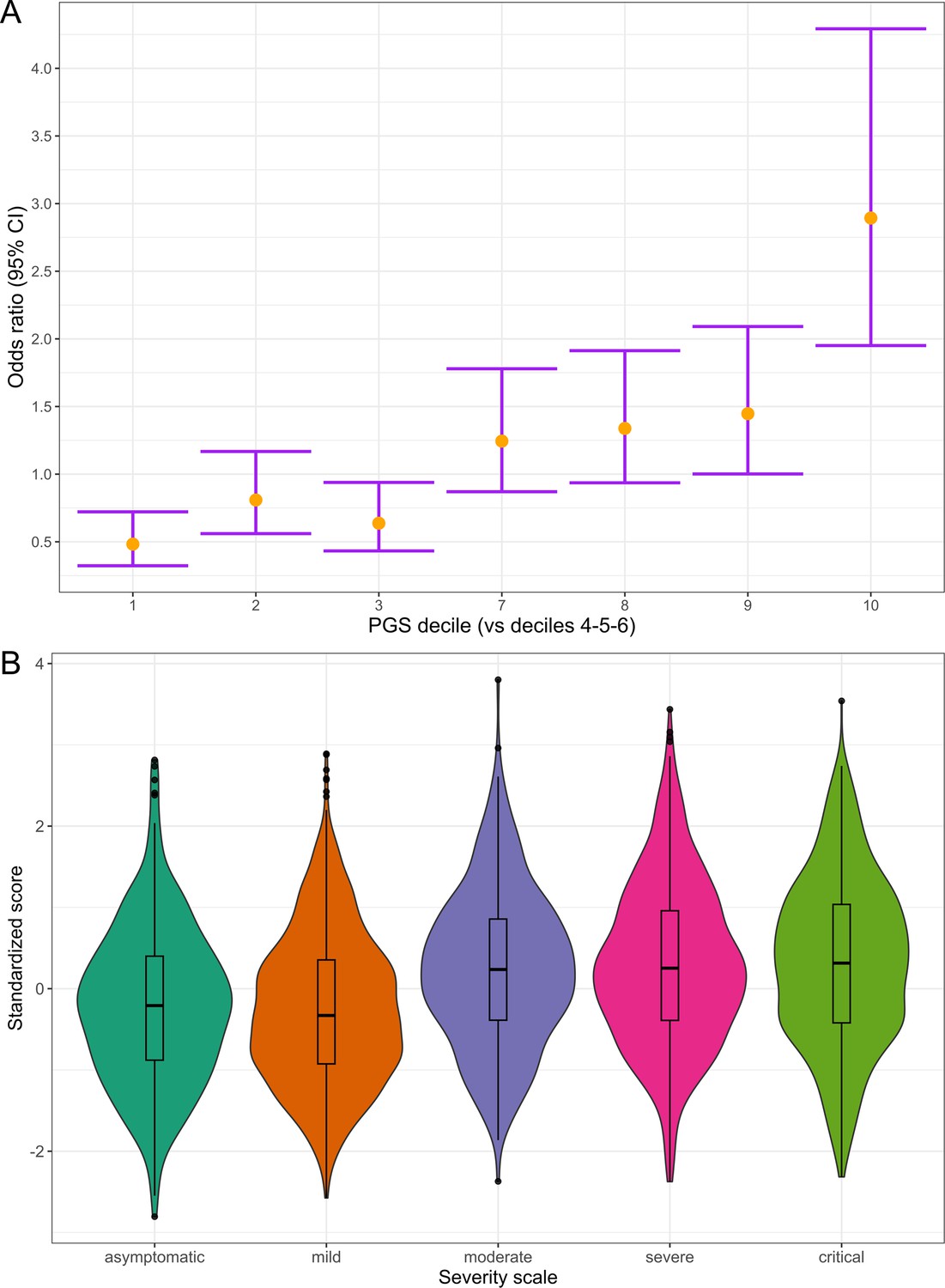
Polygenic risk distribution for COVID-19 hospitalization.
(A) Polygenic risk stratified by polygenic risk score (PGS) deciles comparing each risk group against the lowest risk group (OR–95% CI). (B) Distribution of the PGS in each of the severity scale classes. 0, asymptomatic; 1, mild disease; 2, moderate disease; 3, severe disease; 4, critical disease.
Tables
Table 1
Demographic characteristics of the SCOURGE Latin American cohort.
| Variable | Non-hospitalized (N = 1887) | Hospitalized(N = 1625) | |
|---|---|---|---|
| Age, mean years ±SD | 39.1 ± 11.9 | 54.1 ± 14.5 | |
| Sex, N (%) | |||
| Female (%) | 1253 (66.4) | 668 (41.1) | |
| Global genetic inferred ancestry, % mean ± SD | |||
| European | 54.4 ± 16.2 | 39.4 ± 20.7 | |
| African | 15.3 ± 12.7 | 9.1 ± 11.6 | |
| Native American | 30.3 ± 19.8 | 51.3 ± 26.5 | |
| Comorbidities, N (%) | |||
| Vascular/endocrinological | 488 (25.9) | 888 (64.5) | |
| Cardiac | 60 (3.2) | 151 (9.3) | |
| Nervous | 15 (0.8) | 61 (3.8) | |
| Digestive | 14 (0.7) | 33 (2.0) | |
| Onco-hematological | 21 (1.1) | 48 (3.00) | |
| Respiratory | 76 (4.0) | 118 (7.3) | |
Table 2
Lead independent variants in the admixed AMR genome-wide association studies (GWAS) meta-analysis.
| SNP rsID | chr:pos | EA | NEA | OR (95% CI) | p-Value | EAF cases | EAF controls | Nearest gene | Mamba PPR |
|---|---|---|---|---|---|---|---|---|---|
| rs13003835 | 2:159407982 | T | C | 1.20 (1.12–1.27) | 3.66E-08 | 0.563 | 0.429 | BAZ2B | 0.30 |
| rs35731912 | 3:45848457 | T | C | 1.65 (1.47–1.85) | 6.30E-17 | 0.087 | 0.056 | LZTFL1 | 0.95 |
| rs2477820 | 6:41535254 | A | T | 0.84 (0.79–0.89) | 1.89E-08 | 0.453 | 0.517 | FOXP4-AS1 | 0.18 |
| rs77599934 | 11:82906875 | G | A | 2.27 (1.7–3.04) | 2.26E-08 | 0.016 | 0.011 | DDIAS | 0.95 |
-
EA: effect allele; NEA: noneffect allele; EAF: effect allele frequency in the SCOURGE study; PPR: posterior probability of replicability.
Table 3
Novel variants in the SC-HGIALL and SC-HGI3POP meta-analyses (with respect to HGIv7).
Independent signals after LD clumping.
| SNP rsID | chr:pos | EA | NEA | OR (95% CI) | p-Value | Nearest gene | Analysis |
|---|---|---|---|---|---|---|---|
| rs76564172 | 16:3892266 | T | G | 1.31 (1.19–1.44) | 9.64E-09 | CREBBP | SC-HGI3POP |
| rs66833742 | 19:4063488 | T | C | 0.94 (0.92–0.96) | 1.89E-08 | ZBTB7A | SC-HGI3POP |
| rs66833742 | 19:4063488 | T | C | 0.94 (0.92–0.96) | 2.50E-08 | ZBTB7A | SC-HGIALL |
| rs2876034 | 20:6492834 | A | T | 0.95 (0.93–0.97) | 2.83E-08 | CASC20 | SC-HGIALL |
-
EA: effect allele; NEA: non-effect allele.
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Commercial assay or kit | Chemagic DNA Blood 100 kit | PerkinElmer Chemagen Technologies GmbH | ||
| Software, algorithm | Axiom Analysis Suite | Thermo Fisher Scientific | Version 4.0.3.3 | |
| Software, algorithm | PLINK | Purcell et al., 2007; https://www.cog-genomics.org/plink/ | RRID:SCR_001757 | Version 1.9; v2 |
| Software, algorithm | TOPMed Imputation Server | https://imputation.biodatacatalyst.nhlbi.nih.gov/ | Version 2 | |
| Software, algorithm | ADMIXTURE | Alexander et al., 2009; https://dalexander.github.io/admixture/ | RRID:SCR_001263 | Version 1.3.0 |
| Software, algorithm | SAIGEgds | Zheng and Davis, 2021; https://www.bioconductor.org/packages/release/bioc/html/SAIGEgds.html | Version 1.10.0 | |
| Software, algorithm | METAL | Willer et al., 2010; https://csg.sph.umich.edu/abecasis/metal/ | RRID:SCR_002013 | Version 2011-03-25 |
| Software, algorithm | FUMA | Watanabe et al., 2017; https://fuma.ctglab.nl/ | RRID:SCR_017521 | Version 1.5.2 |
| Software, algorithm | MAMBA | McGuire et al., 2021; https://github.com/dan11mcguire/mamba | Version 1 | |
| Software, algorithm | S-PrediXcan; S-MultiXcan | Barbeira et al., 2018; https://github.com/hakyimlab/MetaXcan | RRID:SCR_016739 | Version 1 |
| Software, algorithm | GTEx v8 mashr prediction models | https://predictdb.org/post/2021/07/21/gtex-v8-models-on-eqtl-and-sqtl/ | ||
| Other | GWAS Catalog | https://www.ebi.ac.uk/gwas/ | RRID:SCR_012745 | Section ‘Definition of the genetic risk loci and putative functional impact’ |
Additional files
-
Supplementary file 1
Participating centers.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp1-v1.xlsx
-
Supplementary file 2
Independent variants with p-value<1 × 10–05 in the SC-HGI_AMR GWAS meta-analysis (hg38).
EA: effect allele; NEA: non-effect allele; EAF: effect allele frequency; EAF_avg: averaged effect allele frequency; FreqSE: standard error of averaged effect allele frequency; SCOURGE_AMR: SCOURGE Latin-America; HGIB2_AMR: HGI meta-analysis of AMR studies.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp2-v1.xlsx
-
Supplementary file 3
Annotated SNPs in moderate-to-strong LD with lead SNPs of the genome-wide significant loci in the SC-HGI_AMR GWAS meta-analysis, with ANNOVAR.
NEA: non-effect allele; EA: effect allele; r2: maximum r2 of the SNP with one of the independent SNPs; IndSigSNP: the independent SNP which has the maximum r2 value with the SNP; dist: distance to the nearest gene; func: functional consequence of the SNP on the gene; CADD: CADD score; RDB: RegulomeDB score; minChrState: the minimum 15-core chromatin state across 127 tissues/cell types; commonChrState: the most common 15-core chromatin state across 127 tissues/cell types; posMapFilt: 1 if the SNP was used for positional mapping, 0 otherwise; eqtlMapFilt: 1 if the SNP was used for eQTL mapping, 0 otherwise.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp3-v1.xlsx
-
Supplementary file 4
Results from the MAGMA gene-based analysis in the SC-HGI_AMR GWAS meta-analysis (hg37).
NSNPS: number of SNPs in the gene; NPARAM: the number of relevant parameters used in the model; ZSTAT: z statistics.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp4-v1.xlsx
-
Supplementary file 5
Prioritized genes by eQTL and positional mapping by FUMA in the SC-HGI_AMR GWAS meta-analysis results (hg37).
HUGO: HGNC gene symbol; pLI: pLI score from ExAC database, probability of being intolerant to loss of function (higher the score, higher the intolerance); ncRVIS: non-coding residual variation intolerance score (higher the score, higher intolerance to non-coding variation); posMapSNPs: number of SNPs mapped by positional mapping; posMapMaxCADD: the maximum CADD score of mapped SNPs by positional mapping; eqtlMapSNPS: the number of SNPs mapped to the genes based on eQTL mapping; eqtlMapminP: the minimum eQTL p-value of mapped SNPs; eqtlMapminQ: the minimum eQTL FDR of mapped SNPs; eqtlMapts: tissue of mapped eQTLs; eqtlDirection: consequential direction of mapped eQTL SNPs after aligning the risk alleles; minGwasP: minimum GWAS p-value of mapped eQTLs; IndSigSNPs: independent SNPs that are in LD with the mapped SNPs.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp5-v1.xlsx
-
Supplementary file 6
Fine-mapped credible set derived with corrcoverage (95%) for the associated region in chromosome 2 (BAZ2B).
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp6-v1.xlsx
-
Supplementary file 7
VEP annotations for the variants included in the fine-mapped credible sets for the novel associated loci in chromosome 2 (hg38).
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp7-v1.xlsx
-
Supplementary file 8
V2G scores for the variants included in the fine-mapped credible sets in the novel risk loci from chromosomes 2 and 16 (hg38).
Shaded in green, the prioritized gene by the V2G score.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp8-v1.xlsx
-
Supplementary file 9
MultiXcan results for the SC-HGI_AMR GWAS meta-analysis.
N: number of tissues available for the gene; n_indep: number of independent components of variation kept among the tissues' predictions; p_i_best: best p-value of single tissue S-prediXcan association; t_i_best: name of best single tissue S-prediXcan association; p_i_worst: worst p-value of single tissue S-prediXcan association; t_i_worst: name of worst single tissue S-prediXcan association; eigen_max: eigenvalue of the top independent component in the SVD decomposition of predicted expression correlation; eigen_min: eigenvalue of the last independent component in the SVD decomposition of predicted expression correlation; eigen_min_kept: eigenvalue of the smallest independent component that was kept in the SVD decomposition of predicted expression correlation; z_min: minimum z-score among single-tissue S-prediXcan associations; z_max: maximum z-score among single-tissue S-prediXcan associations; z_mean: mean z-score among single tissue S-prediXcan associations; z_sd: standard deviation of the mean z-score among single-tissue S-prediXcan associations; tmi: trace of T*T', where T is the correlation of predicted expression levels for different tissues multiplied by its SVD pseudo-inverse and is an estimate for the number of independent components of variation in predicted expression across tissues.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp9-v1.xlsx
-
Supplementary file 10
Top 10 genes for the TWAS trained with the GALA II-SAGE models in admixed Americans.
Bonferroni correction thresholds: Pooled p<4.19E-06; PR p<4.99E-06; MX p<5.19E-06; AA p<4.67E-06. Var_g: variance of the gene expression; pred_perf_r2: cross-validated R2 of tissue model’s correlation to gene’s measured transcriptome; pref_perf_qval: qval of tissue model’s correlation to gene’s measured transcriptome; n_snps_used: number of snps from GWAS used in S-prediXcan analysis; n_snp_in_cov: number of snps in the covariance matrix; n_snps_in_model: number of snps in the model; best_gwas_p: the highest p-value from GWAS snps used in this model; largest_weight: the largest weight in this model.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp10-v1.xlsx
-
Supplementary file 11
Independent variants with p-value<1e-05 in the SC-HGI_ALL GWAS meta-analysis (hg38).
EA: effect allele; NEA: non-effect allele; EAF_avg: averaged effect allele frequency; FreqSE: standard error of averaged effect allele frequency.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp11-v1.xlsx
-
Supplementary file 12
Results of the 40 lead variants associated with COVID-19 hospitalization in the HGIv7 (hg38).
SC-HGI_ALL: meta-analysis SCOURGE-HGI_ALL; SC-HGI_AMR: meta-analysis SCOURGE-HGI_AMR; SC-HGI_3POP: meta-analysis SCOURGE-HGI_3POP.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp12-v1.xlsx
-
Supplementary file 13
Independent variants with p-value<1e-05 in the SC-HGI_3POP GWAS meta-analysis (hg38).
EA: effect allele; NEA: non-effect allele; EAF_avg: average effect allele frequency; FreqSE: standard error of averaged effect allele frequency.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp13-v1.xlsx
-
Supplementary file 14
Instruments used in the polygenic risk score model (hg38).
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp14-v1.xlsx
-
Supplementary file 15
Multinomial regression results.
Reference class for the multinomial regression is ‘asymptomatic’.
- https://cdn.elifesciences.org/articles/93666/elife-93666-supp15-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/93666/elife-93666-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Novel risk loci for COVID-19 hospitalization among admixed American populations
eLife 13:RP93666.
https://doi.org/10.7554/eLife.93666.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}