RNA tertiary structure and conformational dynamics revealed by BASH MaP
- Department of Pharmacology, Weill Medical College, Cornell University, United States
Figures
Figure 1 with 1 supplement
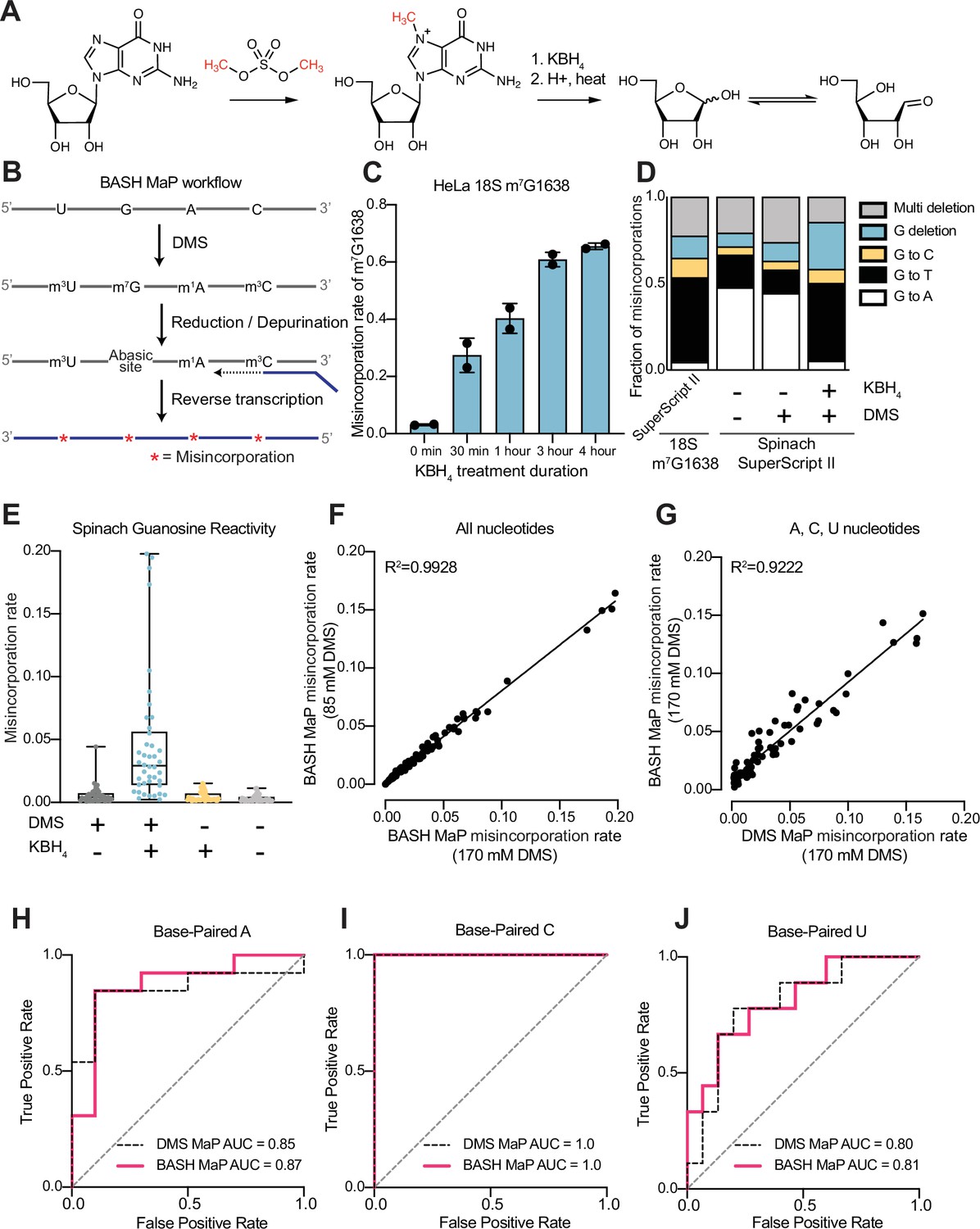
Reduction and depurination of DMS-modified RNA enables detection of N7G-DMS adducts with misincorporations.
(A) Strategy for the misincorporation detection of N7G methylation. N7-methylated G is reduced by potassium borohydride and then heated in acidic conditions to yield an abasic site. The abasic site then proceeds to induce misincorporations in the cDNA following reverse transcription. (B) Overall schematic for the BASH MaP experimental workflow. RNA is first treated with dimethyl sulfate (DMS) which produces the following adducts: m1A, m3C, m7G, and to a lesser extent m1G and m3U. DMS-modified RNA is then subjected to reduction by potassium borohydride (800 mM) for 4 hr at room temperature followed by purification and heating in a pH 2.9 buffer of acetic acid and sodium acetate for 4 hr at 45 °C. RNA is then purified and subjected to reverse transcription with enzymes and buffer conditions which promote cDNA misincorporations at methylated bases. (C) Optimization of reduction duration and efficiency of abasic sites to induce misincorporations. To determine the optimal reduction duration, we treated total HeLa RNA with potassium borohydride (800 mM) for various durations of time, performed depurination for 4 hr in acid buffer, and then prepared RT-PCR amplicons surrounding the endogenously methylated base m7G1638 in the 18 S rRNA. We then quantified the fraction of reads containing a misincorporations at G1638 divided by the total number of reads to yield a misincorporations rate at G1638 following amplicon sequencing. Plots of misincorporations rates revealed that 4 hr of borohydride treatment induced an average misincorporations rate of 70% at G1638. (D) Misincorporation signature of abasic sites under reverse transcription conditions for detection of methylated bases. On the left, quantification of the types of misincorporations at G1638 as described in (C) for SuperScript II, a reverse transcriptase enzyme commonly used to detect methylated bases with cDNA misincorporations. On the right, fraction of each type of misincorporation calculated collectively from all G residues in Spinach following reverse transcription with SuperScript II. Spinach RNA was either modified with DMS (170 mM) for 8 min at 25 °C or treated with an ethanol control. Modified and control RNA was then reduced with potassium borohydride (800 mM) for 4 hr or incubated in water for 4 hr. All three Spinach samples underwent identical heating in acidic buffer conditions before undergoing reverse transcription. Comparison of types of misincorporations shows that reduction of DMS-treated Spinach RNA produces a misincorporation signature at G residues which mirrors the positive control G1638 when reverse transcribed with SuperScript II. (E) Reduction of DMS-treated Spinach RNA produces novel misincorporation data at G bases. To determine if Spinach is highly modified by DMS at N7G, we utilized the experimental data as described in (E) with an additional control group in which DMS was omitted but the sample underwent reduction and depurination. Not shown, all four samples underwent identical heating in acidic buffer prior to reverse transcription. We then plotted the misincorporation rate of each G in Spinach for each experimental condition. This misincorporation rate reveals a dramatic increase in misincorporation rates for G bases in Spinach modified with DMS and reduced with potassium borohydride. (F) Reproducibility of BASH MaP. Spinach RNA was probed with either 85 mM or 170 mM DMS for 8 min at 25 °C and then reduced and depurinated. The misincorporation rate at each position in Spinach was compared between the two samples and a linear regression was performed which showed an R2 of 0.9928 demonstrating high reproducibility. (G) Effect of reduction and depurination on the detection of m1A, m3C, and m3U. To determine whether reduction and depurination of DMS-treated RNA impaired the detection of other methylated bases, we treated Spinach with DMS (170 mM) for 8 min at 25 °C using buffer conditions which promote the methylation of m1G and m3U (Mustoe et al., 2019; Mitchell et al., 2023). Then, DMS-treated Spinach was either directly reverse transcribed (DMS MaP) or subjected to reduction and depurination (BASH MaP) before reverse transcription. We then compared the misincorporation rate at each A, C, and U position in Spinach and performed a linear regression. The R2 of 0.9222 demonstrates that reduction and depurination do not impair the detection of m1A, m3C, and m3U generated by the modification of RNA by DMS. (H–J) Receiver operator characteristic curves demonstrate that BASH MaP identifies single-stranded regions of RNA. To determine if BASH MaP could accurately distinguish single-stranded from base-paired A, C, and U bases, we constructed Receiver Operator Characteristic (ROC) curves for A, C, and U bases for both BASH MaP and DMS MaP as described in (G). The larger the AUC, the better a method is at discriminating paired vs unpaired RNA bases. An AUC = 1.0 demonstrates perfect discrimination ability. Panels (H–J) demonstrate that BASH MaP accurately discriminates between single-stranded and base-paired A, C, and U bases.
Figure 1—figure supplement 1
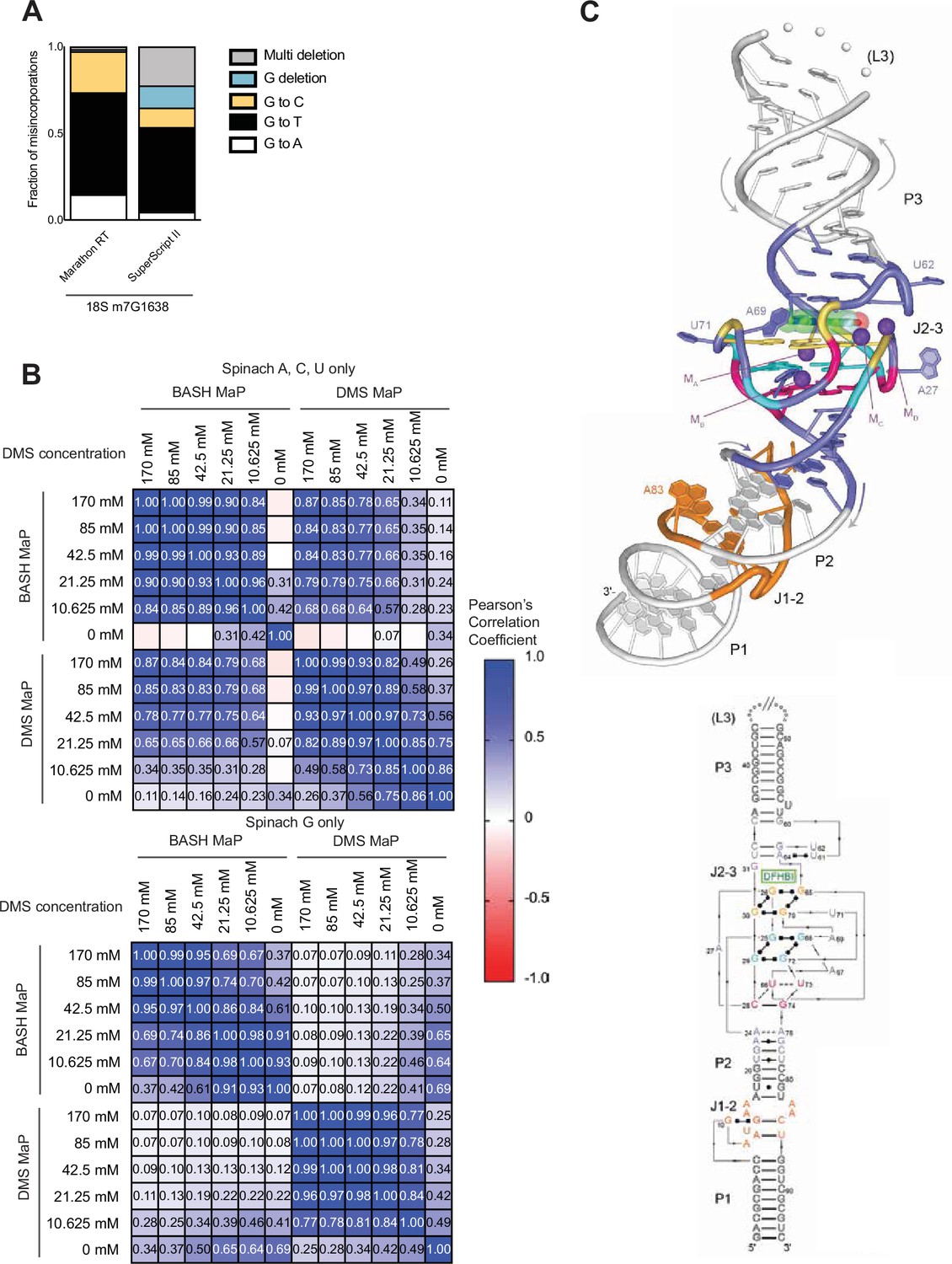
Comparison of reverse transcriptases on the misincorporation signature of abasic sites and BASH MaP reproducibility and correlation with DMS MaP.
(A) The misincorporation signature of abasic sites generated by Marathon RT exhibits fewer deletions than SuperScript II. In Figure 1D, we examined the misincorporation signature of G’s after DMS MaP and BASH MaP with SuperScript II and found high rates of deletion. We therefore asked if the high deletion rate is due to the choice of reverse transcriptase or if it is an intrinsic property of abasic sites during reverse transcription. The mechanism of deletion is important because deletions must be ignored in further data processing steps as these misincorporation are often unable to align to a specific guanine if there are two or more consecutive guanines. We chose to test Marathon RT as this reverse transcriptase was previously shown to induce lower deletion rates than SuperScript II (Guo et al., 2020). As shown, Marathon RT produces predominantly G→T transition and with an overall much lower rate of deletions during reverse transcription of abasic sites. Thus, the deletions seen in SuperScript II BASH MaP data are likely the result of features unique to the SuperScript II enzyme. Future implementations of BASH MaP may benefit from using Marathon RT. (B) BASH MaP and DMS MaP display high reproducibility and show high correlation for A, C, and U bases. In Figure 1F–G, we establish the reproducibility of BASH MaP and show that misincorporation data at A, C, and U bases is not compromised by BASH treatment. Here, we exhaustively examine the reproducibility and correlation between BASH MaP and DMS MaP protocols. Spinach RNA was first probed with the indicated concentrations of DMS for 8 min at 25 °C using bicine (200 mM) pH 7.75 buffer conditions (Mustoe et al., 2019). Then, DMS-treated RNA was split into two fractions. One fraction underwent BASH treatment followed by reverse transcription (BASH MaP), while the other fraction was immediately reverse transcribed with SuperScript II (DMS MaP). We then sequenced the RNA, calculated the misincorporation rate for each nucleotide, and calculated a Pearson’s correlation coefficient between each sample. A, C, and U nucleotide data was analyzed separately from G data. The heatmap of correlation coefficients shows that both BASH MaP and DMS MaP display high reproducibility over a range of DMS concentrations. Furthermore, BASH MaP and DMS MaP display strong correlations for A, C, and U misincorporation data at higher DMS concentrations. As expected, DAGGER MaP and DMS MaP display no correlation among G misincorporation data. (C) Spinach crystal structure 4TS2 (Warner et al., 2014). Top, Spinach crystallographic structure. Bottom, partial secondary structure schematic of the Spinach crystal structure with annotated domains. G-quadruplex G’s are indicated below in orange and blue. The mixed tetrad is indicated below in pink.
Figure 2 with 1 supplement
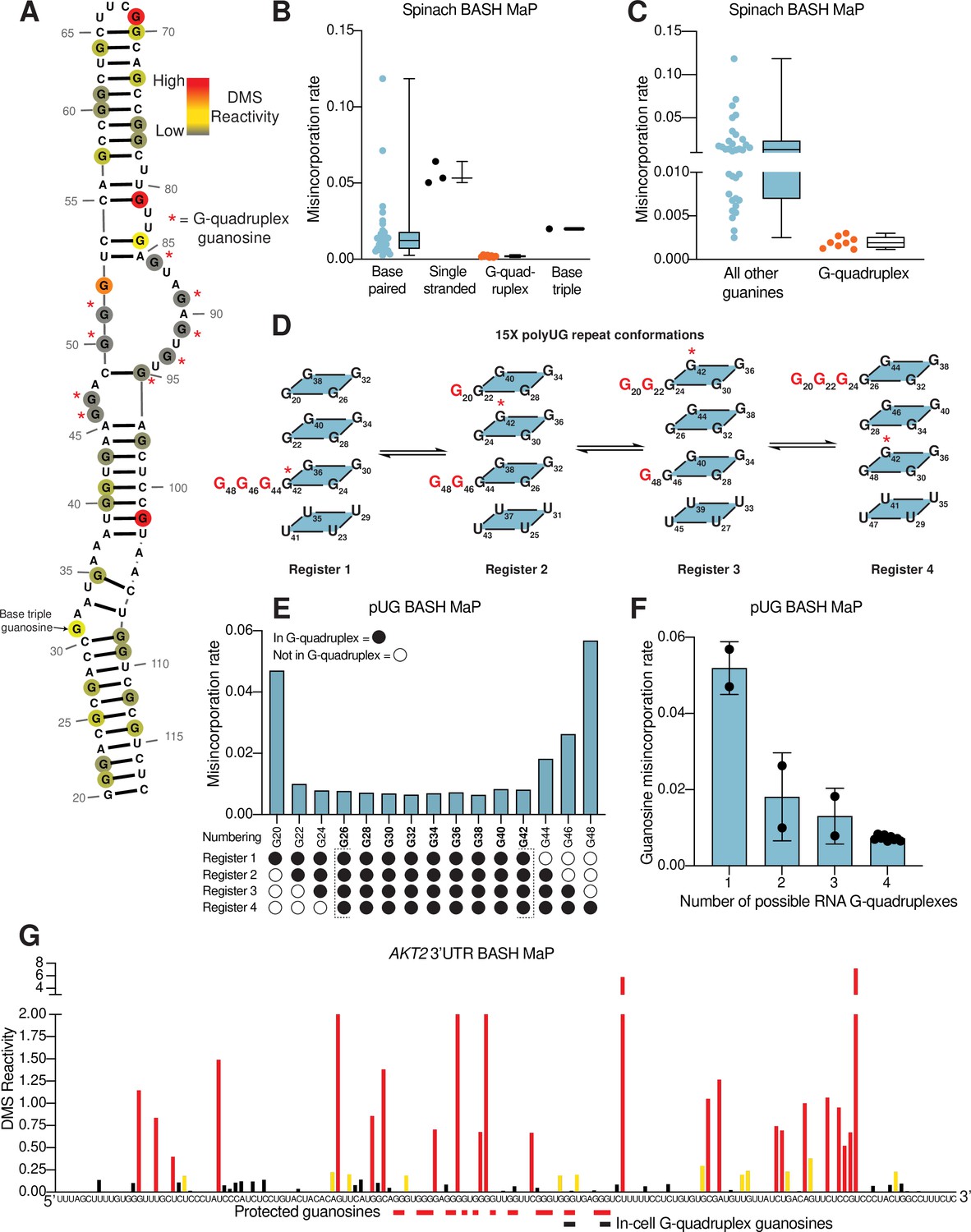
N7G reactivity reveals guanosines involved in tertiary interactions.
(A) N7G reactivity overlaid on the secondary structure model of Spinach as derived from the crystal structure 4TS2 (Warner et al., 2014). G-quadruplex G’s are indicated with a red asterisk. N7G reactivity is colored on a continuous gradient from grey (low reactivity) to yellow (moderate reactivity) to red (high reactivity). N7G reactivities represent normalized values derived from misincorporation rates. Spinach BASH MaP data was obtained as described in Figure 1G and no background subtraction was utilized. (B) Comparison between structural features in Spinach and observed BASH MaP misincorporation rate at G bases. To determine which structural features impact N7G methylation rate we utilized the crystal structure 4TS2, and classified individual G’s as either base-paired, single-stranded, engaged in an RNA G-quadruplex, or engaging in a base triple. This plot reveals single-stranded G’s have relatively high methylation rates whereas RNA G-quadruplex G’s are strongly protected from N7G methylation. (C) Comparison between RNA G-quadruplex G’s and all other G’s in Spinach reveals RNA G-quadruplex G’s are the most protected G’s from N7G methylation in Spinach. (D) Schematic of alternative conformations the 15 x polyUG (pUG) repeat RNA can adopt in solution. The 15 x pUG repeat can adopt four distinct RNA G-quadruplex conformations of 12 consecutive GU repeats. Each of these four distinct conformations is indicated as Register 1 through 4. Each register is uniquely defined by a set of three G’s not engaged in an RNA G-quadruplex (colored red). These G’s are predicted to be single stranded or base-paired and thus display much higher reactivity than the G’s engaged in the RNA G-quadruplex. To show how the G-quadruplex is rearranged in each conformation, G42 is labeled with a red asterisk. (E) BASH MaP misincorporations rate plot of pUG G’s. 15 x pUG RNA was refolded in potassium buffer (100 mM) and modified with DMS (170 mM) for 6 min at 37 °C. G’s engaged in a G-quadruplex for each unique register are indicated at the bottom of the plot. G’s predicted to be engaged in a G-quadruplex in each of the four registers is bolded and bracketed below the plot. The misincorporation rate plot reveals that G’s engaged in a G-quadruplex in all four registers display a strong protection from DMS methylation. Consequently, G20 and G48, which are only predicted to be protected in a single register show the highest misincorporation rates. (F) Quantification of (E) shows the relationship between G inclusion in RNA G-quadruplex registers and measured misincorporation rate. The plot reveals increased protection from N7G methylation for G’s as the number of G-quadruplex conformations increases. (G) BASH MaP of a G-quadruplex in the 3’UTR of the AKT2 mRNA. SH-SY5Y cells were treated with DMS (170 mM) for 6 min at 37 °C. Primers were designed to specifically amplify the putative G-quadruplex region in the AKT2 mRNA. Misincorporation rates were converted to normalized reactivity values as described in METHODS. Putative G-quadruplex G’s as identified in rG4-seq (Warner et al., 2014) are highlighted in red. Red bars below the sequence indicate putative G-quadruplex G’s with strong protections from N7G-DMS methylations. G’s previously identified as engaged in a G-quadruplex in cells are indicated below with a black bar. The normalized reactivity plot reveals N7G protections from DMS at previously identified in cell G-quadruplex G’s as well as other G-tracts. Together, these data support the formation of a G-quadruplex in 3’UTR of the AKT2 mRNA in SH-SY5Y cells.
Figure 2—figure supplement 1
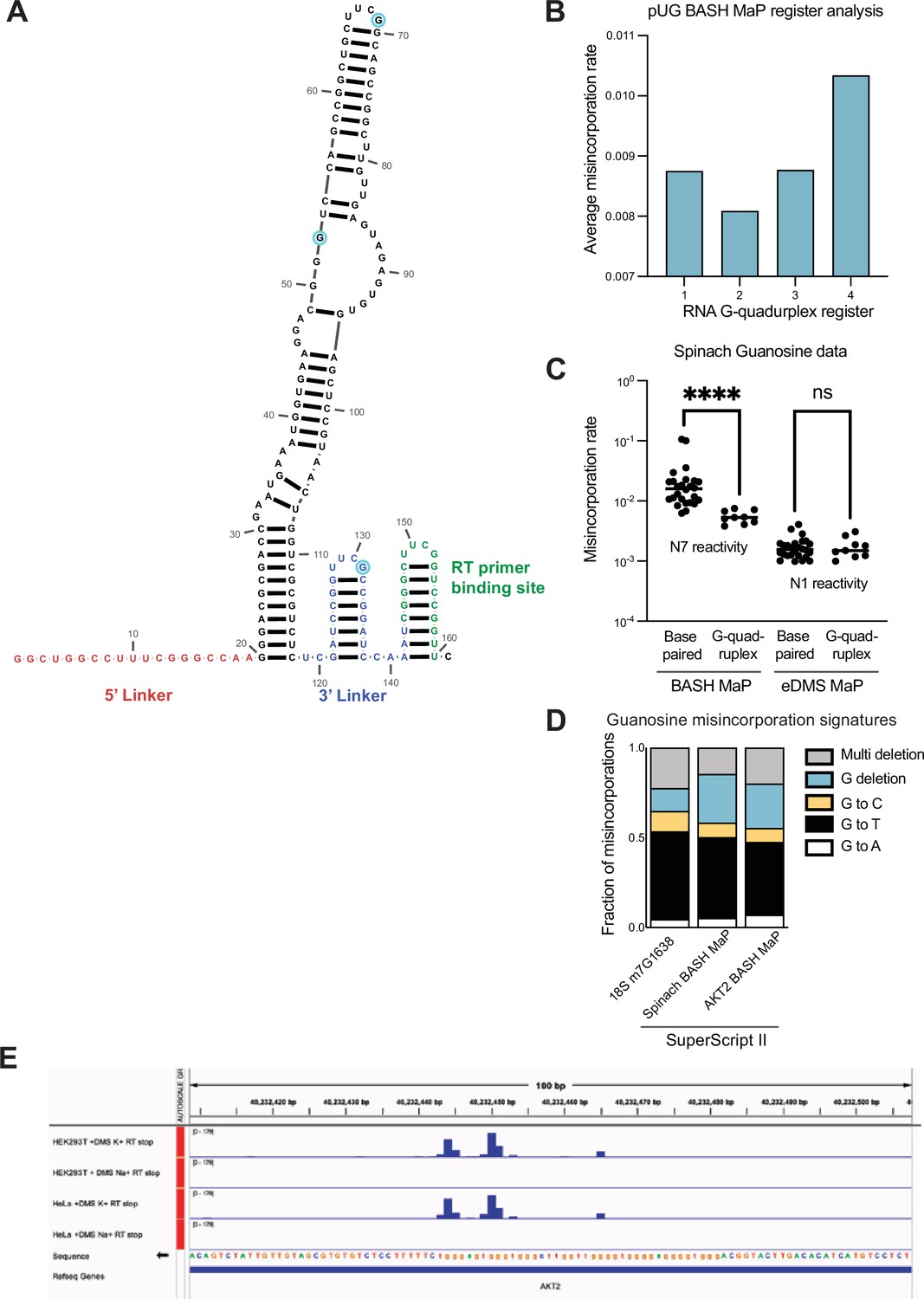
DMS MaP is unable to efficiently detect accessibility of the N7 position in guanine and validation of AKT2 N7G protections in a detected G-quadruplex.
(A) Secondary structure of Spinach RNA model used for in vitro probing experiments. To aid with DMS/BASH MaP library preparation, Spinach was in vitro transcribed with an additional 5’ linker sequence (red) as well as with an additional 3’ linker (blue) and RT primer binding site (green). Due to the use of PCR in library preparation, misincorporation data is unavailable for the 5’ linker and RT primer binding site in the utilized Spinach construct. Three single-stranded G’s in the Spinach structure cassette are highlighted in light blue. The secondary structure model for Spinach was derived from the crystal structure (PDB: 4TS2) and linker regions were folded using mFOLD (Zuker, 2003). (B) Conformational analysis of alternative 15 x polyUG G-quadruplex registers. In Figure 2. (E–F) we found that different G-quadruplex registers can be differentiated by N7G reactivity data. It seemed likely that the 15 x polyUG repeat RNA populates each register with equal stoichiometry. To test this, we reasoned that if each register were equally populated, then the average misincorporation rate of the G’s engaged in a G-quadruplex for each register should be the same. Therefore, we asked whether the average misincorporation rate of the G-quadruplex G’s for each register was the same. Surprisingly, we found that Register 2 displayed the lowest average misincorporation rate among the four registers which suggests that Register 2 is populated with a higher stoichiometry than the other three registers. (C) Comparison of BASH MaP and DMS MaP for discrimination of base-paired G’s versus G’s engaged in a G-quadruplex. Chemical probes that react with the Watson-Crick face of G have previously been used to assess the folding state of G-quadruplexes (Weng et al., 2020). We reasoned that this strategy to profile G-quadruplex folding is prone to false positives because chemical probes that react with the Watson-Crick face, i.e., the N1 position of G, should show reduced chemical reactivity both when a G is base paired and when a G is in a G-quadruplex. Therefore, we asked whether DMS MaP optimized to methylate and detect N1G adducts (eDMS MaP) could differentiate between base-paired G’s and G-quadruplex G’s (Mitchell et al., 2023). To test this, we probed Spinach with DMS (170 mM) for 6 min at 37 °C using bicine (200 mM) pH 8.37 buffer conditions to promote the formation of m1G. To specifically detect m1G adducts, we utilized the property of SuperScript II to specifically encode m1G adducts as G→T and G→C misincorporations. We then prepared a BASH MaP library from the same DMS-treated Spinach sample and compared the two methods for their ability to differentiate G’s engaged in a G-quadruplex. We assigned G’s to either base-paired or G-quadruplex groups, plotted misincorporation rates and performed a Mann-Whitney U test to determine if the groups were statistically different from each other. On the left, BASH MaP, which measures N7 reactivity, clearly differentiates G-quadruplex G’s from base-paired G’s. On the right, eDMS MaP, which measures N1 reactivity, was unable to differentiate G-quadruples G’s from base-paired G’s. Taken together, this data shows that BASH MaP uniquely discriminates G’s engaged in G-quadruplexes from G’s engaged in base-paired interactions. ****p<0.0001, ns p=0.9852, Mann-Whitney U test. (D) Misincorporation signature of G’s in BASH MaP of AKT2. We wanted to validate whether misincorporation data at G’s in BASH MaP of AKT2 produced misincorporations consistent with methylation at N7G. To test this, we compared the misincorporation signature of all G’s in AKT2 with the misincorporation signature of G’s in BASH-treated Spinach and at the 18 S m7G1638 site. All three experiments utilized SuperScript II as the reverse transcriptase. Comparison of misincorporation signatures for the three experiments revealed a common G→T and G deletion misincorporation signature. Consistent misincorporation signatures indicates that misincorporations at G’s in AKT2 represent N7G methylation events. (E) AKT2 3’UTR contains an in cellulo folded G-quadruplex. We wanted to test whether BASH MaP could identify N7G sites with low reactivity in cells. To identify a candidate G-quadruplex that is likely to be folded in cells, we turned to a previously published study which measured in cell G-quadruplex folding (Guo and Bartel, 2016). This study used a method that combined in cell-DMS treatment and subsequent RT stops. In brief, the in-cell DMS treatment methylates all N7 positions not engaged in a G-quadruplex. Then, cellular RNA is purified before undergoing refolding and RT stop profiling in K+vs Na +buffer. The presence of RT stops in K+buffer implies that a G-quadruplex was present in the cell, protected from DMS, and able to refold into a G-quadruplex in the RT step. Shown are potassium-dependent RT stops in both HeLa and HEK293T cells. Thus, this region of the AKT2 3’UTR contains a folded G-quadruplex in cells, of an unknown topology or conformation. The presence of multiple RT stop sites, which occur at the 3’ end of G-quadruplexes, suggests that this region adopts multiple G-quadruplex conformations with different 3’ ends.
Figure 3 with 2 supplements
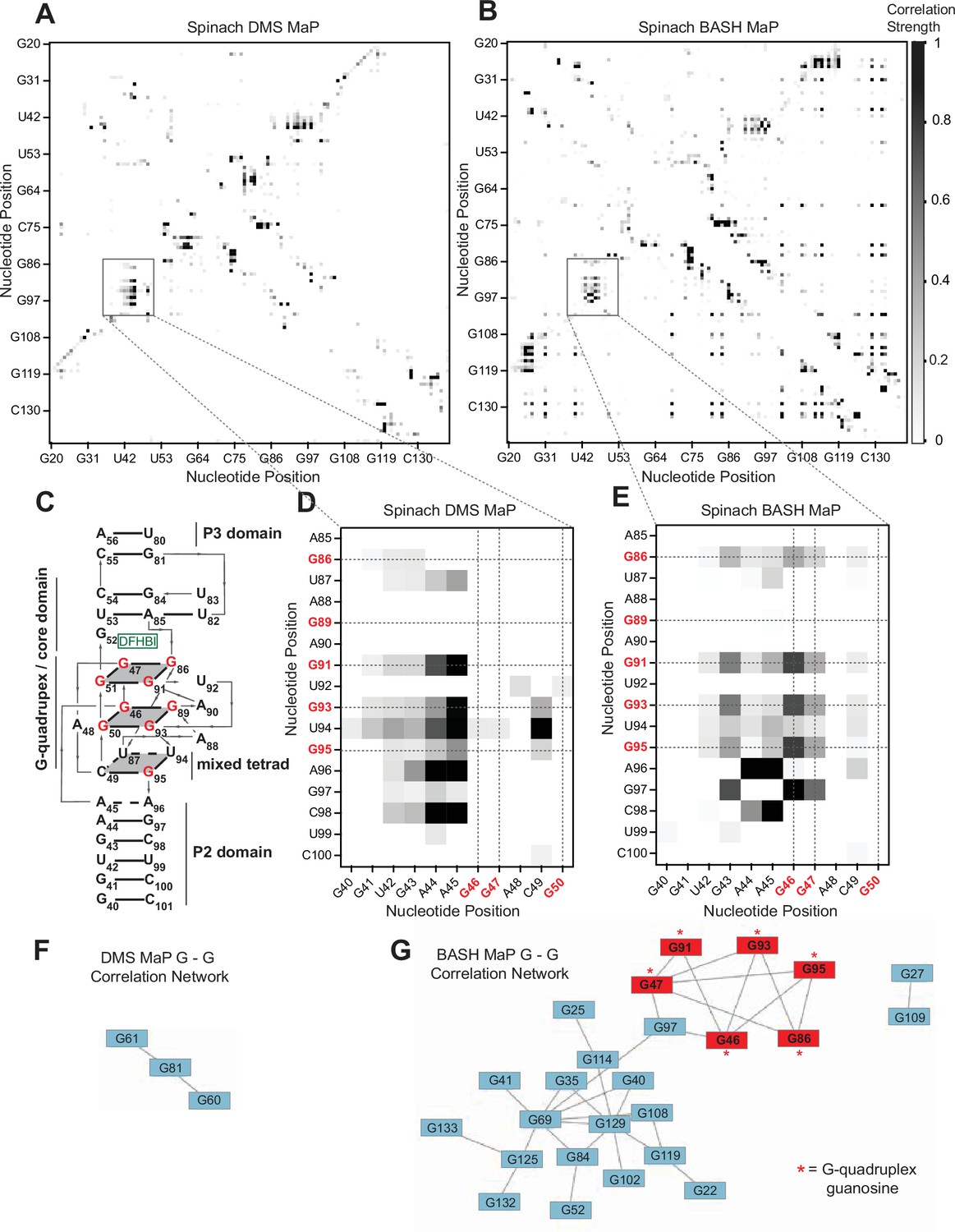
BASH MaP reveals networks of co-occurring modifications in the Spinach G-quadruplex.
(A, B) Heatmap of correlation strength between misincorporation that co-occur on the same sequencing read for Spinach treated with DMS MaP (A) or BASH MaP (B). Spinach was treated with DMS (170 mM) for 8 min at 25 °C. Each point represents a G-test significance value as calculated by RING Mapper which was then scaled to a value between zero and one. Values closer to one appear darker in the correlation heatmap and represent higher G-test correlation strength. (C) Three-dimensional model of Spinach core ligand binding domain with numbering scheme used in (A–D) and (F, G). G-quadruplex and mixed tetrad interactions in Spinach are indicated with a grey plane. The ligand DFHBI-1T interacts with G52 and sits between the upper G-quadruplex tetrad and a U-A-U base triple. Structural domains P2, P3, and the core domain are indicated. (D) Close up of boxed region in (A) shows the pattern of co-occurring misincorporations surrounding the bases involved in the G-quadruplex of Spinach (marked in red). This heatmap displays predominantly co-occurring misincorporations between A – A, A – C, and A – G positions. (E) Close up of boxed region in (B) shows the pattern of co-occurring misincorporations surrounding the bases involved in the G-quadruplex of Spinach (marked in red). This heatmap is enriched in co-occurring misincorporations between G – G positions and displays correlations between G’s involved in the G-quadruplex of Spinach. (F, G) Network analysis of G – G correlations in DMS MaP (F) and BASH MaP (G) of Spinach. A network was constructed by representing G positions in Spinach as vertices and creating edges between G positions that display co-occurring misincorporations above a Z-score threshold of 2.0. BASH MaP of Spinach reveals that G’s form two major clusters. The smaller cluster contains six of nine G’s which comprise the Spinach G-quadruplex.
Figure 3—figure supplement 1
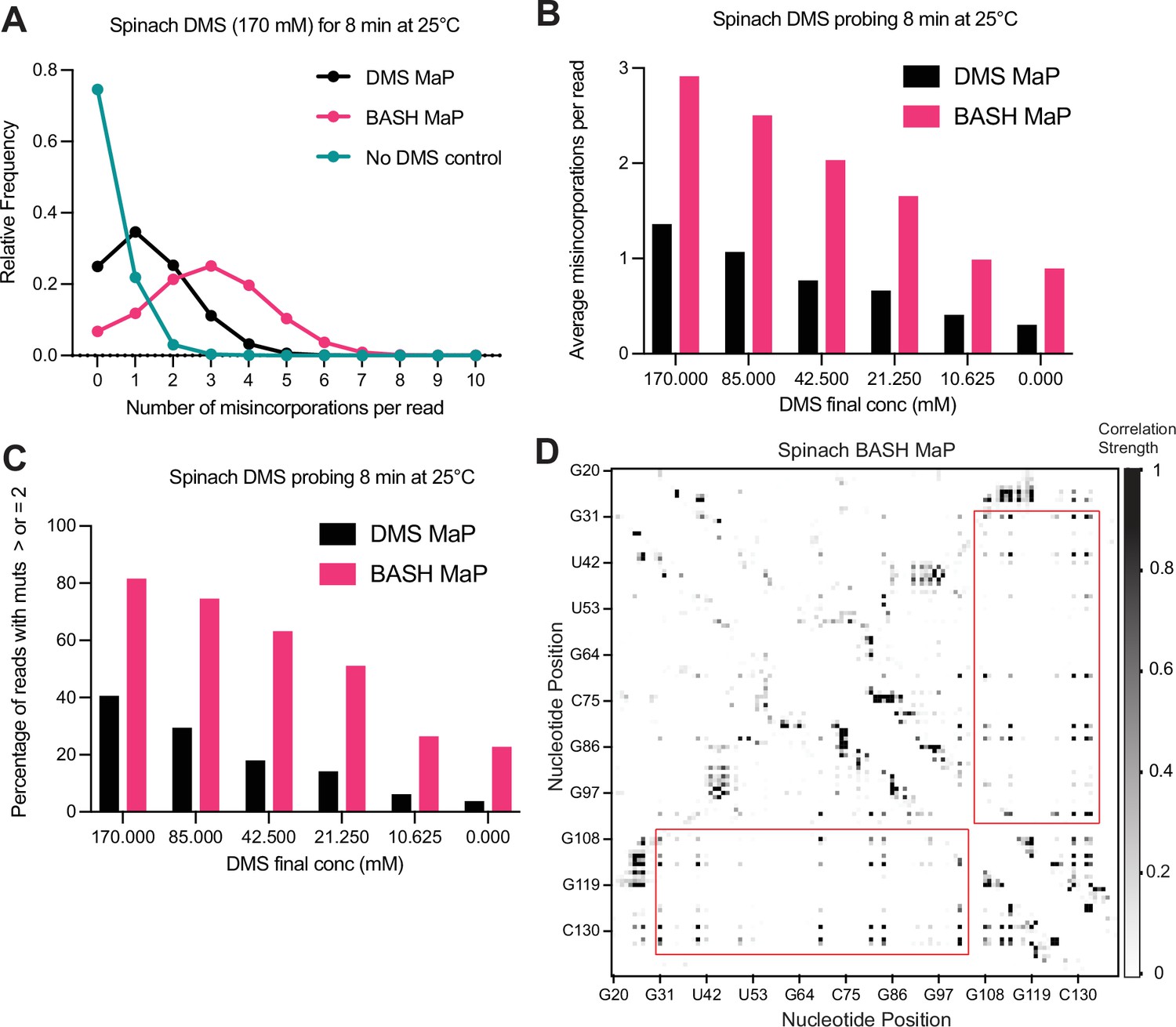
BASH MaP improves single molecule analysis through increased misincorporation density.
(A) Histogram of misincorporation per sequencing read reveals increased misincorporation density in BASH MaP. Single molecule analysis of structure probing data relies on the presence of multiple misincorporations per sequencing read. The number of misincorporations per sequencing read has been limited by the ability to simultaneously detect multiple methylations on a single RNA molecule. Since BASH MaP enables detection of N7G methylation events, we sought to quantify the increase in misincorporations per sequencing read after BASH treatment. We analyzed data from paired DMS MaP and BASH MaP datasets produced from the same pool of DMS-modified RNA and plotted a histogram representing the number of misincorporation per sequencing read. For reference, a full-length sequencing read for the Spinach RNA was 141 nucleotides long. The histogram revealed that the most common sequencing read for the no DMS-modified RNA had zero misincorporation. In contrast, the most common sequencing read for DMS MaP contained one misincorporation whereas the most common sequencing read for BASH MaP contained three misincorporations. Together, this shows that the most common sequencing read for BASH MaP enables observation of three times the number of DMS-methylation events as compared with DMS MaP. (B) Plot of misincorporations per read shows BASH MaP doubles the misincorporations per read as compared to DMS MaP. To further identify whether BASH MaP data is suitable for single molecule analysis, we analyzed the average number of misincorporation per read for a range of DMS concentrations. The plot shows that the number of misincorporation per read is dependent on both DMS concentration and BASH treatment. BASH MaP samples roughly had twice as many misincorporation per sequencing read as DMS MaP libraries prepared from the same RNA. These results suggests that BASH MaP is suitable for single molecule analysis. (C) Quantification of (B) shows BASH MaP doubles the number of sequencing reads which can be used for single molecule analysis. Since single molecule analysis requires two or more misincorporation per read, we quantified the percentage of sequencing reads which meet this criterion for various DMS concentrations. A plot of the percentage of reads which could be used for single molecule analysis reveals that BASH MaP on average doubles the percentage of sequencing reads with two or more misincorporation when compared with paired DMS MaP datasets. (D) Base-paired G’s display low levels of correlated mutations with other base-paired guanines. A heatmap of normalized correlation strength for all possible base-pair combinations in BASH-treated Spinach reveals a checkerboard-like pattern (red boxes). These points correspond to base-paired G’s which appear to co-mutate with other base-paired G’s.
Figure 3—figure supplement 2
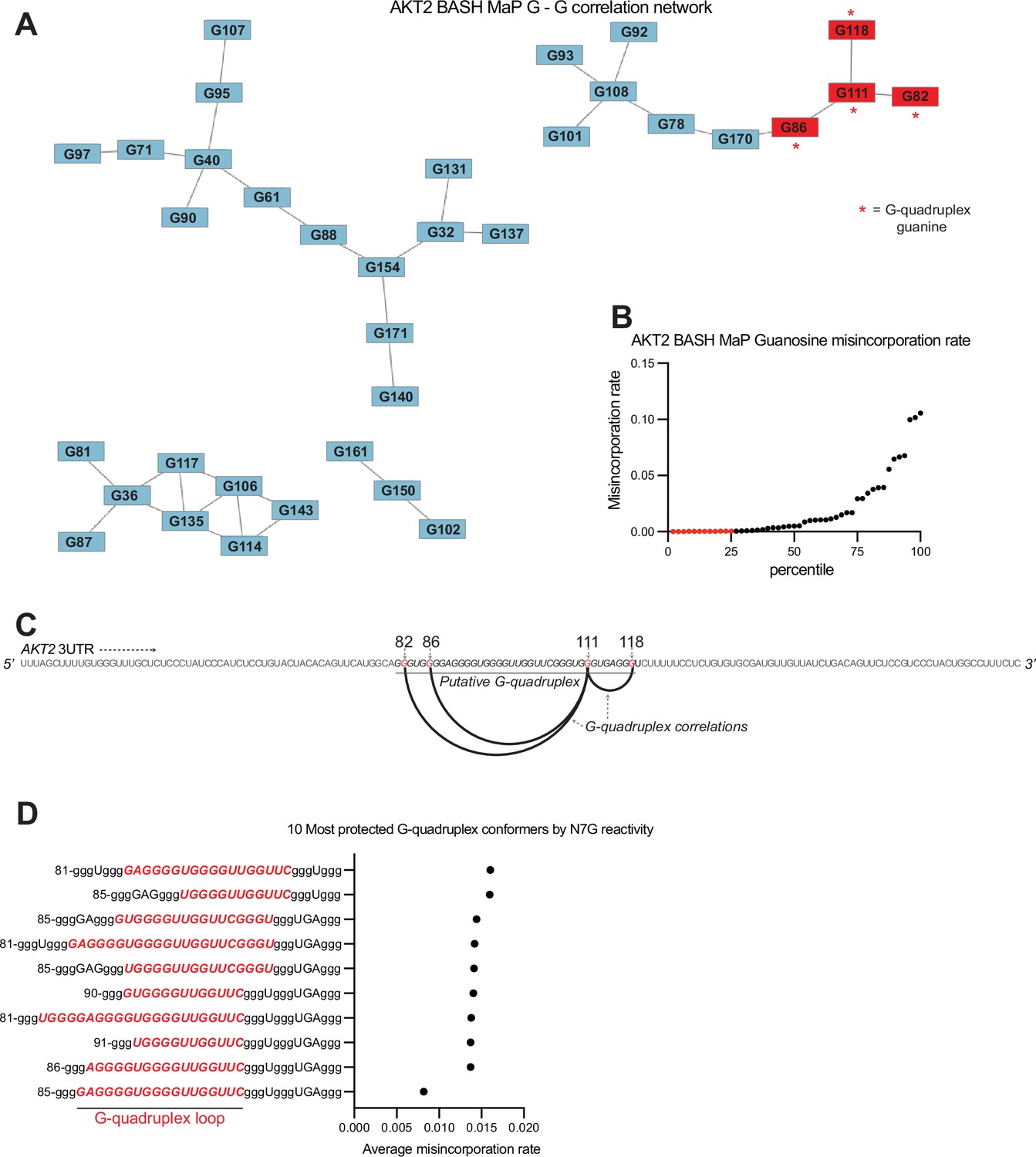
AKT2 3’UTR adopts an atypical G-quadruplex with a long central loop.
(A) G – G correlation network for BASH MaP of AKT2 3UTR. In Figure 3G, we show that network analysis of G – G correlations in Spinach BASH MaP data visualizes the G-quadruplex. We next wanted to know if a similar network analysis could identify a G-quadruplex in the AKT2 3’UTR. Like Spinach prior to its crystallographic structure determination, it is unclear which G’s are engaged in a G-quadruplex, especially since AKT2 displays numerous G’s with low N7G reactivity. The correlation network for AKT2 BASH MaP displays four clusters of G’s. Lowly reactive G’s as identified in (B) display a cluster of four G’s, marked with red asterisks, which appear to be engaged in a G-quadruplex. (B) AKT2 3’UTR N7G reactivity filtering. In Figure 5C, we describe the criteria for identifying a G as engaged in a tertiary interaction. The first step in this process involves identification of the bottom quartile of G’s for N7G reactivity shown as red dots. We then applied this filtering process to the population average reactivity data for AKT2 BASH MaP. From these G’s, we then identified G-quadruplex G’s which were both in the bottom quartile and connected in the network plot and marked these connections with red asterisks in .(A) (C) Arc plot of G-quadruplex G – G correlations in AKT2 BASH MaP data. We first asked whether the G-quadruplex-correlated G’s, as identified in (A), could give insight into which G tracts were likely to be engaged in a G-quadruplex. Interestingly, an arc plot depicting the correlations between identified G-quadruplex G’s suggests that a major conformation of the AKT2 3’UTR G-quadruplex consists of the first two and last two guanine tracts. This finding is surprising because this G-quadruplex conformation would involve a large central loop which is uncommon for G-quadruplexes (Guédin et al., 2010). (D) Conformational analysis of AKT2 3’UTR G-quadruplex suggests a large central loop. Since the AKT2 3’UTR sequence contains seven tracts of three or more guanines, we reasoned that this region may adopt many unique G-quadruplex conformations. This is supported by RT stop profiling which identified at least three distinct 3’ ends of various G-quadruplex conformations (Figure 2—figure supplement 1e). To calculate all possible three-tiered G-quadruplex conformations we utilized the program QGRS Mapper (Kikin et al., 2006). This software identified 129 unique three-tiered G-quadruplex conformations capable of forming in the AKT2 3’UTR. We then asked which conformations were most consistent with the population average N7G reactivity data. We reasoned that we could identify the most populated conformations by calculating the average misincorporation rate of the G-quadruplex G’s for each unique conformation. The conformations with the lowest average misincorporation rates are therefore likely to be the most populated conformations. We calculated the average misincorporation rate of the G-quadruplex G’s and plotted the 10 conformations with the lowest reactivity. The notation in the plot is as follows: the first number indicates the number of the first nucleotide in the G-quadruplex sequence. G’s engaged in a G-quadruplex are identified as a lowercase ‘g’. The 10 G-quadruplex conformations with the lowest N7G reactivity revealed a common large central loop, highlighted in red. Together, these data further support a model in which the AKT2 3’UTR adopts an unusual G-quadruplex conformation with a long central loop.
Figure 4 with 1 supplement
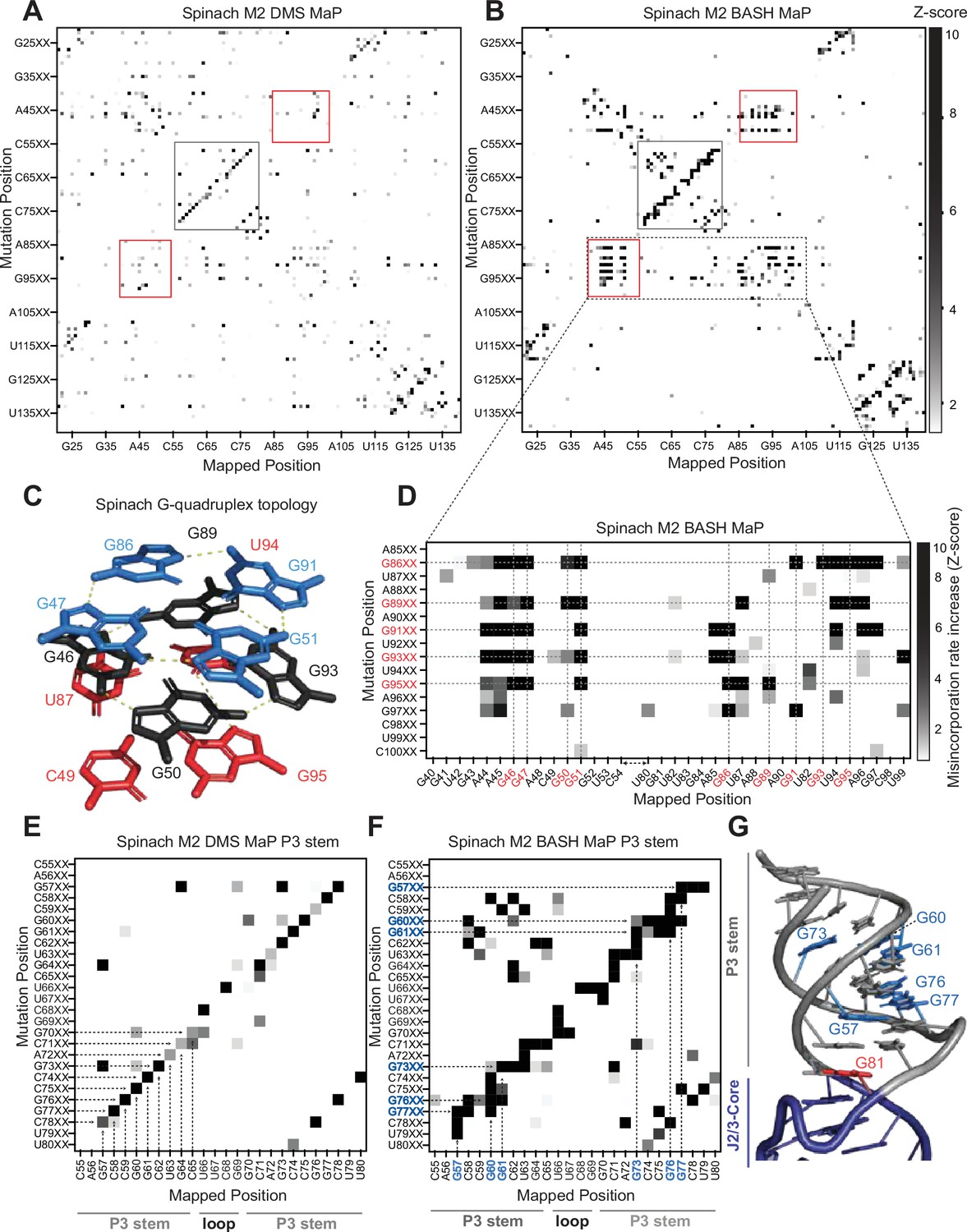
Multiplexed probing of single nucleotide mutants identifies N7G and base stacking interactions.
(A–E) Mutate and Map (M2) heatmaps of DMS MaP (A) and BASH MaP (B) of randomly mutagenized Spinach RNA. A mutate and map heatmap plots the chemical reactivity profile for RNAs with a PCR-derived mutation at a specific position along the length of the RNA. When a position in an RNA is mutated through mutagenic PCR, it is predicted that all interacting nucleotides will display an increase in chemical reactivity. Each row of the heatmap (Mutation Position) represents sequencing reads with a PCR-derived mutation at the indicated position within Spinach. Each column of the heatmap (Mapped Position) represents the misincorporation rate at the indicated position in Spinach. Visualization of changes in reactivity to DMS which are induced by point mutations is enabled by performing Z-score normalizations for each column individually. Only positive Z-scores are plotted to display increases in chemical reactivity due to PCR-derived mutations. M2 BASH MaP displays unique signals in the mutate-and-map heatmap in the G-quadruplex (red box) and P3 (grey box) region of Spinach. (C) G-quadruplex topology of Spinach. Spinach contains a two-tiered G-quadruplex with a mixed tetra stacked below. G-quadruplex numberings and colors are used to denote G-tetrads. The top G-tetrad is colored blue. The middle G-tetrad is colored black. The bottom mixed tetrad is colored red. N7G interactions are denoted with a yellow dotted line. (D) Mutate-and-Map has previously been used to identify base-pair interactions in RNA. To determine whether N7G interactions could be identified by mutate-and-map we examined the mutate-and-map heatmap generated by M2 BASH MaP of Spinach shown in (B) zoomed in on the Spinach G-quadruplex core domain. G-quadruplex G’s are indicated in red. The plot reveals that PCR-derived mutations in G-quadruplex G’s induce increases in N7G reactivity to DMS throughout the entire Spinach core domain. This suggests that PCR-derived mutations within the Spinach G-quadruplex perturb the structure of the entire core domain. (E) Zoom in of the M2 DMS MaP heatmap for the P3 domain of Spinach. P3 stem and loop nucleotides are indicated below the plot. The base-pairing pattern of the P3 domain is clearly identified as a diagonal line in the mutate-and-map heatmap. Base pairs are highlighted with dotted arrows. (F) Zoom in of M2 BASH MaP heatmap for the P3 domain of Spinach. G’s which display long vertical lines in the heatmap and give the P3 region a jagged appearance are highlighted in blue. The vertical lines indicate that PCR-derived mutations at multiple adjacent nucleotides all cause increased reactivity to DMS of highlighted G’s. This suggests that N7G reactivity to DMS is sensitive to local disruption of helix stacking. (G) Crystal structure of the Spinach P3 stem. The P3 stem is denoted in grey. G bases which display signals in the M2 BASH MaP heatmap are highlighted in blue. Hyper-reactive G’s at helix termini such as G81 are colored red.
Figure 4—figure supplement 1
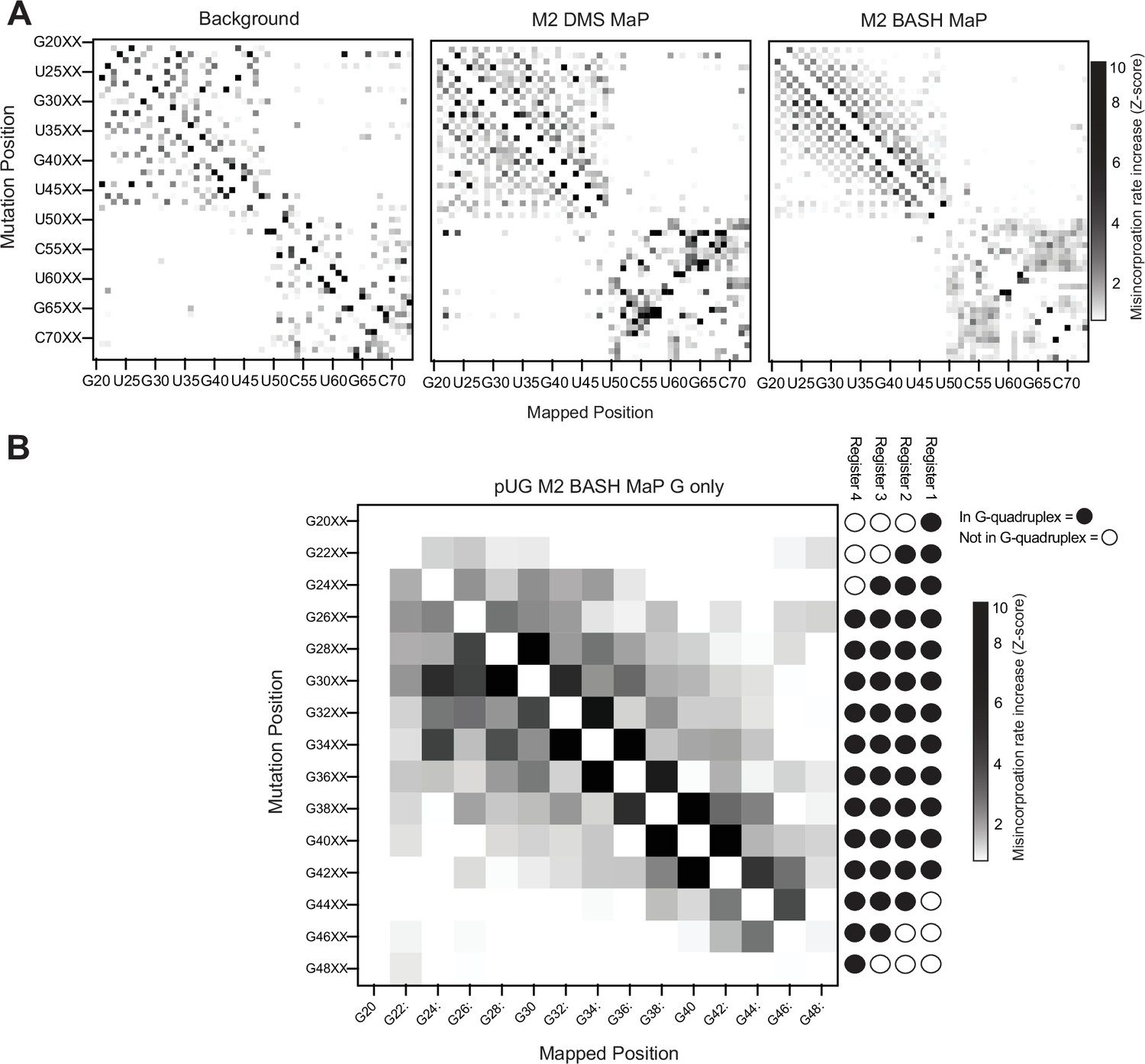
M2 BASH MaP of 15 x polyUG repeat RNA.
(A) M2 BASH MaP of 15 x polyUG repeat RNA reveals mutation-induced G-quadruplex unfolding. We next asked whether PCR-derived point mutations destabilized the G-quadruplex in the 15 x polyUG repeat RNA. To test this, we performed random mutagenesis with PCR (left) and then performed M2 DMS MaP (middle) or M2 BASH MaP (right). Like the heatmap of M2 BASH MaP of Spinach in Figure 4B and M2 BASH MaP of the polyUG RNA displayed a strong checkerboard pattern which occurred at pairs of G’s. As seen in Spinach, mutation of a G in a G-quadruplex appears to induce increased N7G reactivity of adjacent G’s. (B) M2 BASH MaP of 15 x polyUG repeat RNA supports a four-register model of RNA conformation. To validate whether mutations in the G-quadruplex cause global destabilization of the 15 x polyUG repeat RNA, we analyzed how mutations at G-quadruplex G’s affected N7G reactivity of all other G’s. As expected, mutations in G’s that only populate one or two G-quadruplex registers had minimal impact on the global N7G reactivity. In contrast, mutations at G’s expected to populate all four G-quadruplex registers induced global increases in N7G reactivity. Mutations at G’s expected to populate only three G-quadruplex registers only induced local increases in N7G reactivity. The M2 BASH MaP heatmap for the 15 x polyUG repeat RNA further supports the existence of alternative G-quadruplex registers and suggests that point mutations cause global destabilization and unfolding.
Figure 5 with 1 supplement
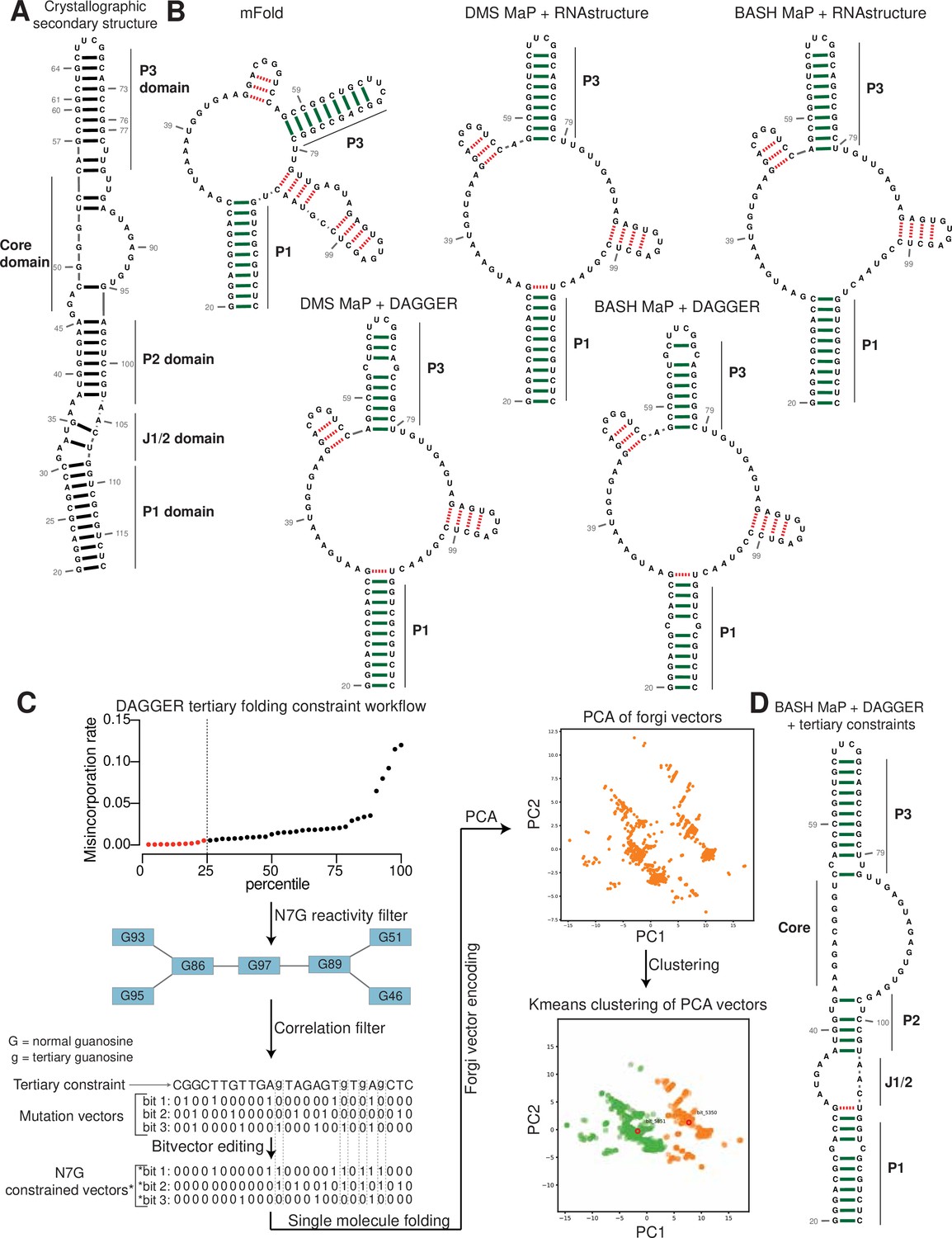
Tertiary folding constraints enable accurate secondary structure modeling of Spinach.
(A) Spinach secondary structure model derived from the crystal structure 4TS2. This secondary structure model was utilized as a benchmark for comparing structure modeling approaches. (B) Spinach secondary structure models generated by the indicated combination of experimental data (DMS MaP or BASH MaP) and folding algorithm (mFold, RNAstructure, DAGGER). To determine whether structure probing data could improve the modeling of Spinach secondary structure, we assessed the sensitivity and specificity of a variety of computational approaches. Base pairs which are correctly predicted are indicated by green bars. Base pairs which are incorrectly predicted are indicated with red dashed lines. A base pair was determined to be correct if the true base pairing partner was within one base (+/-) from the indicated pairing partner. For detailed explanation of settings used for each RNA secondary structure modeling approach see Methods. Incorporation of experimental data improved Spinach secondary structure modeling; however, all structures included false helices and lacked the P2 domain of Spinach. (C) Tertiary-folding constraints derived from N7G-reactivity data are implemented through modification of the DaVinci data analysis pipeline (DAGGER). To generate tertiary constraints, G’s in the bottom quartile of N7G reactivity are first identified. Then, all pairs of bottom quartile G’s which display significant rates of co-occurring misincorporations with each other are identified as likely to be engaged in a tertiary interaction. These positions are indicated by annotating the base as lowercase in the input FASTA file for a modified DaVinci analysis pipeline. Each sequencing read is first converted to a bitvector where a zero represents no misincorporation and a one represents a misincorporation. The DaVinci pipeline forces sites of misincorporations to be single stranded upon subsequent folding. G’s identified as likely to be engaged in a tertiary interaction are forced to be single stranded by editing the bitvectors and setting the value at each tertiary G to one. Sequencing reads with a misincorporation at any G identified as tertiary are treated separately because a modification at these positions indicates a change in tertiary structure. For these bitvectors, tertiary G’s are allowed to be considered for base pairing by ContraFold, the folding engine used in DaVinci. After folding of each unique sequencing read, RNA secondary structures are converted to forgi vectors which utilize the Forgi library to encode RNA structure in a string of numbers. Principle component analysis (PCA) is then performed on the forgi vectors, and the ensemble of RNA structures is visualized through a plot of the first two principle components. Clustering of related RNA structures is performed in the PCA reduced dimensional space using techniques such as Kmeans clustering. Together, this pipeline enables more accurate structure modeling of G-quadruplex containing RNA. (D) Tertiary constraints and DAGGER analysis of BASH MaP-treated Spinach accurately model Spinach secondary structure. To determine whether tertiary folding constraints could improve Spinach structure modeling, we implemented the technique as described in (C) and applied it to Spinach BASH MaP data. Base pairs which are correctly predicted are indicated by green bars. Base pairs which are incorrectly predicted are indicated with red dashed lines. A base pair was determined to be correct if the true base pairing partner was within one base (+/-) from the indicated pairing partner. The resulting secondary structure most closely matches the crystallographic secondary structure through formation of the P2 domain and absence of false helices.
Figure 5—figure supplement 1
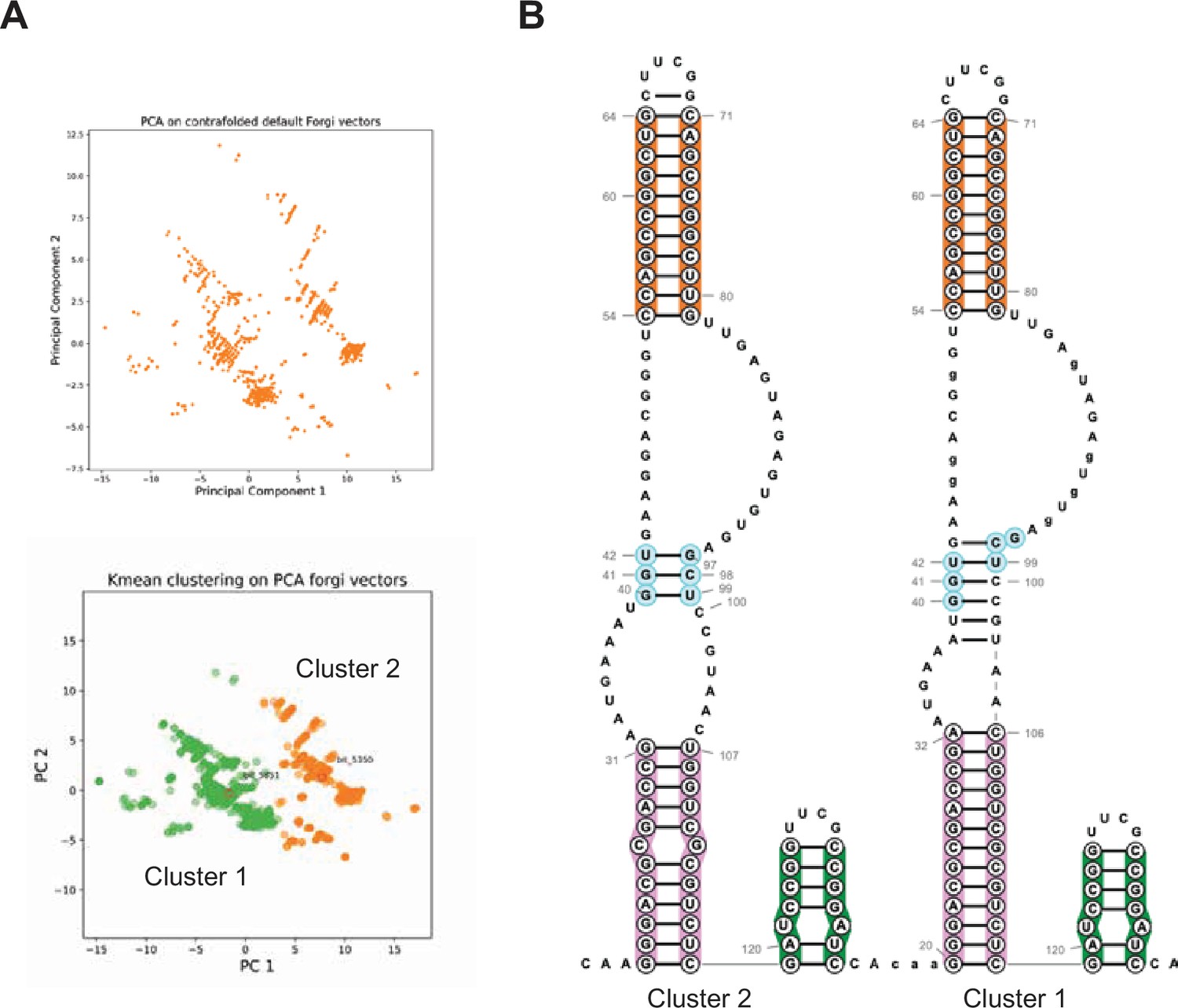
DAGGER clustering of Spinach BASH MaP.
(A) DAGGER clustering of 10,000 reads for BASH-MaP-treated Spinach. Principle component analysis revealed roughly two major clusters of RNA conformation. Kmeans clustering was used to identify the most representative secondary structure for each of the clusters. Cluster 1 is colored green. Cluster 2 is colored orange. (B) Comparison of Cluster 1 and Cluster 2 representative secondary structures reveals alternative base pairing in P2 domain. Enlarged image of Figure 6I depicts overall conservation of P1 and P3 domains between the two clusters. Differences between Cluster 1 and Cluster 2 are depicted in the alternative base pairing pattern in the P2 stem, colored light blue.
Figure 6 with 1 supplement
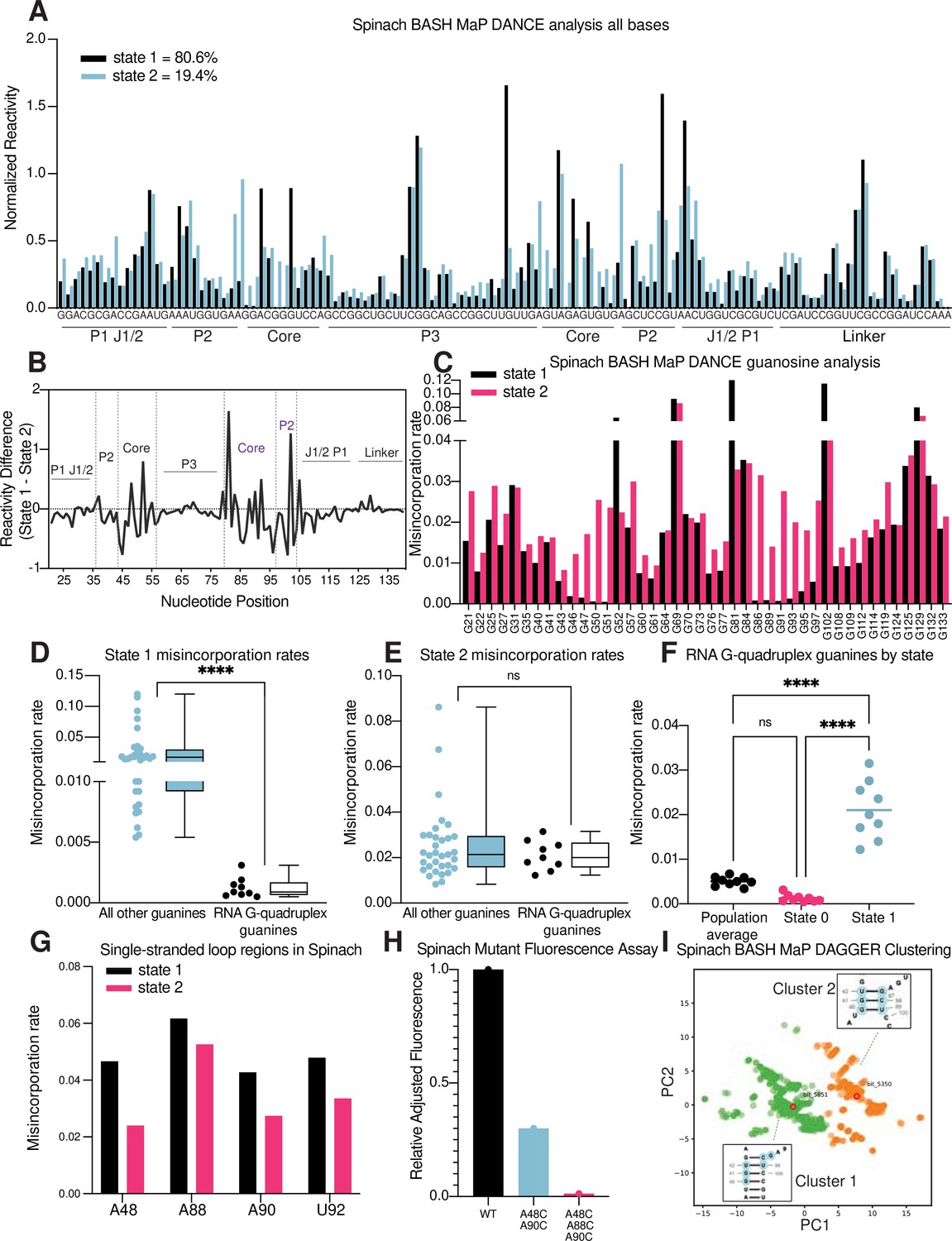
RNA structure deconvolution reveals G-quadruplex and P2 misfolding in Spinach.
(A) RNA structure deconvolution identifies two conformations of Spinach. To identify multiple conformations of Spinach, we applied the program DANCE which utilizes a Bernoulli mixture model to identify mutually exclusive patterns of misincorporation in sequencing data. Spinach was subjected to BASH MaP and the sequencing data was input into the DANCE pipeline. DANCE identified two conformations denoted as State 1 and State 2 of 80.6% and 19.4% abundance. Misincorporation rates for each state were converted into reactivity values through normalization and plotted for comparison (see METHODS). (B) Reactivity differences between DANCE-identified states in Spinach reveal changes in G-quadruplex core and P2 domains. To identify which regions of Spinach adopted different structures between the two states identified by DANCE, we plotted the difference between reactivities of State 1 and State 2. The plot of change in reactivity shows that State 1 and State 2 differ in the core and P2 domains but remain unchanged in stems P1 and P3. (C) Spinach alternative states display differential N7G reactivity at G-quadruplex G’s. To determine whether the alternative states of Spinach display differences in N7G reactivity, we compared the misincorporation rate of each G for State 1 and State 2. G-quadruplex G’s are indicated below the plot in red. The plot shows that most G’s in Spinach display no change in N7G reactivity to DMS whereas G-quadruplex G’s show marked changes in N7G reactivity to DMS. (D, E) Spinach G-quadruplex G’s are differentiated from all other G’s in State 1 (D) ****p<0.0001, unpaired t-test with Welch’s correction. G-quadruplex G’s show no difference in misincorporation rates for State 2 (E) ns p=0.2262, unpaired t-test with Welch’s correction. (F) The Spinach G-quadruplex is unfolded in State 2. To determine whether the G-quadruplex in Spinach was unfolded in State 2 we compared the misincorporation rate of G-quadruplex G’s for the population average and DANCE deconvolved States 1 and 2. The plot shows that G-quadruplex G’s display increased misincorporation rates in State 2 which suggests State 2 consists of an unfolded G-quadruplex. ****p<0.0001, Tukey’s multiple comparison test. (G) Single-stranded loops in the Spinach G-quadruplex show decreased reactivity in State 2. To determine whether the single-stranded loops in the Spinach G-quadruplex core remain unpaired, we compared misincorporation rates between State 1 and State 2. The plot shows that all single-stranded loop residues show reduced reactivity to DMS in State 2 which suggests these positions display increased base-pairing interactions in State 2. (H) Nucleotide substitutions in Spinach G-quadruplex loop residues reduce Spinach fluorescence. To determine whether Spinach G-quadruplex loop residues make base-pair interactions with G-quadruplex G’s in the Spinach alternative conformation, we systematically changed loop residues from A to C which should stabilize any base-pairing interactions with G’s. We quantified Spinach fluorescence through an in-gel fluorescence assay (see Methods). The plot shows that conversion of A residues to C residues in the G-quadruplex loop region induces a progressive loss in Spinach fluorescence. (I) DAGGER clustering with tertiary constraints applied to Spinach BASH MaP data identifies two clusters with altered base pairing in the P2 domain. To identify alternative base pairing conformations of the misfolded Spinach, we utilized the orthogonal single molecule analysis method DaVinci with N7G reactivity data (DAGGER). We incorporated N7G reactivity data to create tertiary folding constraints before DAGGER folding and clustering (see Methods). Dimensional reduction and clustering identified two major clusters denoted Cluster 1 and Cluster 2. The most representative secondary structure of each cluster is boxed and indicated by the bit number. The DaVinci clustering plot reveals that the misfolded Spinach displays a register shift in the P2 domain. Cluster 1, colored green, is consistent with the Spinach crystallographic secondary structure. Cluster 2 is colored orange.
Figure 6—figure supplement 1
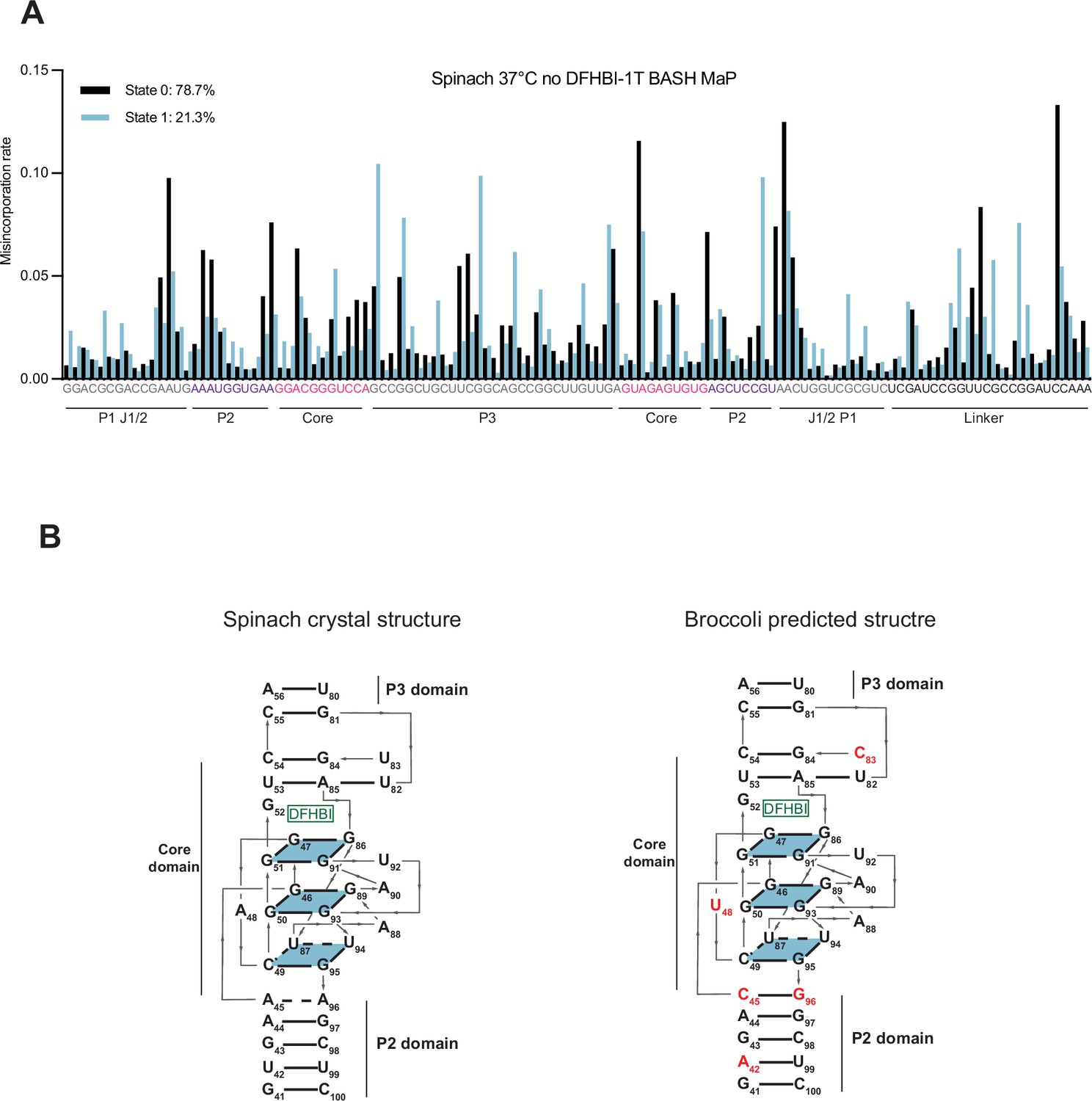
DANCE clustering of Spinach BASH MaP data and comparisons to Broccoli.
(A) DANCE deconvolution of BASH MaP-treated Spinach identifies two conformations. To validate that the DANCE deconvolution of M2 BASH MaP was not due to PCR-derived mutations, we performed BASH MaP on non-mutagenized Spinach at an elevated temperature and without its ligand DFHBI-1T. DANCE clustering identified two conformations with similar abundances to Figure 6A. As seen in the DANCE conformations of M2 BASH MaP, State 0 is consistent with a folded G-quadruplex whereas State 1 suggests a misfolding of the G-quadruplex in the core domain. These results suggests that the DANCE-identified clusters represent true alternative conformations of Spinach and are reproducible under a range of probing conditions. (B) Alignment of Spinach and Broccoli three-dimensional structures. The predicted Broccoli three-dimensional structure displays an identical G-quadruplex topology to Spinach. Nucleotide differences in the Broccoli structure are indicated in red. Broccoli structure numbering was adjusted to align with the Spinach numbering. Key differences involve the strengthening of a C-G-C base triple above the ligand binding pocket, a change of a bulge A between the tetrad and a G-quartet to a U and three nucleotide differences in the P2 domain.
Figure 7 with 2 supplements
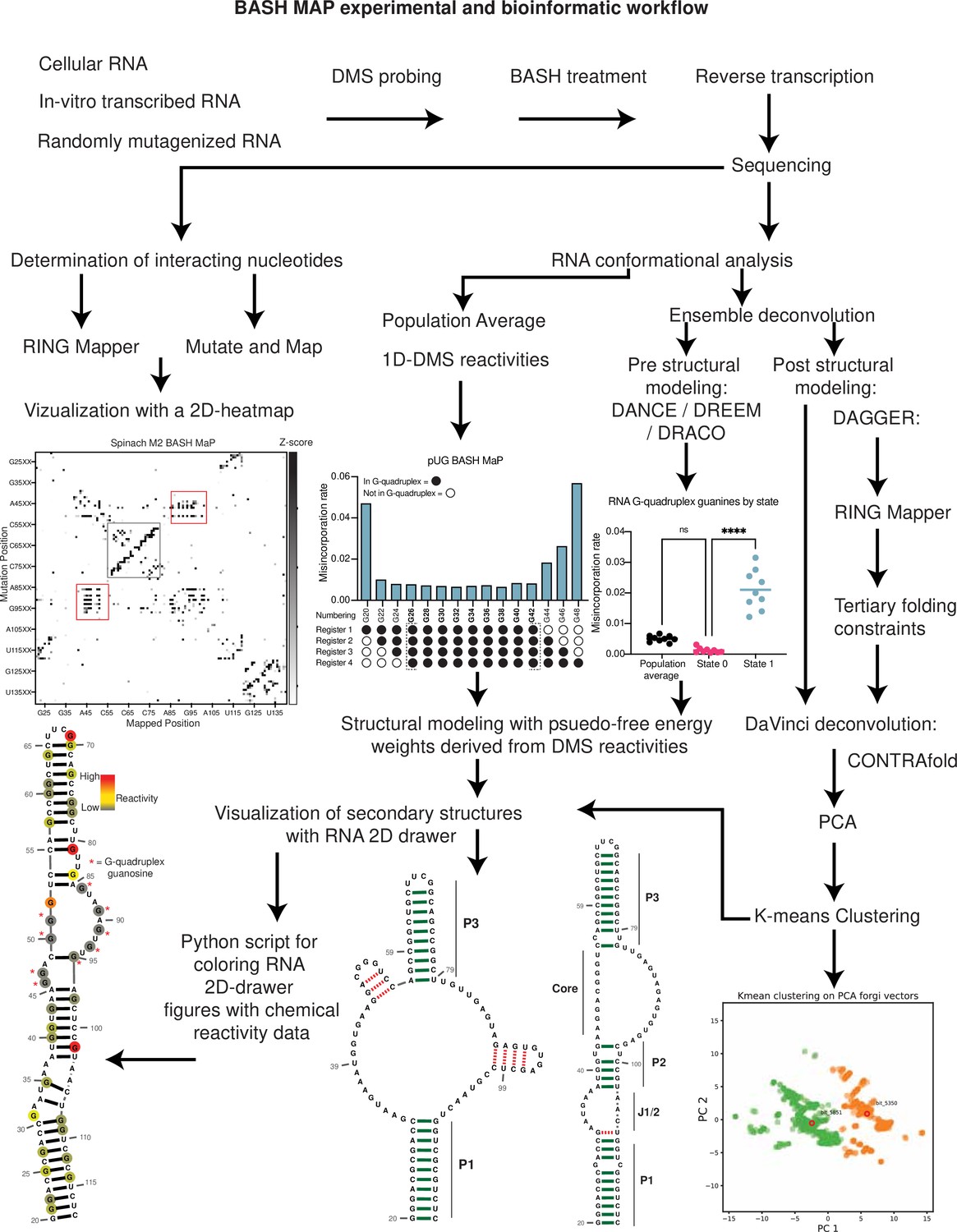
Summary BASH MaP experimental and bioinformatic workflow.
Overview of the BASH MaP experimental and bioinformatic workflow.
Figure 7—figure supplement 1
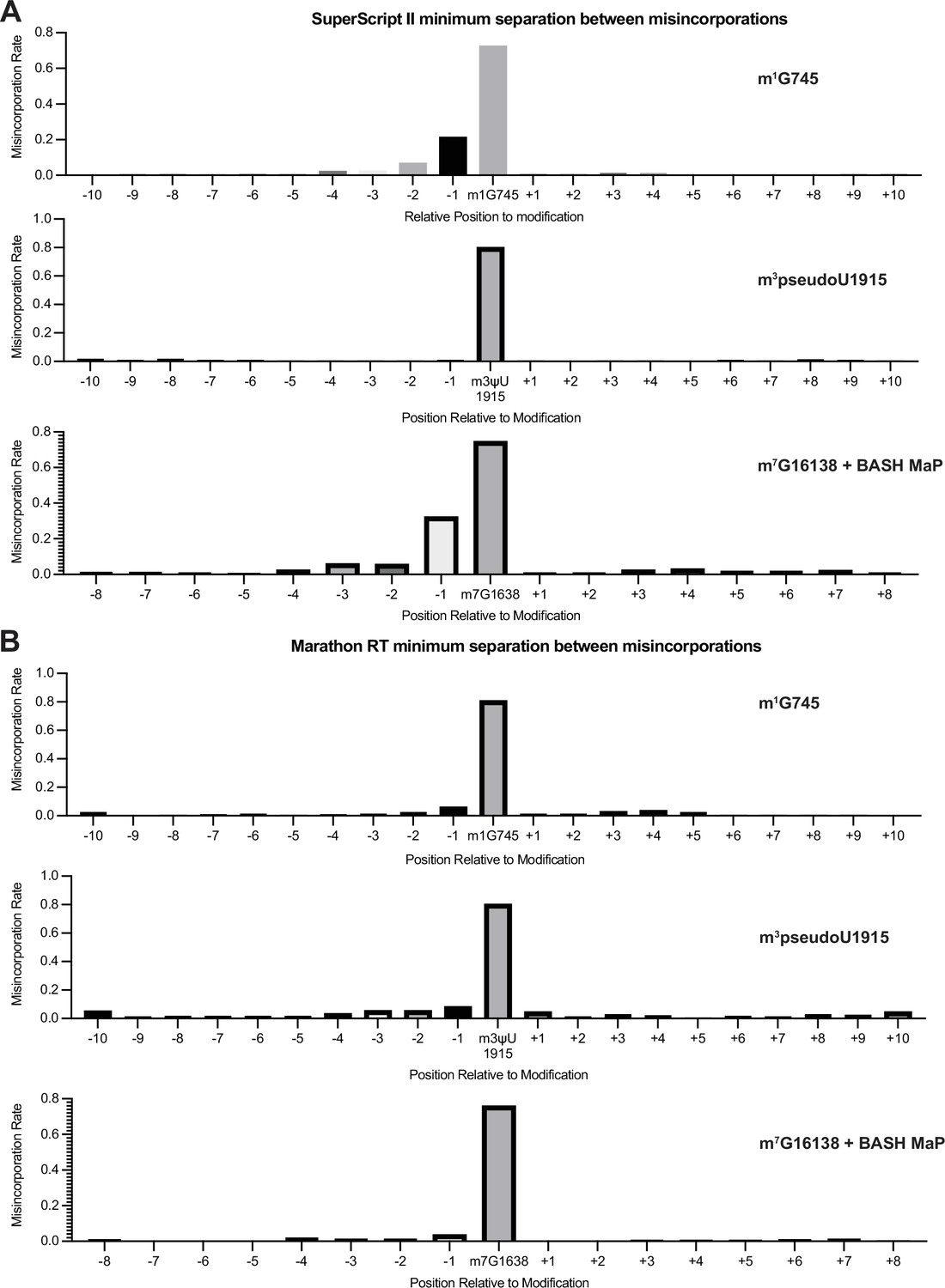
Validation of minimum separation between misincorporations for SuperScript II and Marathon RT.
(A, B) Misincorporations induced by (A) SuperScript II and (B) Marathon RT at m1G, m3psuedoU, and abasic sites. Misincorporations in DNA sequencing can be simple such as a G→T transition. Previous studies found that certain reverse transcriptases produce more complex types of misincorporations such as clusters of misincorporations for a single modified nucleotide (Busan and Weeks, 2018; Tomezsko et al., 2020). To determine whether two misincorporations on a sequencing read represent two methylation events or a single methylation event which induced multiple misincorporations, we analyzed misincorporation data at endogenously methylated bases. We reasoned that we could determine a minimum separation distance between unique methylation events by plotting the misincorporation rate around endogenously methylated bases. If either SuperScript II or Marathon produced multiple misincorporations for a single methylation site, then there would be an increased misincorporation rate at surrounding un-modified nucleotides. These plots show that SuperScript II produces multiple misincorporations at m1G and abasic sites with a complex misincorporation type causing downstream bases to appear highly mutated. Interestingly, SuperScript II encodes m3pseudoU as a single point misincorporation. Furthermore, Marathon RT encodes all three modification types as single point misincorporation. This suggests that each unique combination of reverse transcriptase enzyme and modification type may be associated with a unique misincorporation signature. Together, these data suggest that two misincorporations should be separated by at least two non-misincorporated bases, to be confident that these derive from two separate methylated nucleotides, for BASH MaP with SuperScript II.
Figure 7—figure supplement 2
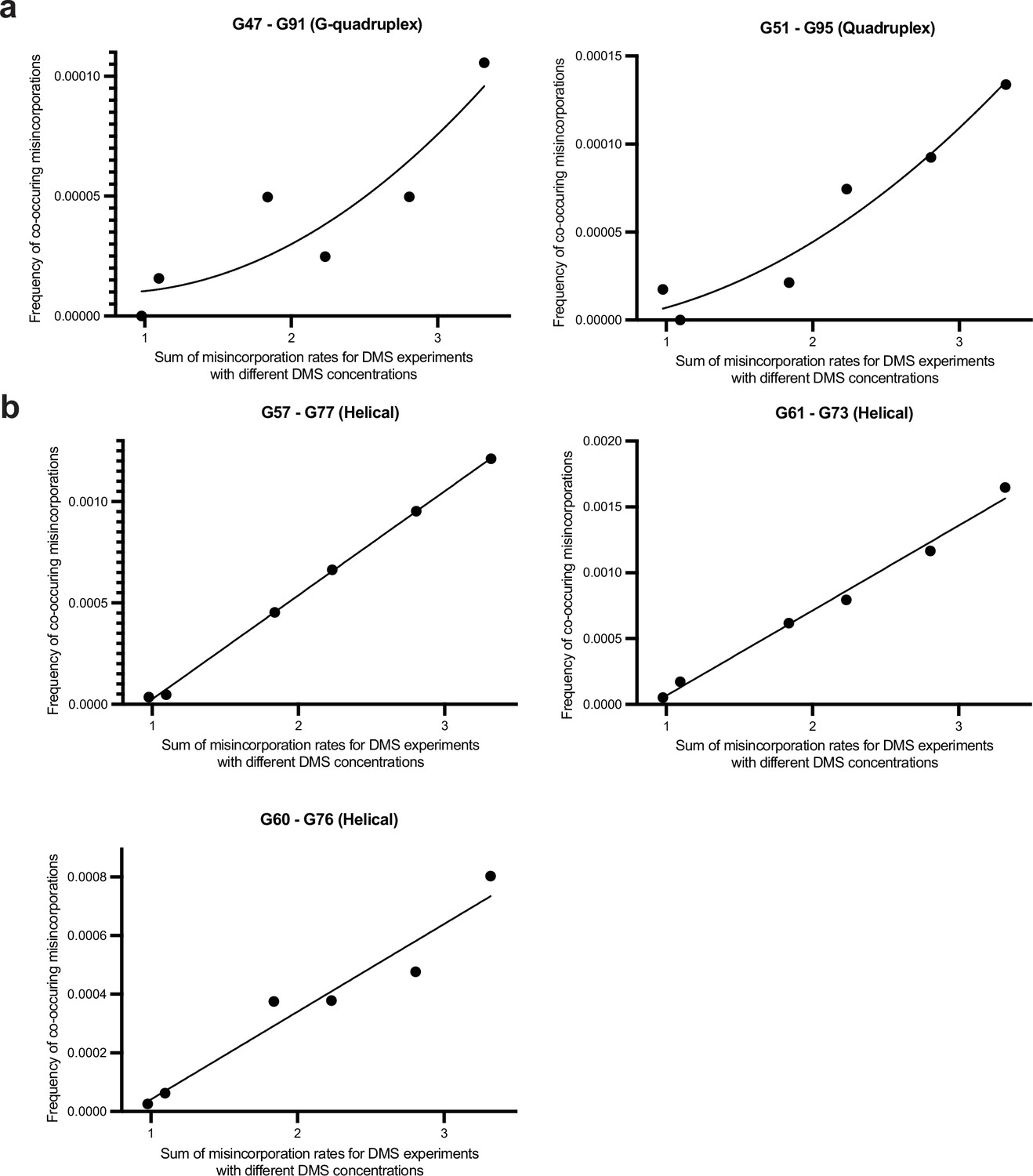
Discrimination between multi-hit versus single-hit mechanisms for co-occurring misincorporations between G’s in Spinach BASH MaP data.
(A, B) BASH MaP was performed on Spinach over a range of DMS concentrations and the frequency of co-occurring misincorporations between specific G residues was plotted. Single-hit versus multi-hit mechanisms for producing co-occurring misincorporations are expected to display different relationships to increasing DMS modification rates. Single-hit mechanisms predict a linear dependence between DMS modification rate and the frequency of co-occurring misincorporations between two locations. In contrast, multi-hit mechanisms predict a quadratic relationship between DMS modification rate and the frequency of co-occurring misincorporations between two locations. Two sets of G-quadruplex G’s display a quadratic relationship while all G’s engaged in Watson-Crick base pairs display a linear relationship.
Tables
Table 1
Step 1 PCR primers.
| RNA sample name | Step 1 forward primer sequence | Step 1 reverse primer sequence |
|---|---|---|
| SSII_HeLa_18 s_0 min_1 | ACACGACGCTCTTCCGATCTNNNNNATATC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTAAGGCG AGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_0 min_2 | ACACGACGCTCTTCCGATCTNNNNNCGCG CCCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTAAG GCGAGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_30 min_1 | ACACGACGCTCTTCCGATCTNNNNNATAT CCCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNCGTA CTAGGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_30 min_2 | ACACGACGCTCTTCCGATCTNNNNNCGCG CCCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNCGTAC TAGGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_1 hour_1 | ACACGACGCTCTTCCGATCTNNNNNATATC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNAGGCA GAAGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_1 hour_2 | ACACGACGCTCTTCCGATCTNNNNNCGCG CCCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNAGGC AGAAGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_2 hour_1 | ACACGACGCTCTTCCGATCTNNNNNATAT CCCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTCCTGA GCGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_2 hour_2 | ACACGACGCTCTTCCGATCTNNNNNCGCGC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTCCTG AGCGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_3 hour_1 | ACACGACGCTCTTCCGATCTNNNNNATATC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNGGACTC CTGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_3 hour_2 | ACACGACGCTCTTCCGATCTNNNNNCGCGC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNGGACTCC TGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_4 hour_1 | ACACGACGCTCTTCCGATCTNNNNNATATCC CGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTAGGCAT GGTGTGTACAAAGGGCAGGGAC |
| SSII_HeLa_18 s_4 hour_2 | ACACGACGCTCTTCCGATCTNNNNNCGCGC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTAGGCATG GTGTGTACAAAGGGCAGGGAC |
| MaRT_HeLa_18 s_4 hour_1 | ACACGACGCTCTTCCGATCTNNNNNATATCC CGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTAGGCAT GGTGTGTACAAAGGGCAGGGACCATGCCTA |
| MaRT_HeLa_18 s_4 hour_2 | ACACGACGCTCTTCCGATCTNNNNNCGCGC CCGTTGAACCCCATTCGTGA | GACGTGTGCTCTTCCGATCTNNNNNTAGGCAT GGTGTGTACAAAGGGCAGGGACCATGCCTA |
| Structure Cassette Universal | ACACGACGCTCTTCCGATCTNNNNNGGC TGGCCTTTCGGGCCAA | GACGTGTGCTCTTCCGATCTNNNNNGAACCG GACCGAAGCCCG |
| AKT2 3UTR | ACACGACGCTCTTCCGATCTNNNNNAACACC TCTGGGTGTTTGGAGTTTAGC | GACGTGTGCTCTTCCGATCTNNNNNCCGTAC AAATATGAAGACGAGGAGAAAGGC |
Table 2
Oligos and primers.
| Oligos | Primers |
|---|---|
| Spinach oligo | GTATAATACGACTCACTATAGGGCTGGCCTTTCGGGCCAAGGGACGCGACCGAATGAAATGGTGAAGGACGGGTCCAGCCGGCTGCTTCGGCAGCCGGCTTGTTGAGTAGAGTGTGAGCTCCGTAACTGGTCGCGTCTCGATCCGGTTCGCCGGATCCAAATCGGGCTTCGGTCCGGTTC |
| pUG oligo | GGCTGGCCTTTCGGGCCAAGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTTCGATCCGGTTCGCCGGATCCAAAT |
| Structure Cassette T7 Forward | GTATAATACGACTCACTATAGGGCTGGCCTTTCGGGCCAA |
| Structure Cassette T7 Reverse | GAACCGGACCGAAGCCCGATTTGGATCCGGCGAACCGGAT |
| Structure Cassette RT primer | GAACCGGACCGAAGCCCGA |
| Spinach A48C, A90C template | ACGGGCCAGATATACGCGTAGTTCCTGCTATAATTAGCCTTCCTCATAAGTTGCACTGCTCCAGGTGATAGTGCGGGAACCTCGATGGTCTTCACACTTTACTTCAGCGTCtggtaggcgtgtacggtgggaggcctatataagcagagctTCTGGCTAACTAGGCTGGCCTTTCGGGCCAAGGGACGCGACCGAATGAAATGGTGAAGGCCGGGTCCAGCCGGCTGCTTCGGCAGCCGGCTTGTTGAGTAGCGTGTGAGCTCCGTAACTGGTCGCGTCTCGATCCGGTTCGCCGGATCCAAAT |
| Spinach A48C, A88C, A90C template | ACGGGCCAGATATACGCGTAGTTCCTGCTATAATTAGCCTTCCTCATAAGTTGCACTGCTCCAGGTGATAGTGCGGGAACCTCGATGGTCTTCACACTTTACTTCAGCGTCtggtaggcgtgtacggtgggaggcctatataagcagagctTCTGGCTAACTAGGCTGGCCTTTCGGGCCAAGGGACGCGACCGAATGAAATGGTGAAGGCCGGGTCCAGCCGGCTGCTTCGGCAGCCGGCTTGTTGAGTCGCGTGTGAGCTCCGTAACTGGTCGCGTCTCGATCCGGTTCGCCGGATCCAAAT |
Additional files
-
MDAR checklist
- https://cdn.elifesciences.org/articles/98540/elife-98540-mdarchecklist1-v1.pdf
-
Supplementary file 1
Spinach G-quadruplex false positive rate estimation for various BASH MaP data processing approaches.
- https://cdn.elifesciences.org/articles/98540/elife-98540-supp1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
RNA tertiary structure and conformational dynamics revealed by BASH MaP
eLife 13:RP98540.
https://doi.org/10.7554/eLife.98540.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}