Consistent global structures of complex RNA states through multidimensional chemical mapping
- Stanford University, United States
Figures
Figure 1 with 2 supplements
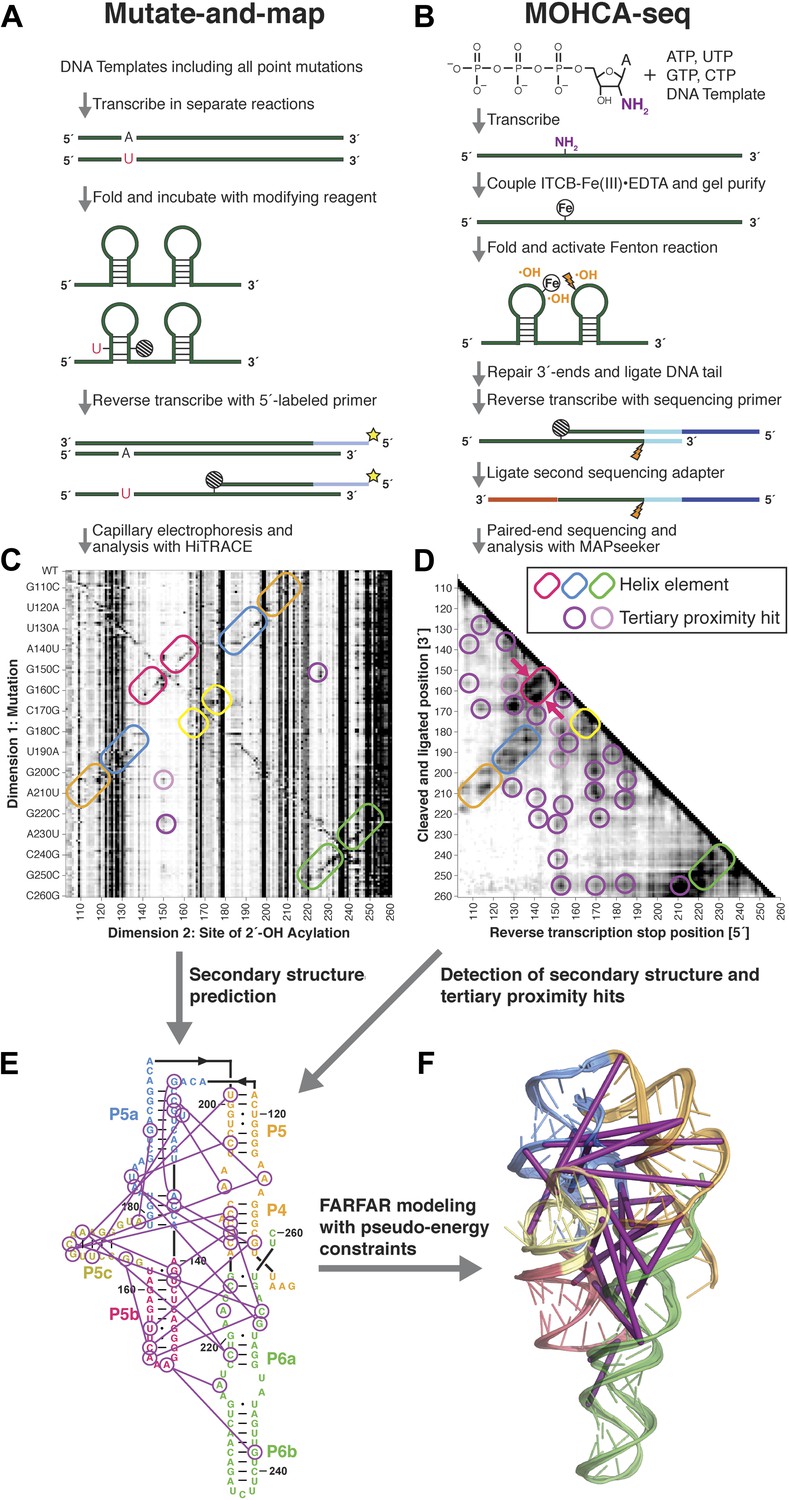
A multidimensional chemical mapping (MCM) pipeline to infer non-coding RNA 3D folds.
(A) Schematic of mutate-and-map (M2) workflow, showing transcription of comprehensive point mutant library and chemical mapping of library with reverse transcription readout (Kladwang et al., 2011a). (B) Schematic of Multiplexed •OH (hydroxyl radical) Cleavage Analysis (MOHCA)-seq workflow, showing random incorporation of radical sources, fragmentation, adapter ligations, and analysis by sequencing. (C) M2 data set for P4–P6 domain of Tetrahymena group I ribozyme. (D) MOHCA-seq data set (proximity map) for P4–P6. In (C) and (D), rounded rectangles indicate helix elements with colors matching helices in (E–F), purple circles indicate hits corresponding to < 30 Å pairwise distance in the crystal structure, and pink circles indicate hits corresponding to > 30 Å pairwise distance in the crystal structure. In (D), magenta arrows indicate MOHCA-seq hits due to diffusion across the major (top arrow) or minor (bottom arrow) grooves from radical sources located in P5b. (E–F) Representation of MOHCA-seq tertiary proximities on M2-guided secondary structure (E) and on final single Rosetta model (F). Purple lines indicate MOHCA-seq hits corresponding to < 30 Å pairwise distance in the crystal structure. Figure 1—figure supplement 1 shows stages of MOHCA-seq data analysis in the MAPseeker software package (accessible through the RNA Mapping Database server at http://rmdb.stanford.edu/tools/). Figure 1—figure supplement 2 shows the pseudo-energy potential used for pairwise MOHCA-seq constraints in Rosetta modeling and plots of MOHCA-seq signal vs pairwise distance.
Figure 1—figure supplement 1
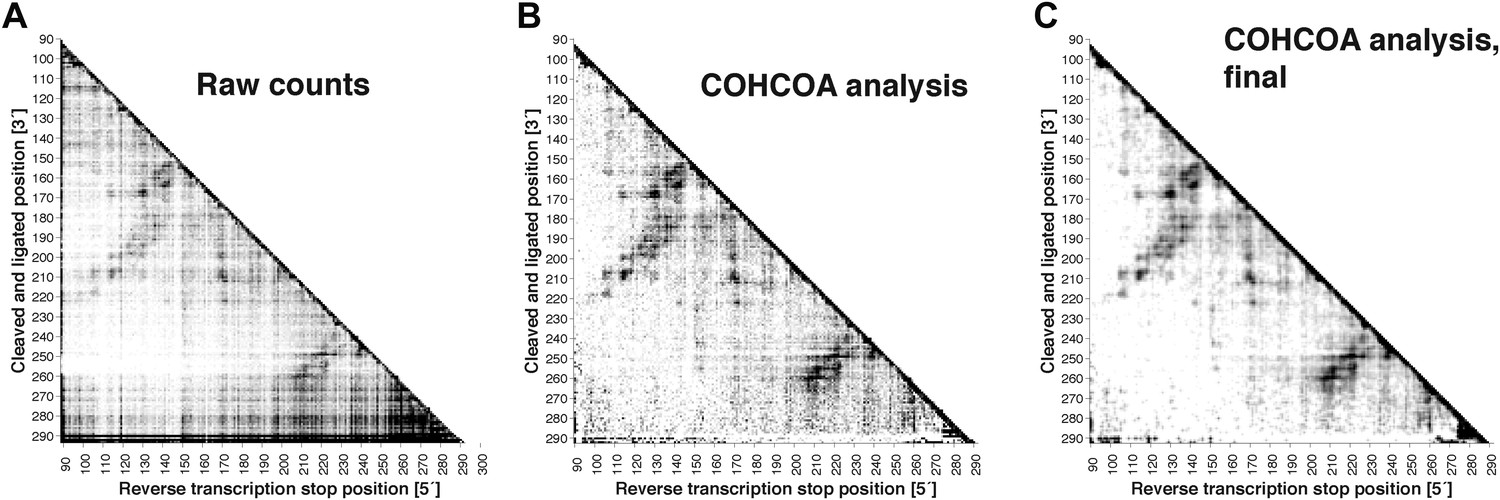
MOHCA-seq data analysis.
(A) Raw counts for a single P4–P6 MOHCA-seq data set. Following paired-end sequencing, the MAPseeker software is used to align the reads to the sequence of the RNA that was probed. (B) Closure-based •OH COrrelation Analysis (COHCOA) after 40 iterations on P4–P6 data set. (C) Final analyzed P4–P6 proximity map. A filter is applied to remove points with signal-to-noise ratio < 1, a 2D smoothing algorithm aids visualization of the strongest features, and the data are scaled using the mean of the data. A full description of the analysis is given in the ‘Materials and methods’.
Figure 1—figure supplement 2
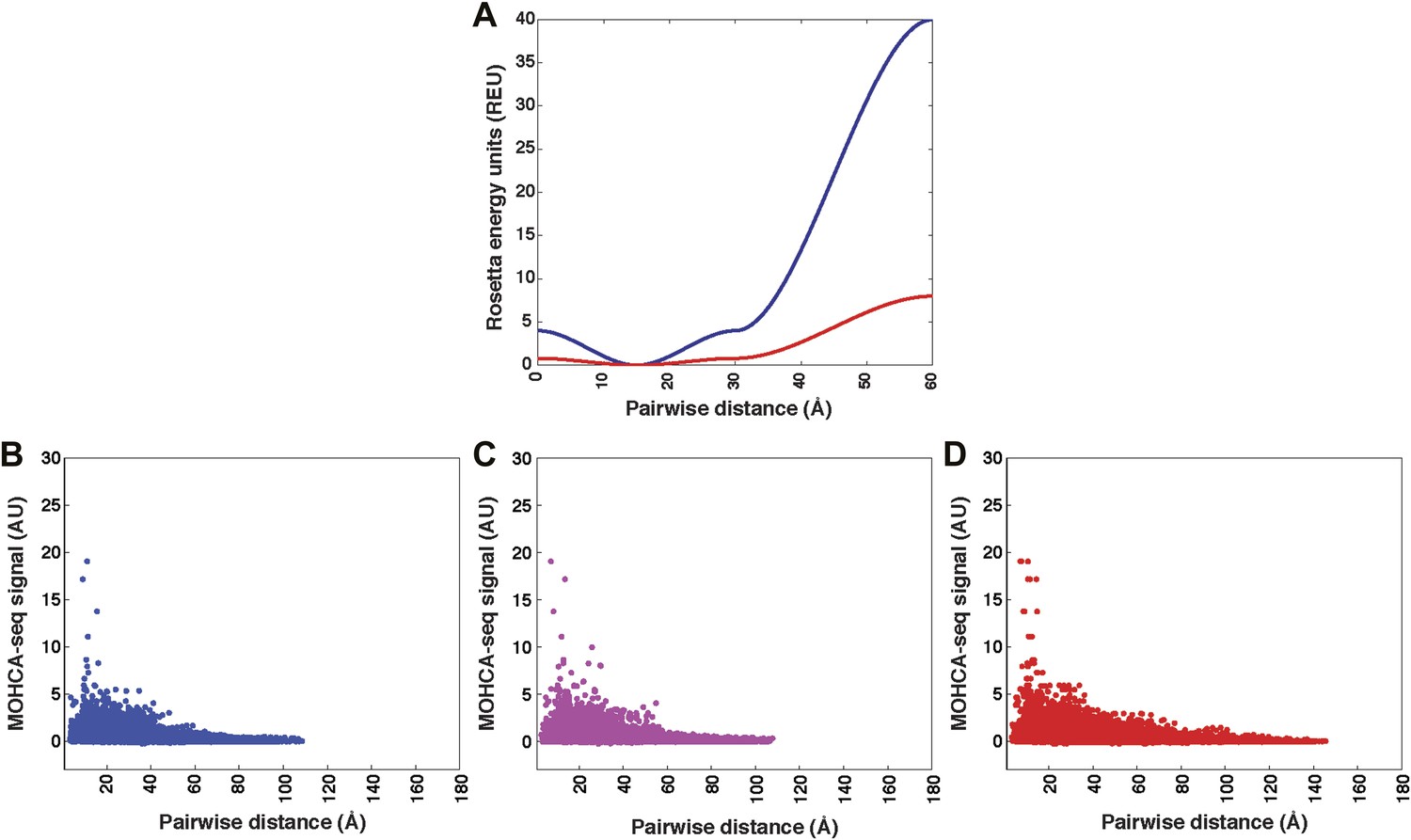
Pseudo-energy potential used to incorporate MOHCA-seq constraints in Rosetta modeling, and plots of MOHCA-seq signal vs pairwise distance.
(A) Pseudo-energy potentials for strong (blue) and weak (red) MOHCA-seq constraints. The potential was generated using the smoothstep function (see ‘Materials and methods’). (B) MOHCA-seq signal vs pairwise distance plot for crystal structures of RNAs listed in Table 1. (C) MOHCA-seq signal vs pairwise distance plot for MCM models of RNAs listed in Table 1 and HoxA9 IRES domain. (D) MOHCA-seq signal vs pairwise distance plot for ligand-free MCM models of c-di-GMP, glycine, and AdoCbl riboswitch aptamers shown in Figures 4C, 5C, 6B. Pairwise distances are calculated between 2′-OH of first residue (‘radical source location’) and C4′ of second residue (‘cleavage location’). MOHCA-seq signal is from final COHCOA data sets and ignores pairs of residues separated by fewer than 7 nucleotides due to ambiguities in aligning short reads to the RNA sequence.
Figure 2 with 2 supplements
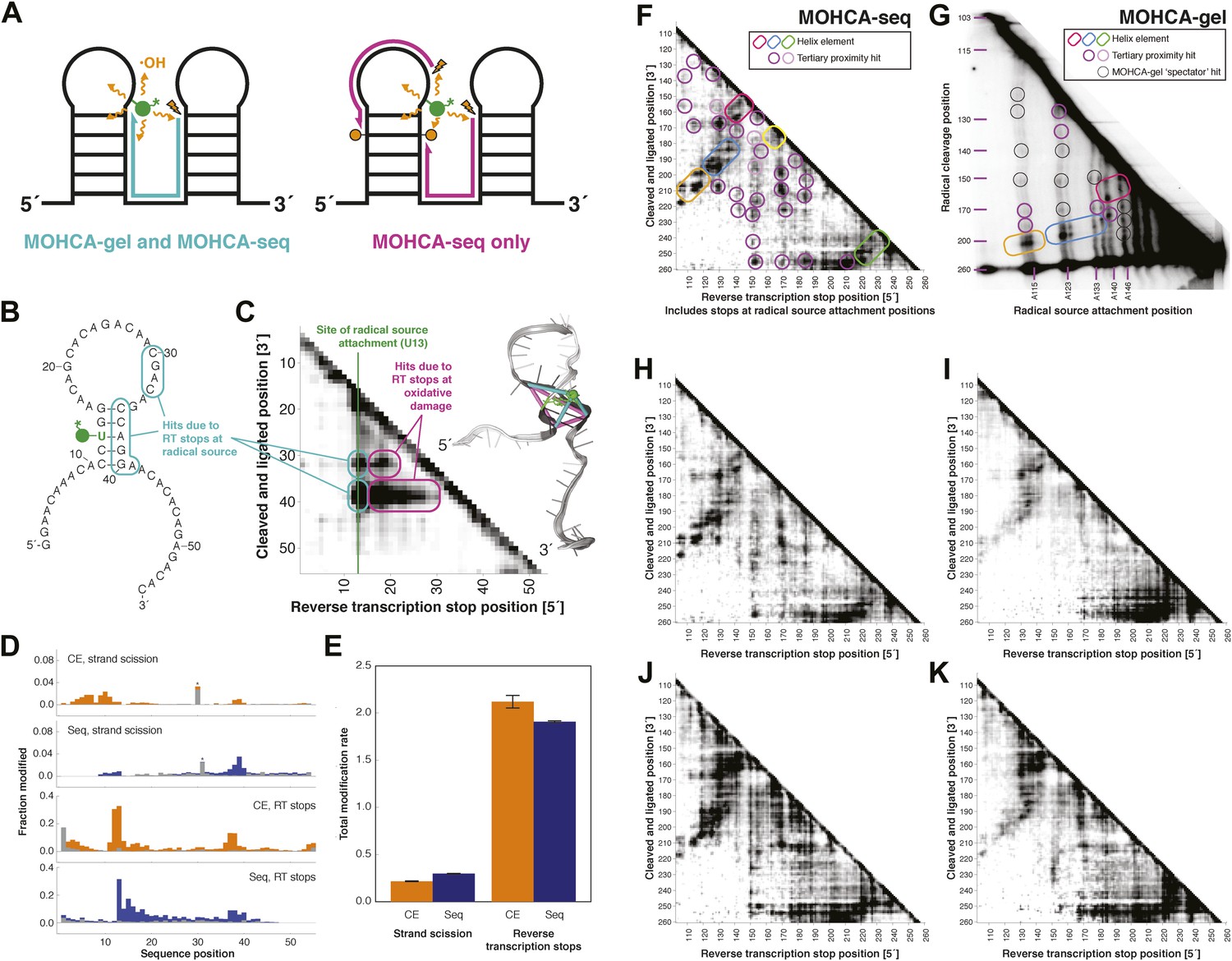
MOHCA-seq detects rich pairwise proximity information in single experiments and is robust to changes in the experimental protocol.
(A) Schematic illustrating RNA fragments detectable by MOHCA-seq. At left, reverse transcription from a strand scission position (orange bolt) terminates at the radical source (in green); these fragments are also detectable by MOHCA-gel. At right, reverse transcription from strand scission positions can also terminate at additional oxidative damage events (orange circles) caused by the same radical source; these fragments are only detectable by MOHCA-seq. (B) Sequence and secondary structure of a proof-of-concept RNA with one radical source attachment site (U13, in green). Cyan rounded rectangles indicate expected MOHCA-seq hits due to RT stops at the radical source after cleavage at the circled residues. (C) MOHCA-seq proximity map showing expected hits (cyan rounded rectangles), as well as additional reverse transcription stops occurring 3′ of the radical source position but 5′ of the cleaved and ligated position, due to oxidative damage events 3′ of the radical source that are spatially correlated with the radical source (magenta rounded rectangles). At right, strand scission rates detected by paired-end sequencing (see (D)) are shown on a scale from white (low) to black (high) on a visualization-only model of the proof-of-concept RNA. Pairwise hits detectable by MOHCA-gel and MOHCA-seq (as in (A), left) are shown as cyan lines; hits detectable by MOHCA-seq only (as in (A), right) are shown as magenta lines. (D) Quantified strand scission and reverse transcription-terminating modification rates (note difference in scale) in the proof-of-concept RNA from capillary electrophoresis (CE) or paired-end sequencing data (Seq). Blue and orange bars are data for a MOHCA-seq sample with a tethered radical source, and gray bars are data for an identically treated control sample without a radical source. A ‘*’ indicates a data point that was not included in calculation of the total rate of strand scission due to high background or bleed-through of a fluorescent reference ladder peak. (E) Total modification rates for strand scission or reverse transcription stops calculated from CE or sequencing data. (F–G) MOHCA-seq (F) and MOHCA-gel (G) proximity maps of P4–P6 from single experiments. MOHCA-gel data were collected previously according to the published method, with 5′-32P-labeled P4–P6 RNA (Das et al., 2008). Rounded rectangles indicate helix elements, purple circles indicate hits corresponding to < 30 Å pairwise distance in the crystal structure, pink circles indicate hits corresponding to > 30 Å pairwise distance in the crystal structure, and black circles indicate MOHCA-gel ‘spectator hits’, which appear due to lack of rigorous background subtraction (Das et al., 2008; Kim et al., 2011). The pink and purple circles in (C) correspond to the hits annotated in Figure 1D, which shows the average of four data replicates; features that are not circled disappear after averaging. Note distortion of MOHCA-gel compared to MOHCA-seq due to nonlinearity of gel electrophoresis rates with RNA length. (H–K) MOHCA-seq proximity maps for P4–P6 with alternative 2′-NH2-modified nucleotides incorporated during transcription. Modified nucleotide triphosphate included in transcription reaction at molar ratio of 0.5 to unmodified NTP: (H) 2′-NH2-2′-dATP; (I) 2′-NH2-2′-dUTP; (J) 2′-NH2-2′-dGTP; (K) 2′-NH2-2′-dCTP. All fragmentation reactions were performed for 30 min. All four data sets were collected in one Illumina MiSeq run using a 50-cycle MiSeq Reagent Kit v2. Additional variations of the MOHCA-seq protocol are shown in Figure 2—figure supplement 1 (variation of radical source incorporation rate) and Figure 2—figure supplement 2 (variation of fragmentation reaction time).
Figure 2—figure supplement 1
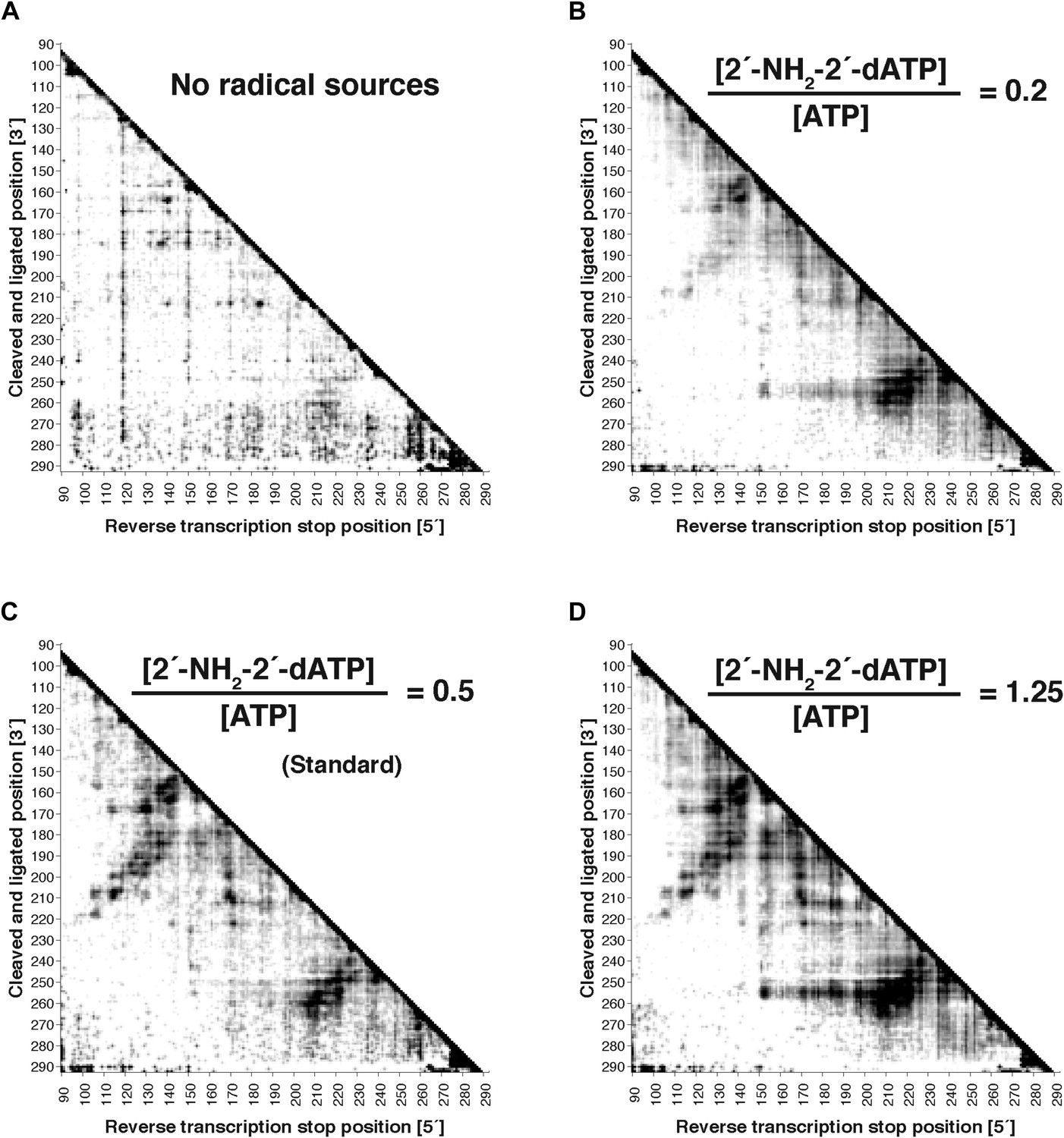
Variation of radical source incorporation rate.
MOHCA-seq proximity maps for P4–P6. Ratio of concentrations of 2′-NH2-2′-dATP to ATP in transcription reaction: (A) 0; (B) 0.2; (C) 0.5 (standard); (D) 1.25. All fragmentation reactions were performed for 10 min. Note that ‘hits’ in (A) were weak but are accentuated on the proximity map by scaling (see ‘Materials and methods’).
Figure 2—figure supplement 2
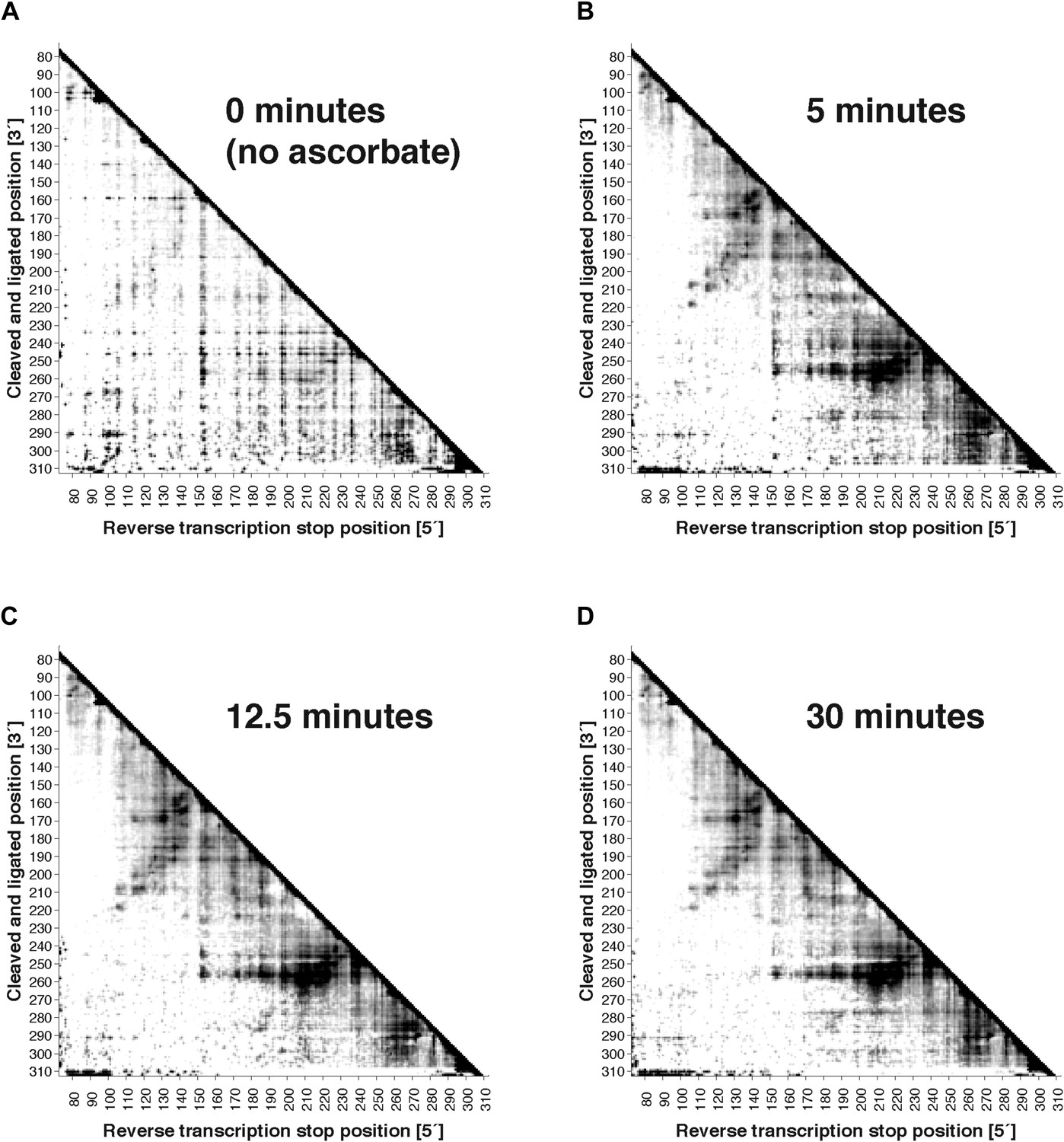
Variation of fragmentation reaction time.
MOHCA-seq proximity maps for P4–P6. Fragmentation reaction time: (A) 0 min (no ascorbate added); (B) 5 min; (C) 12.5 min; (D) 30 min. All fragmentations reactions used a 2′-NH2-2′-dATP:ATP ratio of 0.5. The P4–P6 construct used for this experiment included flanking hairpins 5′ and 3′ reference hairpins used for standardizing chemical mapping data (Kladwang et al., 2014) (sequence listed in Supplementary file 1). All four data sets were collected in one Illumina MiSeq run using a 50-cycle MiSeq Reagent Kit v2. Note that ‘hits’ in (A) were weak but are accentuated on the proximity map by scaling (see ‘Materials and methods’).
Figure 3 with 7 supplements
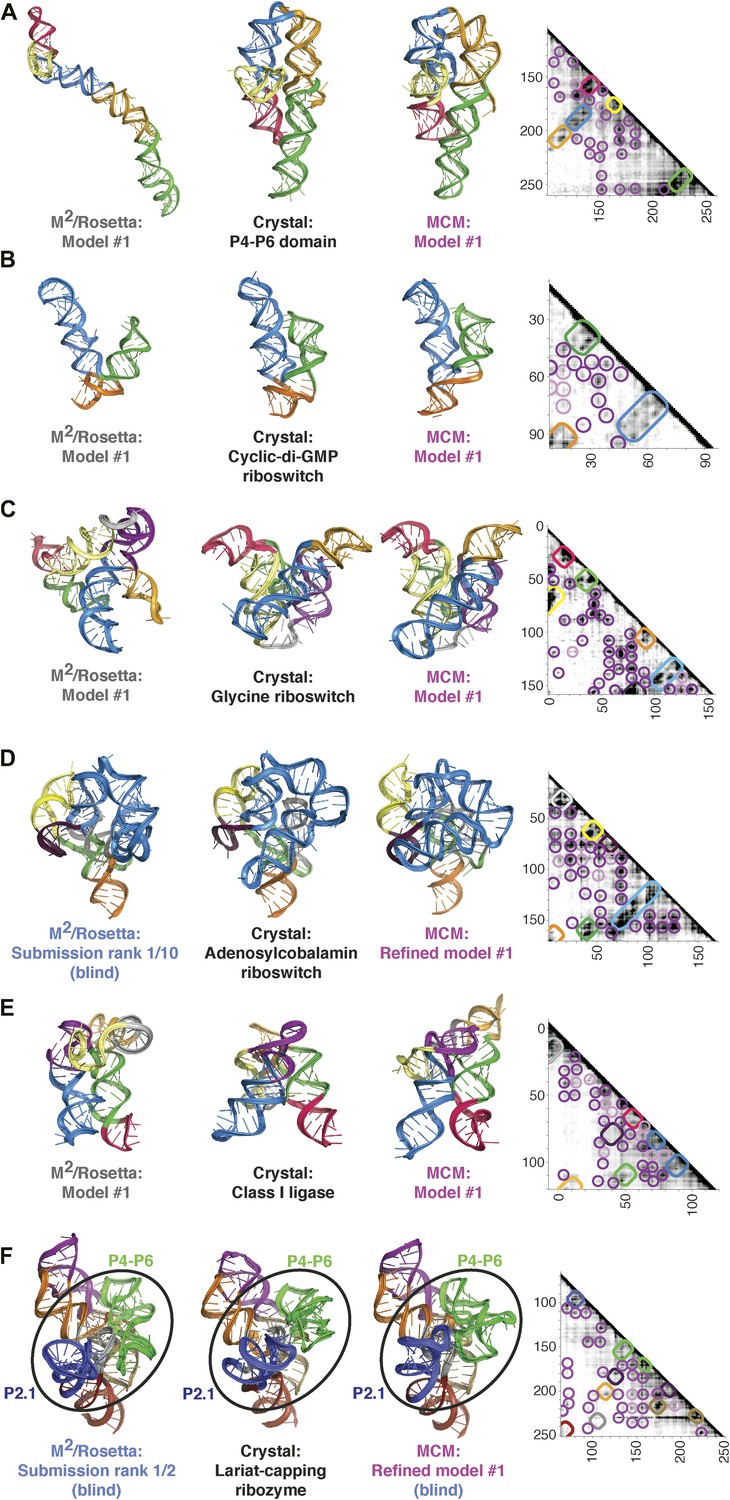
MCM achieves 1-nm resolution models of complex RNA folds.
(A) P4–P6 domain: M2/Rosetta model (left), 38.3 Å root-mean-squared-deviation (RMSD); crystal structure (PDB ID 1GID, center left); M2/MOHCA-seq/Rosetta (MCM) model (center right), 8.6 Å RMSD. (B) V. cholerae cyclic-di-GMP riboswitch aptamer: M2/Rosetta model (left), 11.3 Å RMSD; crystal structure (PDB ID 3IRW, center left); MCM model (center right), 7.6 Å RMSD. (C) F. nucleatum double glycine riboswitch ligand-binding domain: M2/Rosetta model (left), 30.5 Å RMSD; crystal structure (PDB ID 3P49, center left); MCM model (center right), 7.9 Å RMSD. (D) S. thermophilum adenosylcobalamin (AdoCbl) riboswitch aptamer: M2/Rosetta submission rank 1 of 10 for RNA-puzzle 6 (left), 17.1 Å RMSD; crystal structure (PDB ID 4GXY, center left); MCM model (center right), 11.9 Å RMSD. (E) Class I ligase: M2/Rosetta model (left), 26.3 Å global RMSD, and 14.0 Å core RMSD; crystal structure (PDB ID 3HHN, center left); MCM model (center right), 14.5 Å global RMSD and 11.1 Å core RMSD. (F) D. iridis lariat-capping ribozyme: M2/Rosetta submission rank 1 of 2 for RNA-puzzle 5 (left), 17.0 Å P2.1/P4–P6 RMSD and 9.6 Å global RMSD; crystal structure (PDB ID 4P8Z, center left); MCM model (center right), 11.2 Å P2.1/P4–P6 RMSD and 8.2 Å global RMSD. In (A–F), MOHCA-seq proximity maps with annotated helix elements (rounded rectangles) and tertiary hits (purple and pink circles) as in Figure 1D are shown at right. Full-size proximity maps, including 5′- and 3′-flanking sequences outside the region of interest, are shown in Figure 3—figure supplement 3. M2 analyses of AdoCbl riboswitch aptamer, class I ligase, and lariat-capping ribozyme are shown in Figure 3—figure supplements 1, 2, 7. Figure 3—figure supplement 4 shows comparisons of models generated by different computational methods for RNA-puzzle 6. Figure 3—figure supplements 5, 6 show comparisons of additional modeling runs for class I ligase and AdoCbl riboswitch aptamer to crystal structures.
Figure 3—figure supplement 1
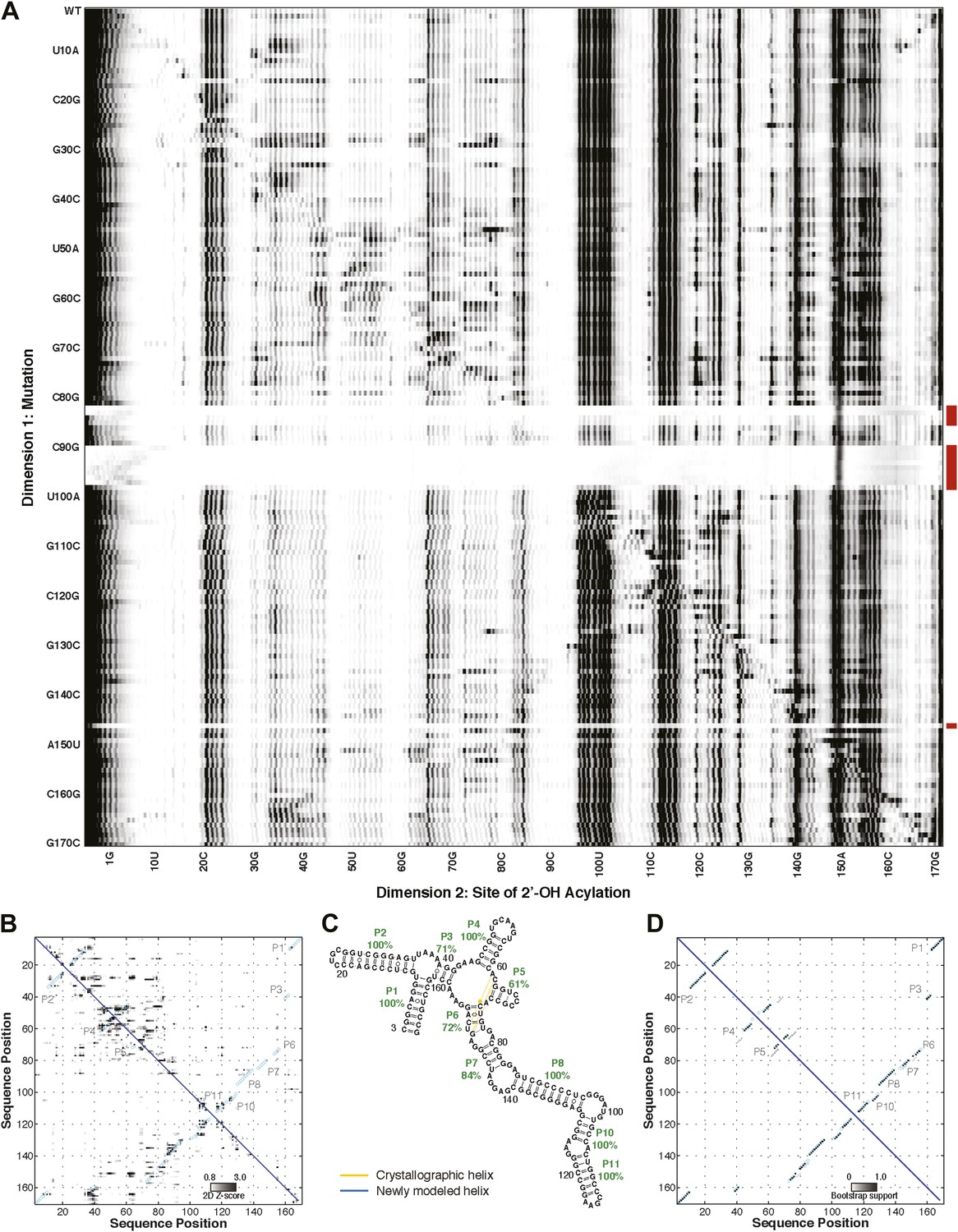
M2 analysis of the S. thermophilum adenosylcobalamin riboswitch aptamer, performed during the sixth RNA-puzzles structure prediction trial.
(A) M2 data set for 1M7 modification across 168 single mutations along the AdoCbl riboswitch aptamer sequence in the presence of 60 µM AdoCbl ligand. Mutants showing poor data quality are marked by red bars. (B) Z-score contact map extracted from (A). (C) Secondary structure prediction and (D) bootstrap support matrix using M2 data. In (B) and (D), the crystallographic secondary structure is overlaid as cyan circles.
Figure 3—figure supplement 2
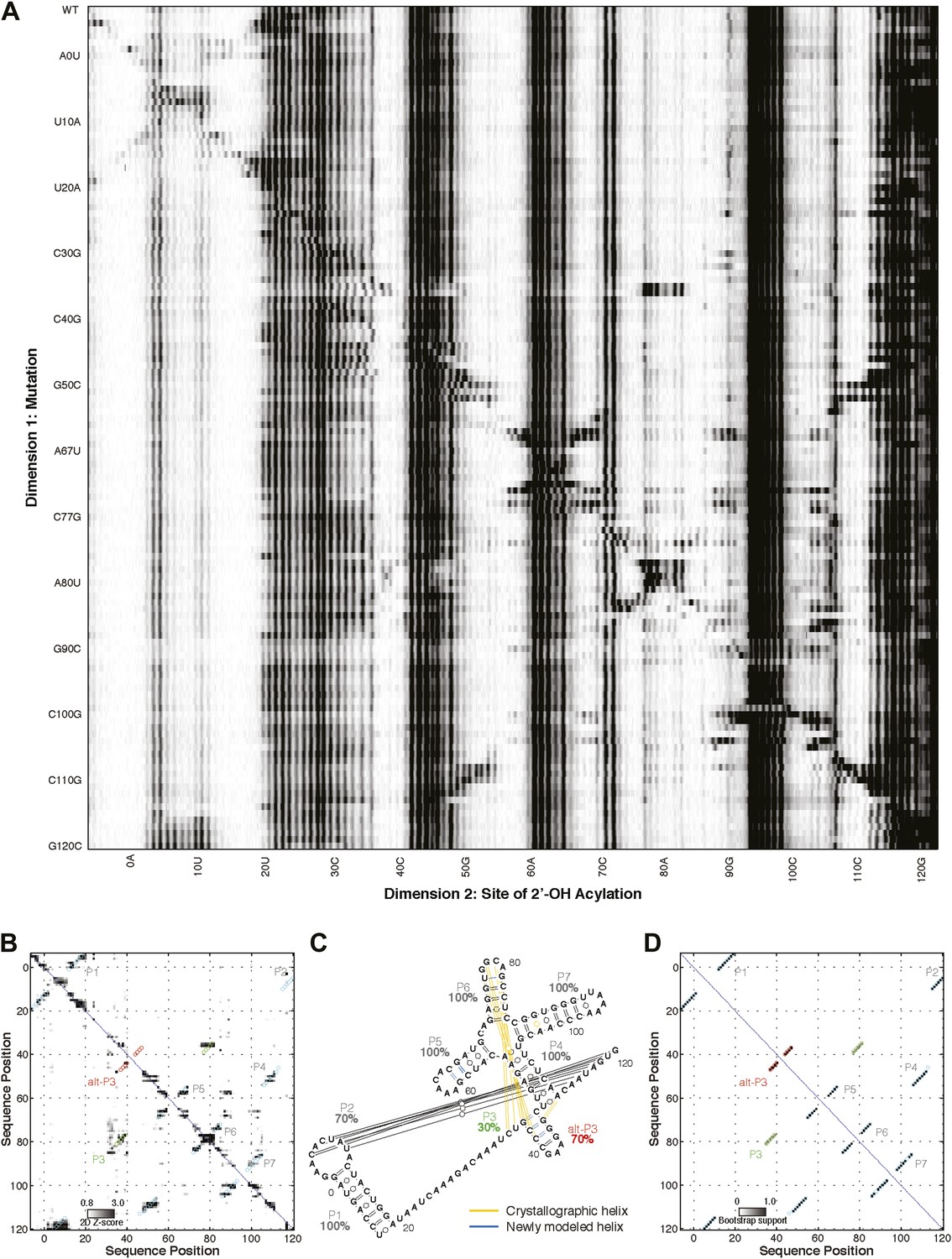
M2 analysis of class I ligase.
(A) M2 data set for 1M7 modification across 127 single mutations along the class I ligase sequence. (B) Z-score contact map extracted from (A). (C) Secondary structure prediction and (D) bootstrap support matrix using M2 data. In (B) and (D), the crystallographic secondary structure is overlaid as cyan and green circles, with an alternative P3 helix predicted by M2 data overlaid as red circles. When SHAPEknots (Hajdin et al., 2013) was used to predict the pseudoknots in the full sequence, only the P2 pseudoknot was recovered. However, SHAPEknots successfully predicted the P3 helix when the P1 helix was omitted.
Figure 3—figure supplement 3
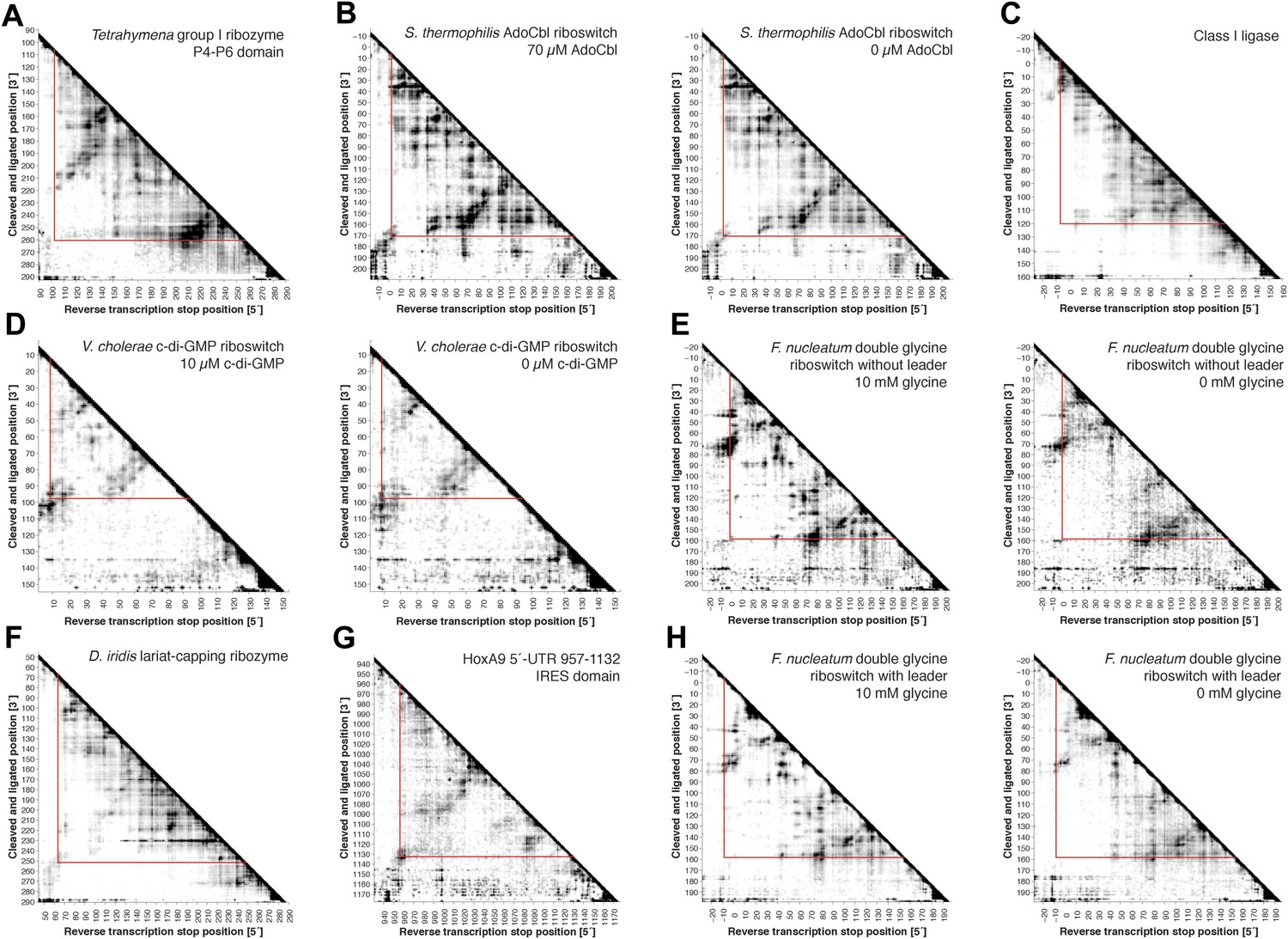
Full MOHCA-seq proximity maps, including 5′- and 3′-flanking regions.
Full COHCOA-analyzed MOHCA-seq proximity maps of (A) P4–P6, (B) S. thermophilum adenosylcobalamin riboswitch aptamer in the presence (left) or absence (right) of 70 µM AdoCbl, (C) D. iridis lariat-capping ribozyme, (D) V. cholerae cyclic-di-GMP riboswitch aptamer in the presence (left) or absence (right) of 10 µM c-di-GMP, (E) F. nucleatum glycine riboswitch ligand-binding domain without leader sequence in the presence (left) or absence (right) of 10 mM glycine, (F) class I ligase, (G) HoxA9 5′-UTR 957–1132 IRES domain, and (H) F. nucleatum glycine riboswitch ligand-binding domain with leader sequence (Kladwang et al., 2012) in the presence (left) or absence (right) of 10 mM glycine. On all maps, the region of interest is enclosed by red lines. All RNAs except for P4–P6 include 5′ and 3′ reference hairpins used for normalizing chemical mapping data from other techniques (Kladwang et al., 2014) (sequences listed in Supplementary file 1).
Figure 3—figure supplement 4
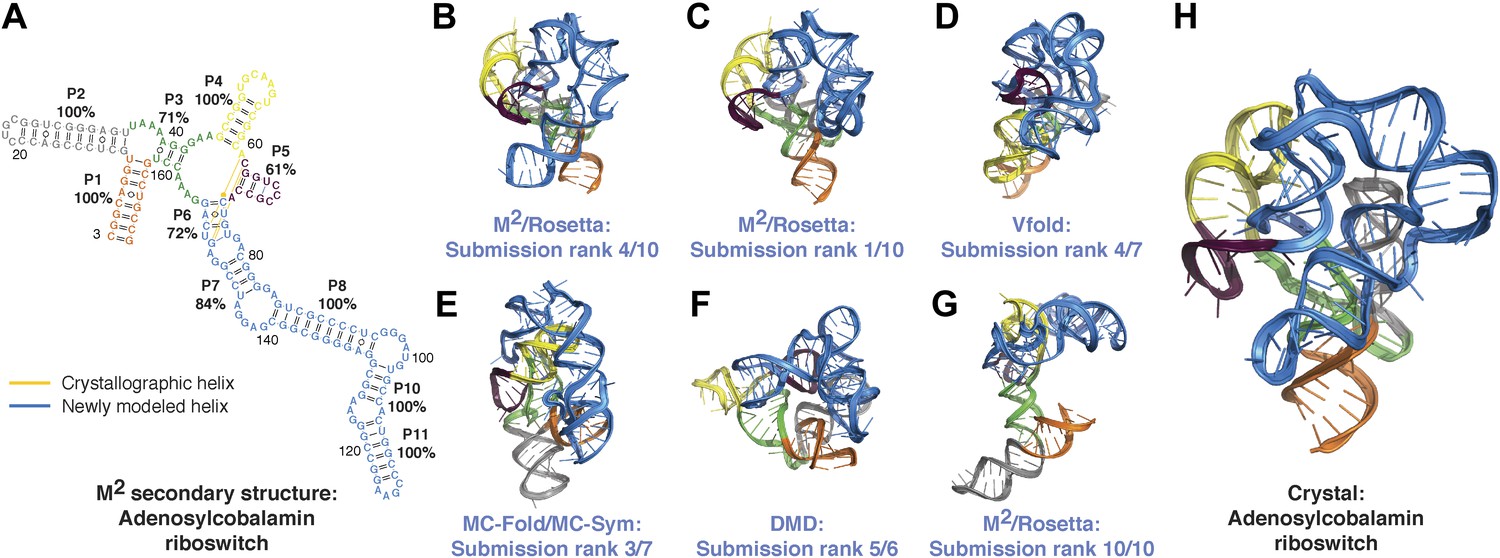
Blind models generated for RNA-puzzles can attain 1-nm resolution or better but cannot predict the most accurate models.
(A) M2-derived secondary structure of S. thermophilus adenosylcobalamin riboswitch aptamer, generated for the RNA-puzzle 6 challenge (Miao et al., 2015). Bootstrap support values for each helix are shown as percentages. Crystallographic Watson–Crick base pairs missing in the secondary structure are connected by yellow lines, and non-crystallographic Watson–Crick base pairs predicted in the secondary structure are connected by blue lines. (B–G) Blind 3D models of the S. thermophilus adenosylcobalamin riboswitch aptamer generated for the RNA-puzzle 6 challenge. Models were generated using (B) M2/Rosetta, 12.1 Å all-heavy-atom RMSD to crystal; (C) M2/Rosetta, 17.1 Å RMSD; (D) Vfold, 22.1 Å RMSD; (E) MC-Fold and MC-Sym, 23.4 Å RMSD; (F) DMD, 24.0 Å RMSD; (G) M2/Rosetta, 32.8 Å RMSD. Modeling methods were previously described in detail (Miao et al., 2015). For each modeling method, the most accurate submitted model to the crystal structure is shown (B, D–F). The ranks predicted by the modelers of their own submissions (‘submission rank’) are given below each model. (H) Crystal structure of the S. thermophilus adenosylcobalamin riboswitch aptamer (PDB ID 4GXY).
Figure 3—figure supplement 5
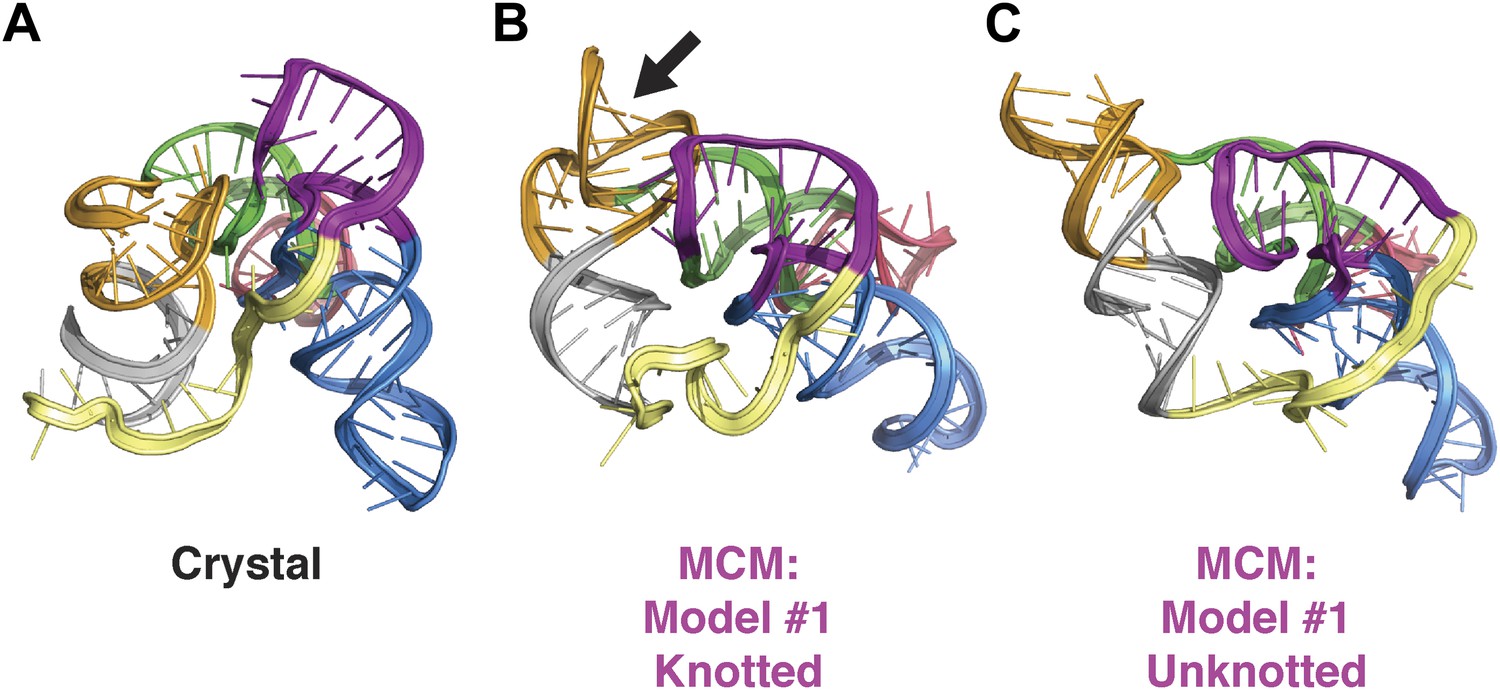
Comparison of class I ligase crystal structure and knotted and unknotted MCM models.
(A) Crystal structure of the class I ligase. (B) Final MCM model from a cluster with knotted 3′-end (black arrow). (C) Final MCM model from a cluster without knotted 3′-end.
Figure 3—figure supplement 6
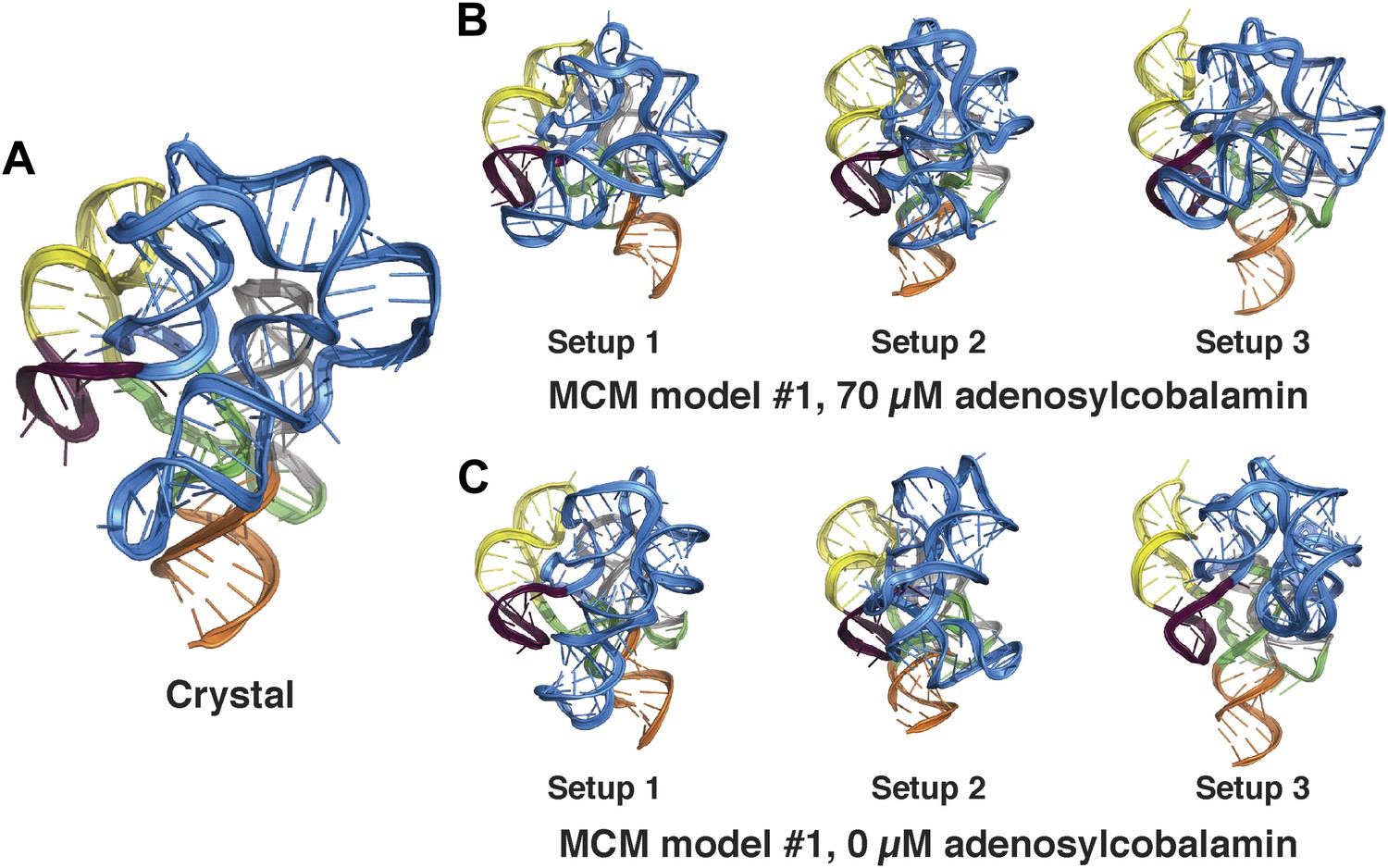
Comparison between MCM models of the adenosylcobalamin riboswitch aptamer using different initial RNA fragment sets.
(A) Crystal structure of S. thermophilum AdoCbl riboswitch aptamer (PDB ID 4GXY) and MCM models using MOHCA-seq data collected in the presence of (B) 70 µM AdoCbl ligand or (C) 0 µM AdoCbl ligand. MCM models were generated using three distinct sets of prebuilt RNA fragments prepared during RNA-puzzle modeling (labeled setups 1–3; see ‘Materials and methods’).
Figure 3—figure supplement 7
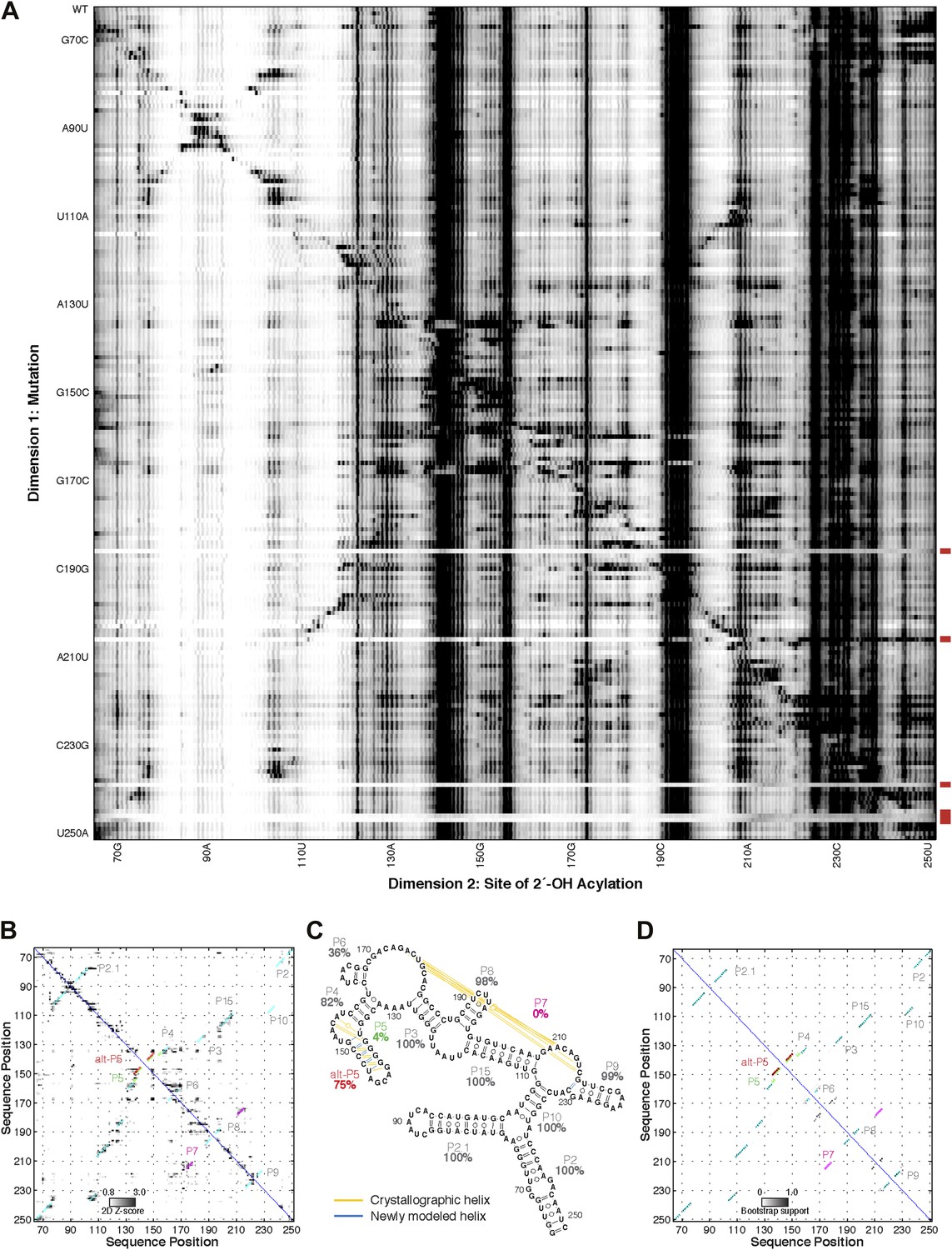
M2 analysis of the GIR1 lariat-capping ribozyme, performed during the fifth RNA-puzzles structure prediction trial.
(A) M2 data set for 1M7 modification across 188 single mutations along the GIR1 ribozyme sequence. Mutants showing poor data quality are marked by red bars. (B) Z-score contact map extracted from (A). (C) Secondary structure prediction and (D) bootstrap support matrix using M2 data. In (B) and (D), the crystallographic secondary structure is overlaid as cyan circles.
Figure 4
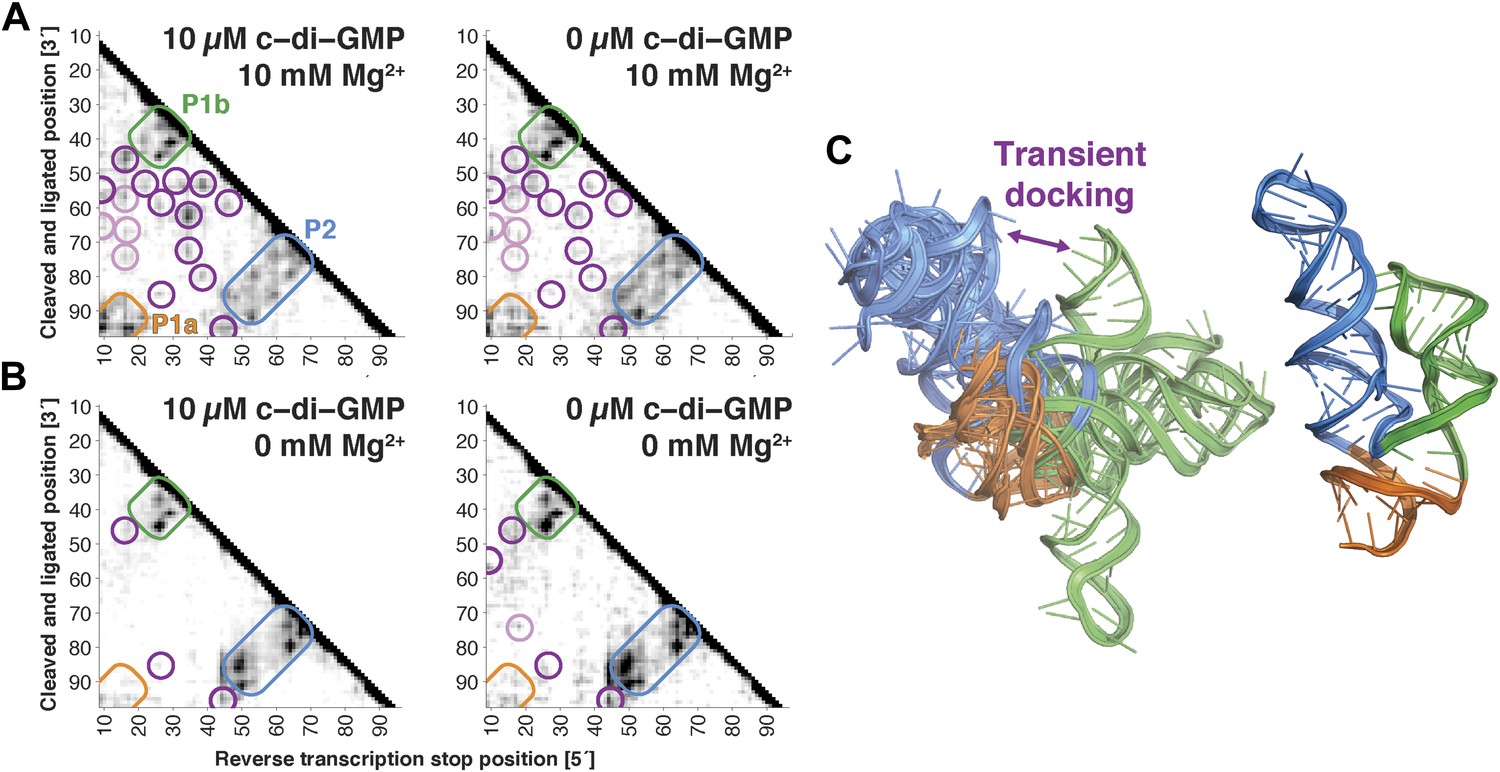
The V. cholerae cyclic-di-GMP riboswitch aptamer exhibits Mg2+-dependent preformed tertiary structure in its ligand-free state.
(A) Proximity maps of c-di-GMP riboswitch aptamer in 10 µM (left) or 0 µM (right) c-di-GMP, folded in 10 mM Mg2+, with annotated helix elements (rounded rectangles) and tertiary hits (purple and pink circles). (B) Proximity maps of c-di-GMP riboswitch aptamer in 10 µM (left) or 0 µM (right) c-di-GMP, folded in 0 mM Mg2+, showing absence of tertiary proximities that are present for the aptamer folded in 10 mM Mg2+. (C) Five MCM models with lowest Rosetta energy of c-di-GMP riboswitch aptamer give an initial visualization of ligand-free ensemble (left); modeling included pseudo-energy constraints from 0 µM c-di-GMP and 10 mM Mg2+ proximity map. The crystal structure is shown at right for comparison.
Figure 5
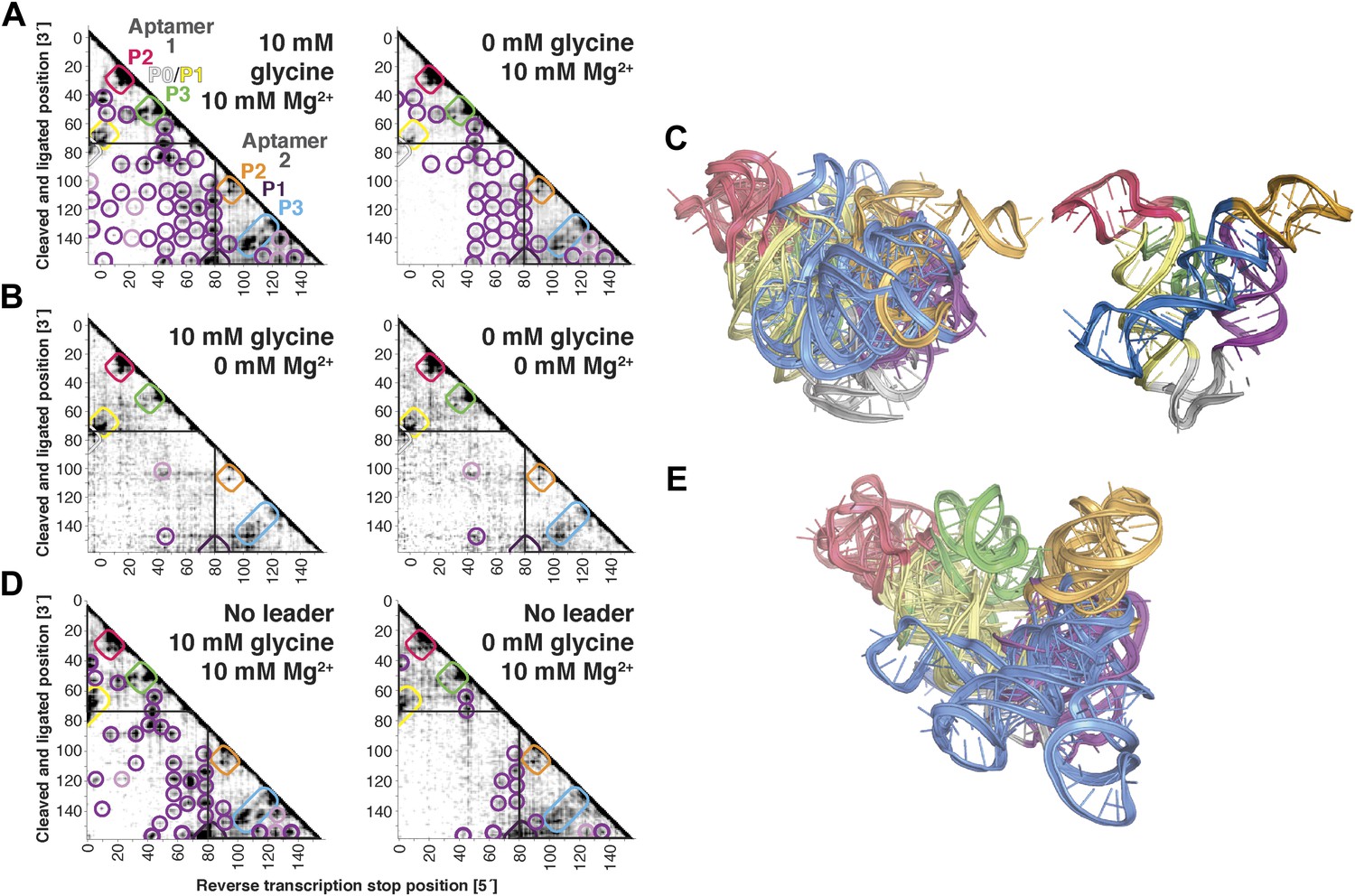
The F. nucleatum double glycine riboswitch ligand-binding domain retains preformed tertiary structure in its ligand-free state, with a requirement of a leader sequence.
(A) Proximity maps of glycine riboswitch ligand-binding domain in 10 mM (left) or 0 mM (right) glycine, folded in 10 mM Mg2+, with annotated helix elements (rounded rectangles) and tertiary hits (purple and pink circles). A black box encloses each glycine-binding aptamer. (B) Proximity maps of glycine riboswitch ligand-binding domain in 10 mM (left) or 0 mM (right) glycine, folded in 0 mM Mg2+, showing absence of tertiary proximities both within and between aptamers that are present for the ligand-binding domain folded in 10 mM Mg2+. (C) Five MCM models with lowest Rosetta energy of glycine riboswitch ligand-binding domain give an initial visualization of ligand-free ensemble (left); modeling included pseudo-energy constraints from 0 mM glycine and 10 mM Mg2+ proximity map. The crystal structure, with kink-turn linker grafted from PDB ID 3CC2 (Kladwang et al., 2012a), is shown at right for comparison. (D) Proximity maps of glycine riboswitch ligand-binding domain without leader sequence in 10 mM (left) or 0 mM (right) glycine, folded in 10 mM Mg2+, showing absence of tertiary proximities between aptamers even in presence of Mg2+. (E) Five MCM models with lowest Rosetta energy of glycine riboswitch ligand-binding domain without leader sequence give an initial visualization of ligand-free ensemble (left); modeling included pseudo-energy constraints from 0 mM glycine and 10 mM Mg2+ proximity map.
Figure 6
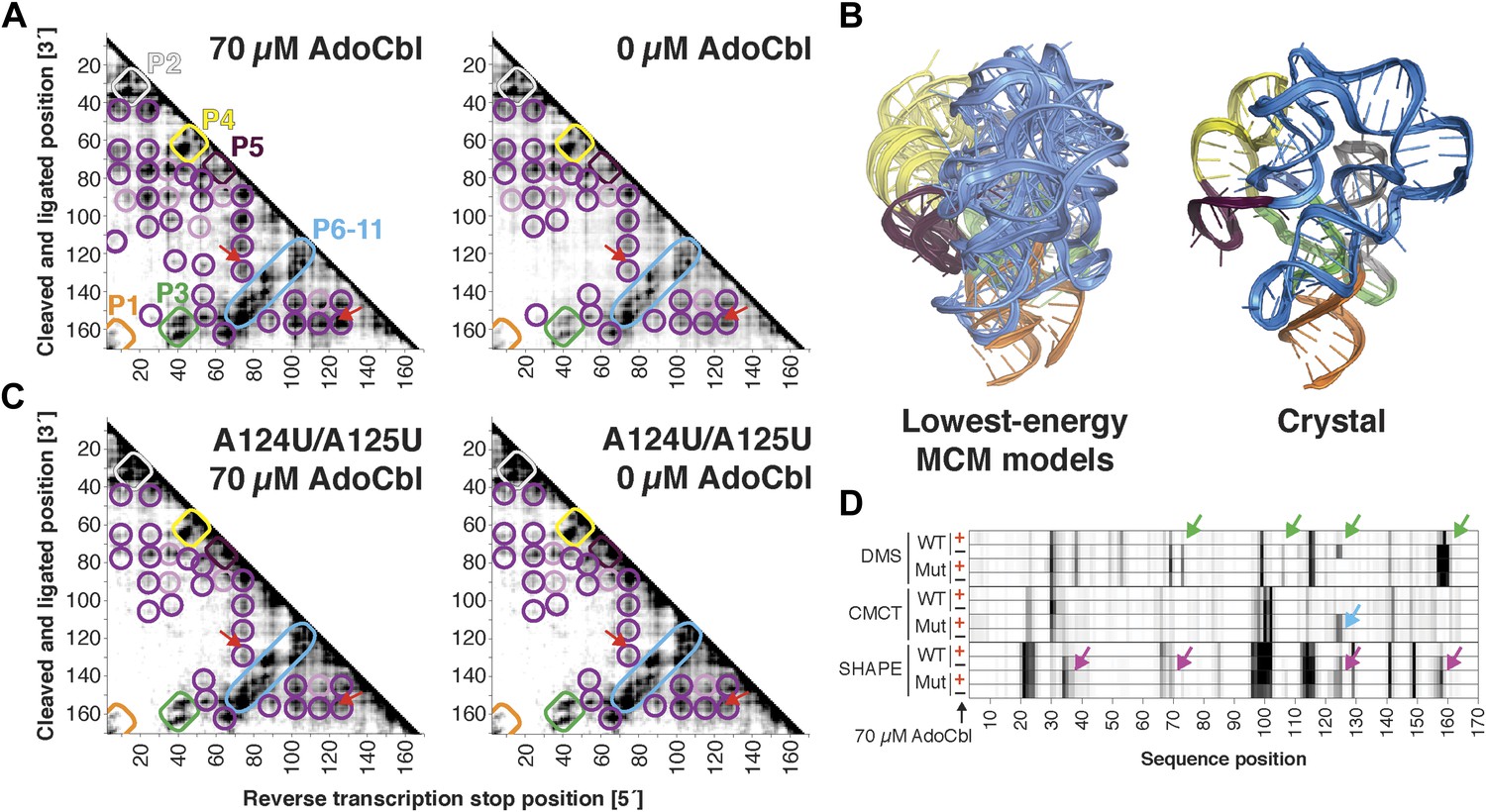
The S. thermophilus adenosylcobalamin riboswitch aptamer retains preformed tertiary structure without binding ligand.
(A) Proximity maps of AdoCbl riboswitch aptamer in 70 µM (left) or 0 µM (right) AdoCbl with annotated helix elements (rounded rectangles) and tertiary hits (purple and pink circles). Red arrows indicate regions of proximity between J11-10 and helix P6 in the crystal structure. (B) Three MCM models with lowest Rosetta energy of AdoCbl riboswitch aptamer give an initial visualization of ligand-free ensemble (left); modeling included pseudo-energy constraints from 0 µM AdoCbl proximity map. The crystal structure is shown at right for comparison. (C–D) Mutation of A124 and A125 to U disrupts ligand-binding but not preformed tertiary structure. (C) Proximity maps of A124U/A125U mutant AdoCbl riboswitch aptamer in 70 µM (left) or 0 µM (right) AdoCbl, showing retention of tertiary proximity hits similar to the wild-type aptamer in 0 µM AdoCbl. (D) 1D chemical mapping reactivities of wild-type and A124U/A125U (‘Mut’) AdoCbl riboswitch aptamer. Green arrows in DMS reactivity lanes indicate locations of reduced reactivity in the presence of ligand in the wild type only. Residues 124 and 125 are unreactive to DMS in the A124U/A125U mutant because DMS does not modify U residues. Blue arrow in CMCT reactivity lanes indicates lack of protection of U124 and U125 in the mutant in the presence of ligand. Magenta arrows in SHAPE reactivity indicate locations of reduced/altered reactivity in the presence of ligand in the wild type but not in the mutant. All ligand-dependent protections are located in or near the AdoCbl-binding site in the riboswitch aptamer (Peselis & Serganov, 2012).
Figure 7
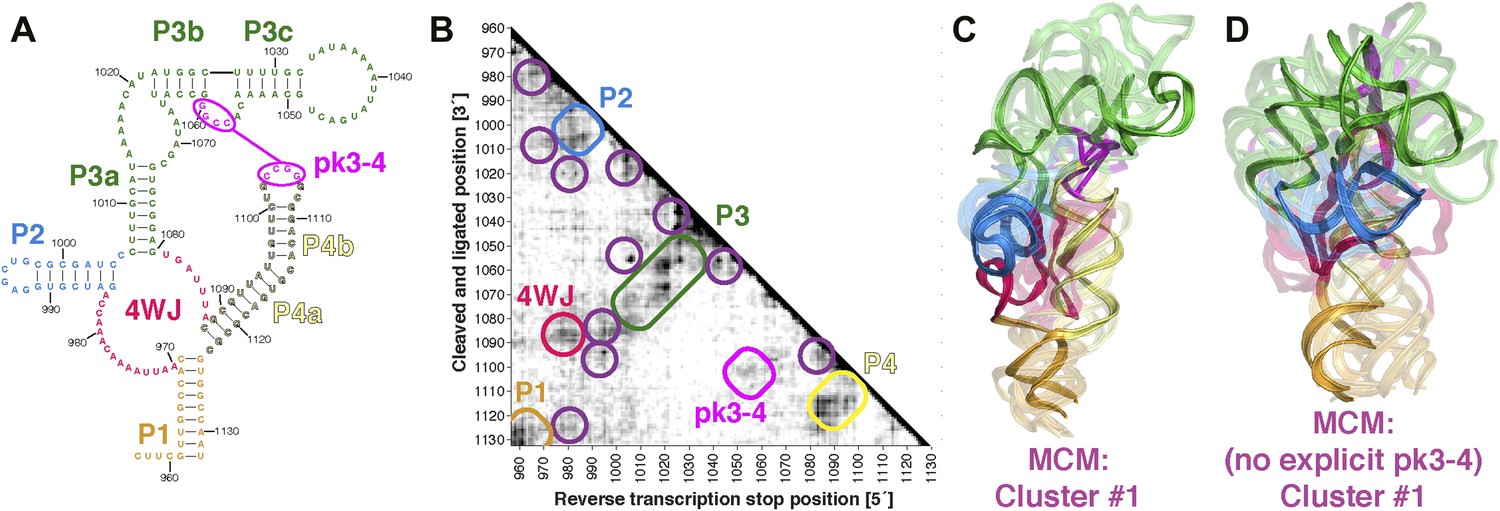
MCM analysis of a recently discovered human cellular mRNA IRES domain.
(A) M2-derived secondary structure model of HoxA9 5′-UTR 957–1132 internal ribosome entry site (IRES) domain. (B) MOHCA-seq proximity map of HoxA9 IRES domain, with evidence for secondary structure elements and pseudoknot predicted by M2 and validated by mutate-and-rescue experiments (Xue et al., 2015) (rounded rectangles). Tertiary hits are indicated by purple circles. (C) MCM model of HoxA9 IRES domain with explicit pseudoknot pk3-4. (D) MCM model of HoxA9 IRES domain without explicit pseudoknot pk3-4. In (C–D), cluster center (opaque) and four additional models (semi-transparent) from the top cluster are shown. Magenta lines connect base pairs of pk3-4 in each cluster center model. Top cluster models for both modeling setups are available in Source code 1.
Tables
Table 1
Benchmark of MCM on RNAs with crystal structures
| RNA | Length | M2/Rosetta (no MOHCA, control) | MCM | ||
|---|---|---|---|---|---|
| RMSD to crystal (Å) (accuracy) | p-value§ | RMSD to crystal (Å) (accuracy) | p-value§ | ||
| Tetrahymena ribozyme P4–P6 domain | 158 | 38.3 | >0.9 | 8.6 | <1.0 × 10−16 |
| V. cholerae cyclic-di-GMP riboswitch aptamer, ligand-bound* | 89 | 11.3 | 2.6 × 10−3 | 7.6 | 6.3 × 10−7 |
| F. nucleatum double glycine riboswitch ligand-binding domain, ligand-bound* | 159 | 30.5 | >0.9 | 7.9 | <1.0 × 10−16 |
| S. thermophilum adenosylcobalamin riboswitch aptamer, ligand-bound* | 168 | 17.1† | 5.3 × 10−7 | 11.9 | 4.0 × 10−15 |
| Class I ligase | 127 | 26.3 | >0.9 | 14.5 | 6.8 × 10−5 |
| Class I ligase, core domain‡ | 87 | 14.0 | 0.13 | 11.1 | 3.1 × 10−3 |
| D. iridis lariat-capping ribozyme | 188 | 9.6† | <1.0 × 10−16 | 8.2 | <1.0 × 10−16 |
| D. iridis lariat-capping ribozyme, MCM refined regions‡ | 69 | 17.0† | n.a.§ | 11.2 | n.a.§ |
-
*
MCM modeling was performed with MOHCA-seq constraints from data sets collected on the ligand-bound state; ligands were not included during Rosetta modeling.
-
†
M2/Rosetta statistics are reported for RNA-puzzle submission rank 1 models, which included subdomains built by homology modeling.
-
‡
Calculated over core domain residues or refined regions after alignment using MAMMOTH; (Ortiz et al., 2002) see ‘Materials and methods’.
-
§
p-value computed using analytical formula for secondary-structure-constrained 3D modeling in ref. (Hajdin et al., 2010); it is not applicable to peripheral domains. Values above 0.9 are not well-determined and are presented as > 0.9.
-
MCM: multidimensional chemical mapping; RMSD: root-mean-squared-deviation.
Table 2
Precision and constraint statistics of MCM modeling, including conformationally heterogeneous states
| RNA | Length | Number of models | Size of largest cluster | RMSD to crystal, cluster center (Å) (accuracy) | In situ RMSD estimate (Å) (precision) | Total constraints | Percent strong constraints satisfied,* cluster center | Percent strong constraints satisfied, crystal | Percent weak constraints satisfied, cluster center | Percent weak constraints satisfied, crystal |
|---|---|---|---|---|---|---|---|---|---|---|
| Tetrahymena ribozyme P4–P6 domain | 158 | 61,115 | 48 | 8.6 | 16.5 | 35 | 65.4 | 69.2 | 55.6 | 44.4 |
| V. cholerae cyclic-di-GMP riboswitch aptamer, bound | 89 | 15,632 | 10 | 7.6 | 6.8 | 19 | 100.0 | 100.0 | 100.0 | 75.0 |
| V. cholerae cyclic-di-GMP riboswitch aptamer, unbound† | 89 | 26,421 | 5 | (26.0)‡ | 9.8 | 14 | 100.0 | 83.3 | 75.0 | 37.5 |
| F. nucleatum double glycine riboswitch ligand-binding domain, bound | 159 | 16,506 | 14 | 7.9 | 10.0 | 31 | 95.8 | 100.0 | 57.1 | 42.9 |
| F. nucleatum double glycine riboswitch ligand-binding domain, unbound† | 159 | 15,771 | 12 | (25.4)‡ | 26.7 | 12 | N/A§ | N/A§ | 58.3 | 83.3 |
| F. nucleatum double glycine riboswitch ligand-binding domain with leader, bound | 167 | 23,153 | 19 | 11.8 | 10.6 | 49 | 89.5 | 84.2 | 83.3 | 83.3 |
| F. nucleatum double glycine riboswitch ligand-binding domain with leader, unbound† | 167 | 18,096 | 15 | (15.6)‡ | 14.7 | 34 | 77.8 | 33.3 | 64.0 | 68.0 |
| S. thermophilum adenosylcobalamin riboswitch aptamer, bound# | 168 | 14,219 | 12 | 11.9 | 13.9 | 38 | 84.0 | 76.0 | 84.6 | 69.2 |
| S. thermophilum adenosylcobalamin riboswitch aptamer, unbound†,# | 168 | 11,980 | 10 | (17.3)‡ | 19.7 | 33 | 83.3 | 94.4 | 73.3 | 73.3 |
| Class I ligase (unknotted) | 127 | 17,881 | 7 | 14.5 | 11.6 | 24 | 86.7 | 53.3 | 22.2 | 77.8 |
| Class I ligase (unknotted), core domain¶ | 87 | 17,881 | 7 | 11.1 | 12.0 | 18 | 91.7 | 58.3 | 33.3 | 50.0 |
| Class I ligase (knotted) | 127 | 17,881 | 14 | 16.1 | 11.6 | 24 | 86.7 | 53.3 | 55.6 | 77.8 |
| D. iridis lariat capping ribozyme | 188 | 19,741 | 16 | 8.2 | 5.7 | 32 | 90.0 | 70.0 | 54.5 | 54.5 |
| D. iridis lariat capping ribozyme, RNA-puzzles submission model #1 (2012) | 188 | – | – | 9.6 | – | – | 60.0 | 70.0 | 59.1 | 54.5 |
| D. iridis lariat capping ribozyme, MCM refined regions¶ | 69 | 19,741 | 16 | 11.2 | 10.5 | 32 | 87.5 | 62.5 | 46.2 | 38.5 |
| D. iridis lariat capping ribozyme, RNA-puzzles submission model #1 (2012), MCM refined regions# | 69 | – | – | 17.0 | – | – | 50.0 | 62.5 | 46.2 | 38.5 |
| HoxA9 5′-UTR 957–1132 domain, pseudoknot | 176 | 15,580 | 12 | – | 19.5 | 20 | 41.7 | – | 50.0 | – |
| HoxA9 5′-UTR 957–1132 domain, no pseudoknot | 176 | 11,234 | 8 | – | 19.8 | 21 | 33.3 | – | 22.2 | – |
-
*
Constraints were considered satisfied if the O2′ of the 5′-residue was less than 30 Å from the C4′ of the 3′-residue (Supplementary file 2).
-
†
For ligand-free states of the adenosylcobalamin riboswitch aptamer, cyclic-di-GMP riboswitch aptamer, and glycine riboswitch ligand-binding domain, RMSDs and percent constraints satisfied were calculated for gold-standard crystal structures solved in the presence of ligand.
-
‡
For states not expected to agree with crystal structures, RMSD accuracy is given in parentheses.
-
§
No strong constraints were selected for the unbound state of the glycine riboswitch ligand-binding domain without leader sequence.
-
#
Three separate modeling runs were performed for each ligand-binding state of the riboswitch aptamer, using distinct sets of prebuilt fragments prepared for RNA-puzzle 6; see ‘Materials and methods’. The number of models, size of largest cluster, cluster center RMSD (accuracy), and constraint satisfaction percentages are representative data from the modeling run with the most models generated. The intra-cluster RMSD used to estimate the in situ RMSD is calculated as the mean pairwise RMSD between the cluster centers of the top cluster from each of the three modeling runs.
-
¶
Calculated over refined regions or core domain residues after alignment using MAMMOTH; (Ortiz et al., 2002) see ‘Materials and methods’.
-
MCM: multidimensional chemical mapping; RMSD: root-mean-squared-deviation.
Additional files
-
Supplementary file 1
Sequences of RNAs, single-stranded DNA ligation adapters, and sequencing primers.
- https://doi.org/10.7554/eLife.07600.023
-
Supplementary file 2
Pairwise MOHCA-seq constraints used for MCM modeling.
- https://doi.org/10.7554/eLife.07600.024
-
Source code 1
Models of HoxA9 5′-UTR 957–1132 IRES domain generated by MCM.
- https://doi.org/10.7554/eLife.07600.025
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Consistent global structures of complex RNA states through multidimensional chemical mapping
eLife 4:e07600.
https://doi.org/10.7554/eLife.07600




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}