Clusters of polymorphic transmembrane genes control resistance to schistosomes in snail vectors
- Department of Immunology and Infectious Diseases, Harvard T. H. Chan School of Public Health, United States
- Department of Integrative Biology, Oregon State University, United States
- Center for Genome Research and Biocomputing, Oregon State University, United States
Figures
Figure 1 with 2 supplements

A region on Linkage Group XII displays a major association with infection risk (Figure 1—source data 1).
(A) Genetic divergence (FST) between infected and uninfected snail pools in 10 kb windows, for variants on contigs arranged by linkage map position (Tennessen et al., 2017). The strongest signal is from PTC2 on Linkage Group XII, far exceeding the signal of known regions RADres and sod1 which reflect wide haplotype blocks (Tennessen et al., 2015a). (B) PTC2 genotypes are associated with infection outcome (Source code 1). Genotypes are displayed for all three pairs of alleles, revealing a substantial difference between R and S1 (center panel), and an intermediate signal of S2 relative to the others (left and right panels).
-
Figure 1—source data 1
Genetic divergence between infected and uninfected snail pools in 10 kb windows.
- https://cdn.elifesciences.org/articles/59395/elife-59395-fig1-data1-v2.txt.zip
Figure 1—figure supplement 1
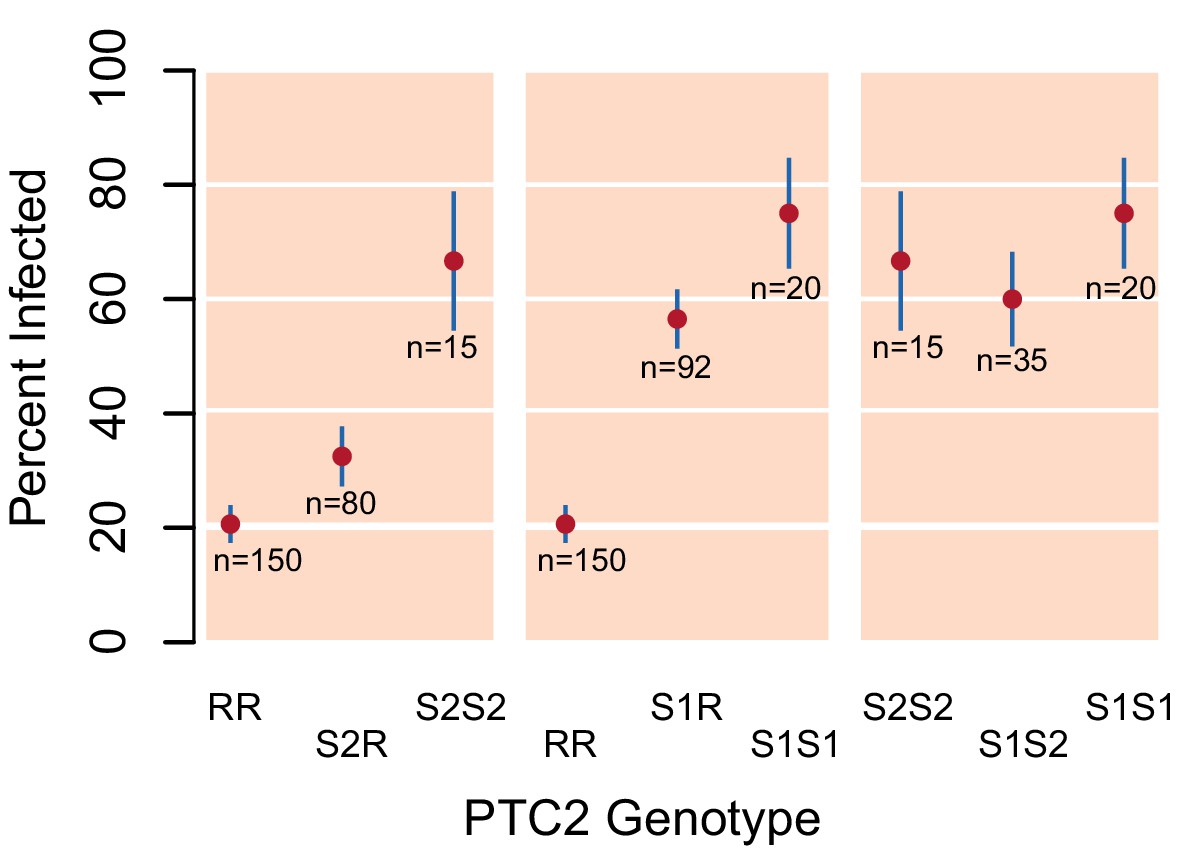
Validation of PTC2 association with infection (Source code 1).
Among 392 snails that had previously been parasite-challenged and phenotyped, we see a significant association with PTC2, independently confirming the signal in our main dataset (Figure 1B).
Figure 1—figure supplement 2
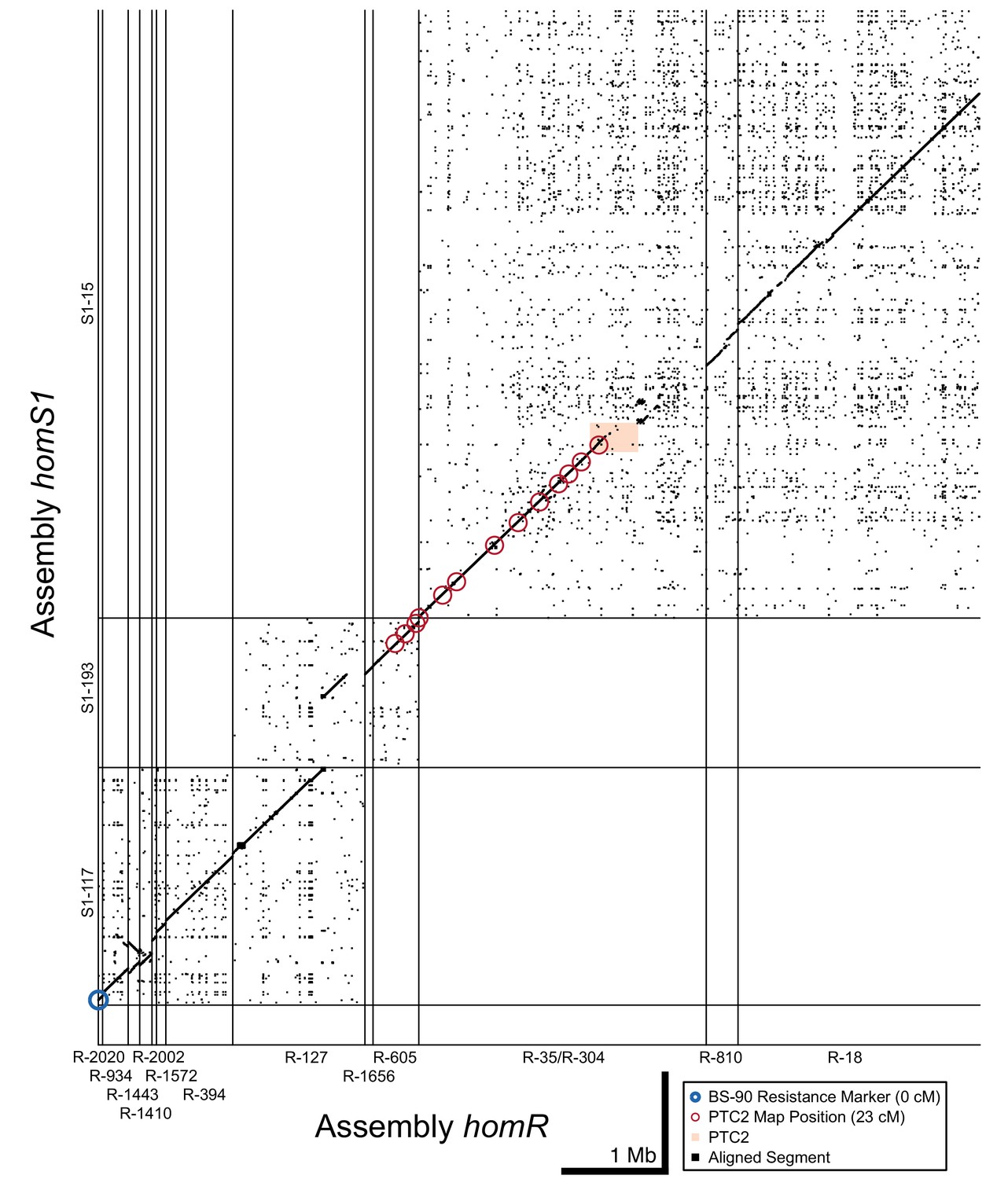
Dot plot of tiled contigs from assemblies homR and homS1 at the start of LG XII.
These contigs bridge the span between PTC2 and the BS-90 RAPD marker (blue circle), suggesting a physical distance of 5 Mb between them. While there is no overlapping sequence between S1-193/R-605 and S1-15/R-35, aligned linkage map markers which all share the same linkage map position as PTC2 (red circles) suggest these blocks are very proximate and occur within a region of relatively low recombination. homR contig R-127 overlaps homS1 contigs S1-193 and S1-117, confirming that they are adjacent and harbor a region of relatively high recombination between PTC2 and the BS-90 RAPD marker 23 cM away.
Figure 2 with 4 supplements
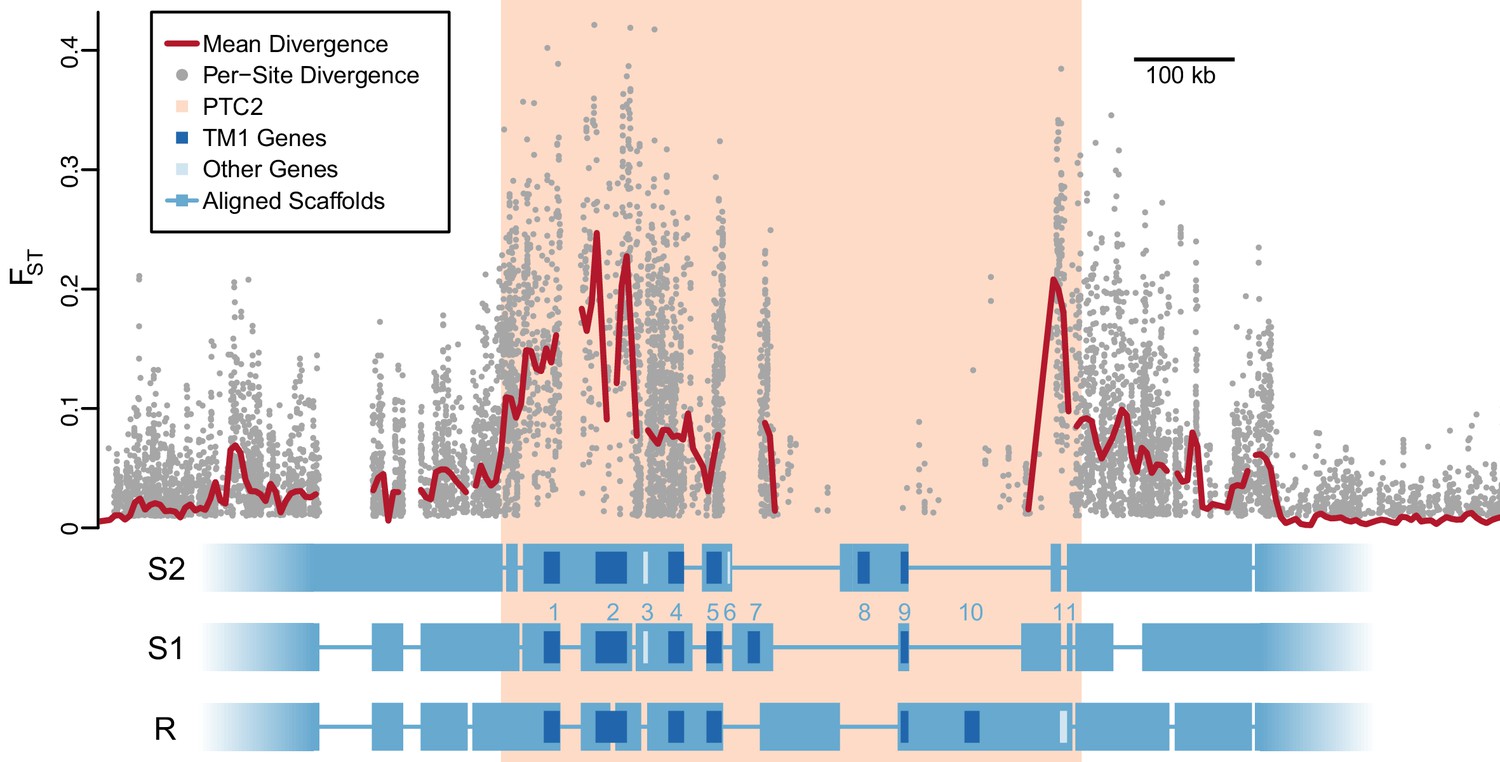
Divergent haplotypes of Polymorphic Transmembrane Complex 2 (PTC2).
As in Figure 1A, genetic divergence (FST) between infected and uninfected snail pools is shown, here both for individual variants (grey circles, only FST ≥0.01 shown) and as mean values for sliding windows of 10 kb (red line). FST is undefined for regions present on only one haplotype. PTC2 (peach) is defined as the 450 kb region containing all windows for which mean FST exceeds 0.1. Within PTC2, the three haplotypes (R, S1, and S2) are aligned with multi-kilobase indels and genes indicated. Assemblies are comprehensive and alignment gaps represent annotated indels, not missing data. PTC2 is characterized by extensive sequence divergence, including large indels containing entire genes (numbered), and is enriched for single-pass transmembrane (TM1) loci (dark blue).
Figure 2—figure supplement 1
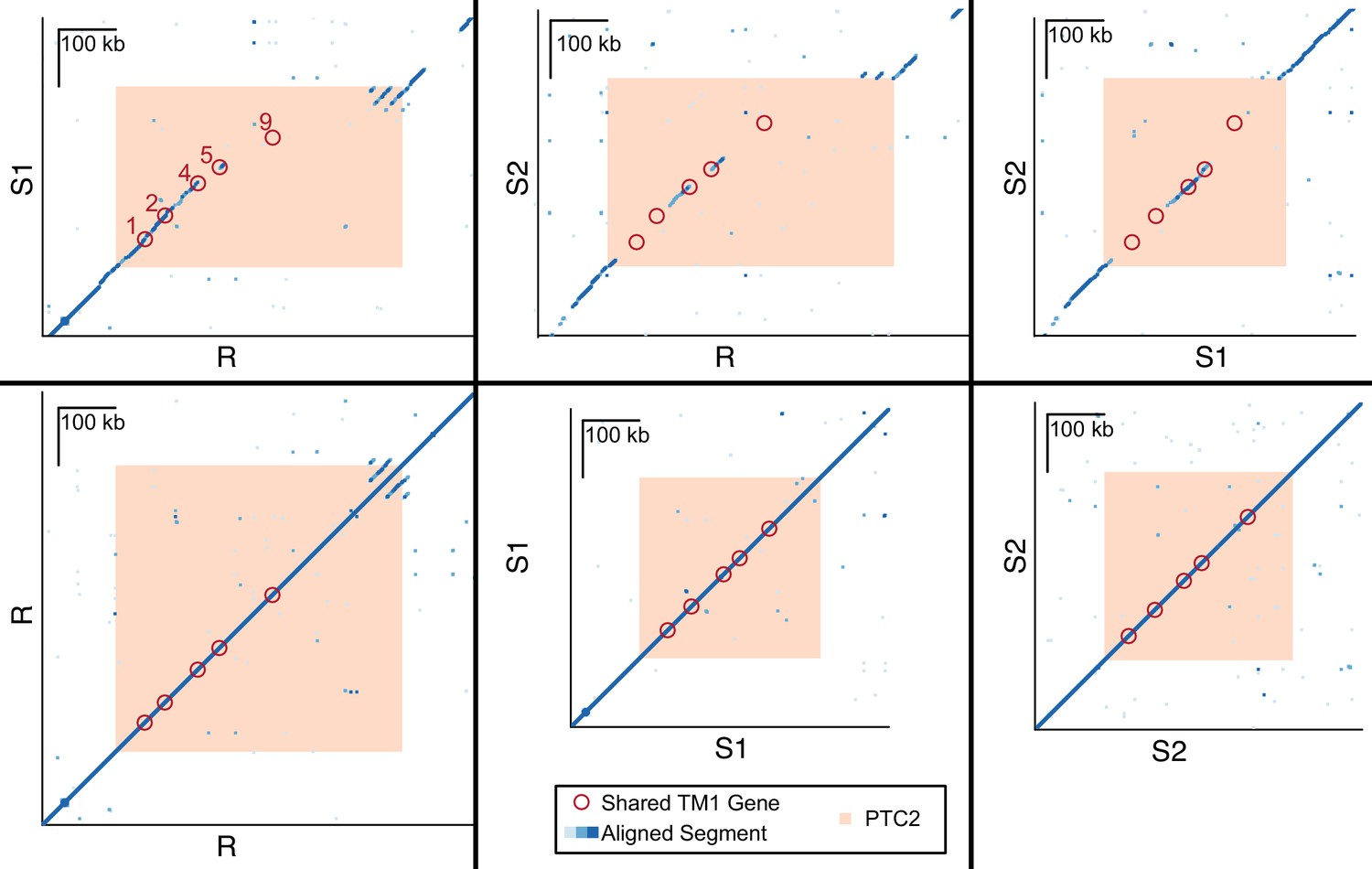
Dot plots for haplotypes R and S1, R and S2, S1 and S2, and each haplotype against itself.
Each dot represents a 600 bp segment, and color indicates sequence similarity (light blue = 50–90% similarity; medium blue = 90–97.5% similarity; dark blue = 97.5–100% similarity). Of the five TM1 genes that are shared across all three haplotypes (red circles, numbered) within PTC2 (peach), several appear to exist in regions of no homology (no dots), because the alleles share such low DNA sequence similarity.
Figure 2—figure supplement 2
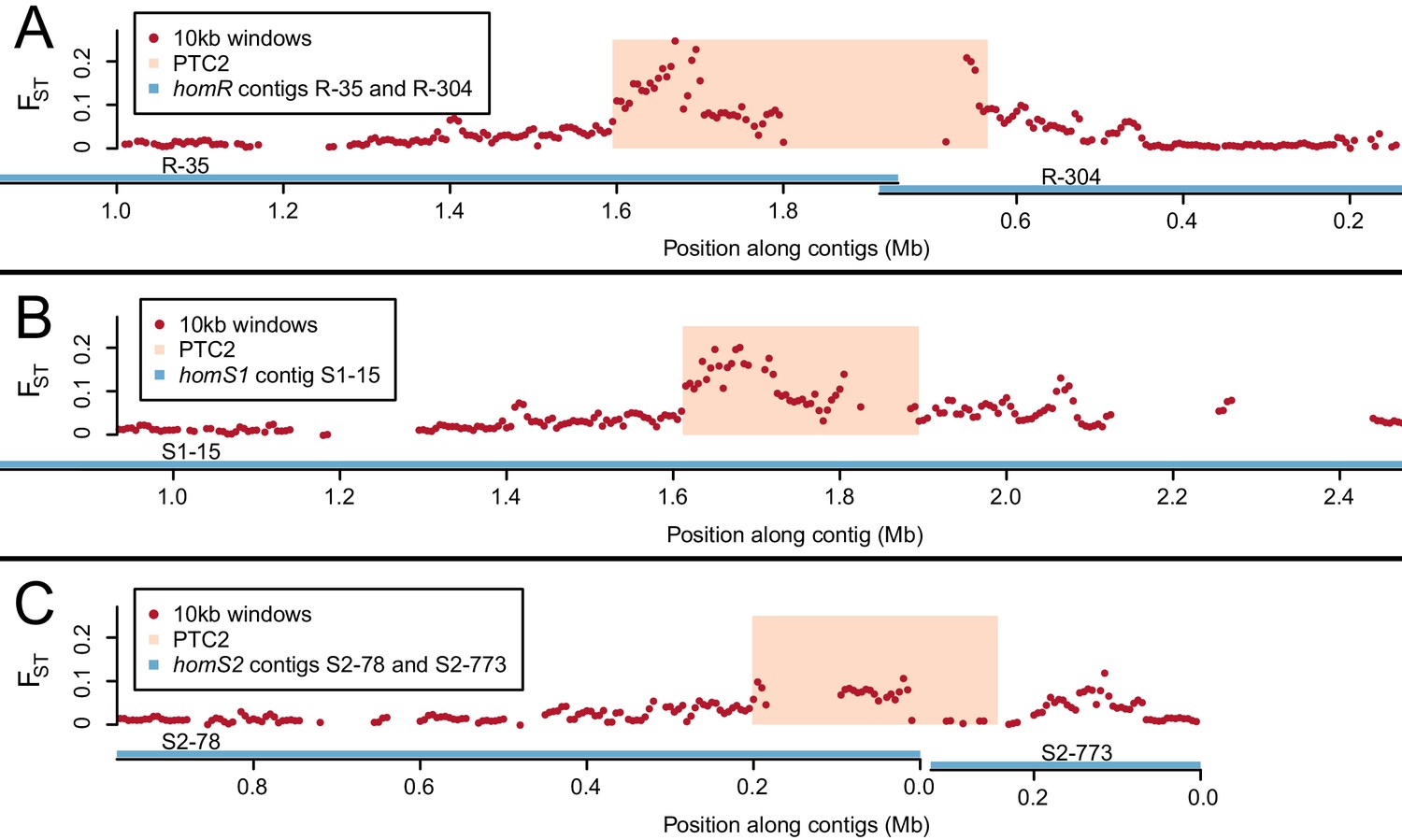
Genetic divergence (FST) between infected and uninfected snail pools in 10 kb windows (red dots), in the vicinity of PTC2 (peach box), for reads aligned to all three genome assemblies (contigs = blue lines).
PTC2 is narrower in S1 and S2 relative to R due to different sets of indels (Figure 2). The FST signal varies by genome assembly because the high sequence divergence only allows a subset of reads to align, which differs for each assembly. The signal is weakest for S2, possibly because the greatest phenotypic contrast can be inferred when (highest-susceptibility) S1 and (lowest-susceptibility) R reads align to the same location, which is least likely to occur with this assembly. We focus on the region of strongest association, defined here as PTC2 encompassing all windows over FST = 0.1 on any homR contig and all windows over FST = 0.15 on any contig across all three assemblies. Other peaks also occur on either side of PTC2, reflecting either decaying linkage disequilibrium with this locus or possibly additional minor causal loci. (A) homR (B) homS1 (C) homS2.
Figure 2—figure supplement 3
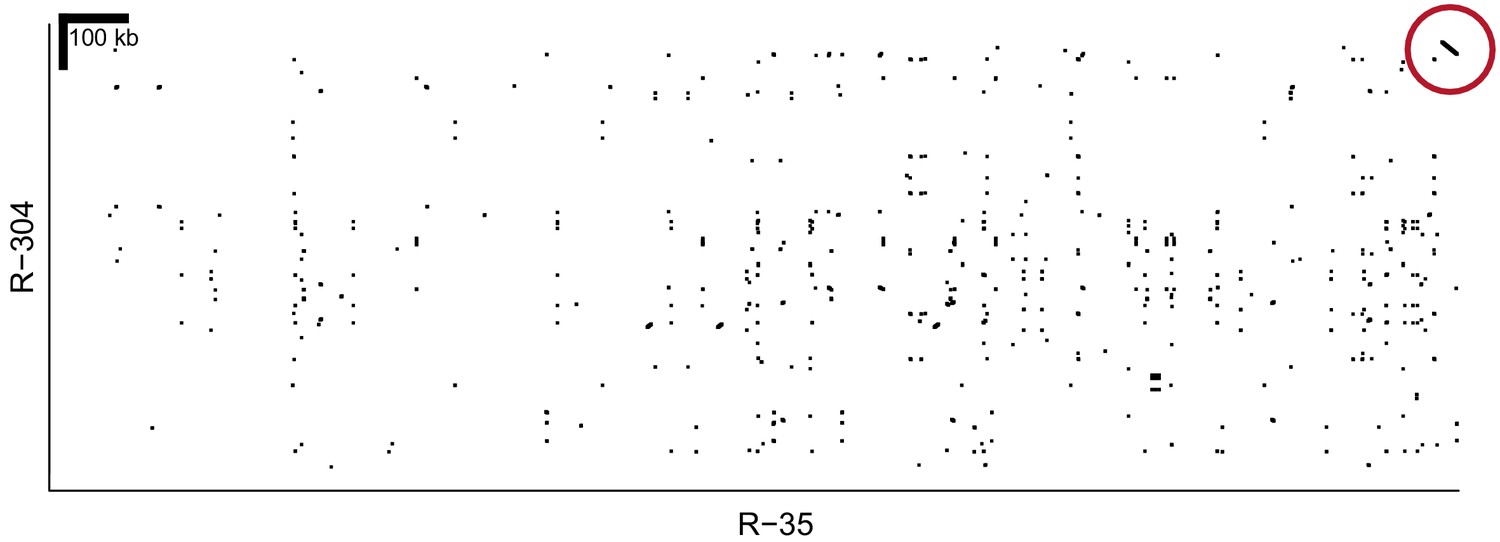
Alignment of homR contigs R-35 and R-304.
The final 22 kb of both contigs show 99.9% similarity in reverse orientation (red circle). These two contigs align to different portions of reference genome BglaB1 contig 13, and both share the association with phenotype (Figure 2), All of this evidence indicates that they are adjacent and partially overlapping. Thus, R-35 and R-304 have been combined into a single haplotype throughout our analysis (e.g. Figure 2).
Figure 2—figure supplement 4
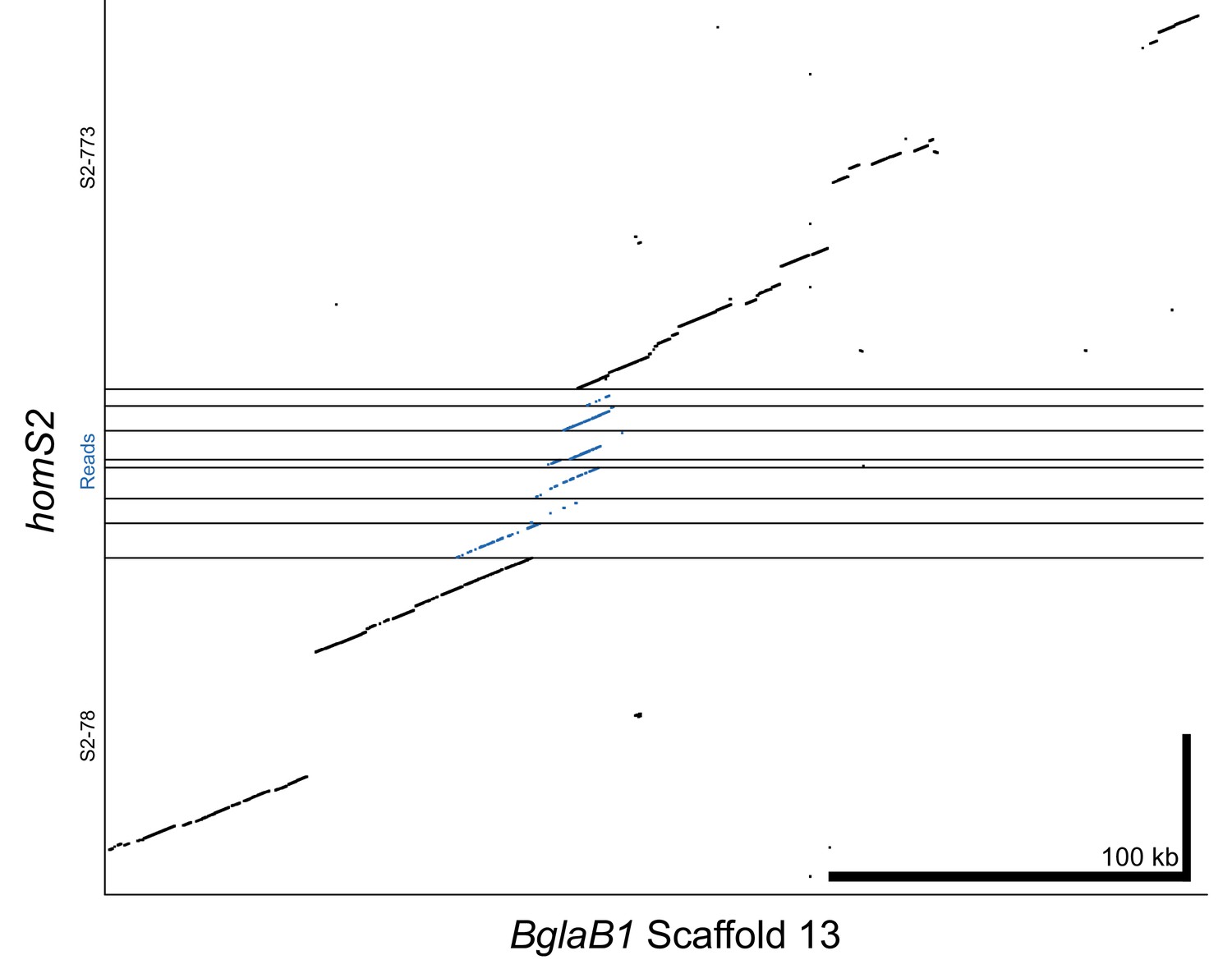
Alignment of homS2 contigs and raw PacBio reads to reference genome BglaB1 contig 13.
Raw reads span the 12.6 kb gap between contigs, which is further confirmed by the high similarity between homS2 and BglaB1. Thus, S2-78 and S2-773 have been combined into a single haplotype throughout our analysis (e.g. Figure 2).
Figure 3 with 2 supplements
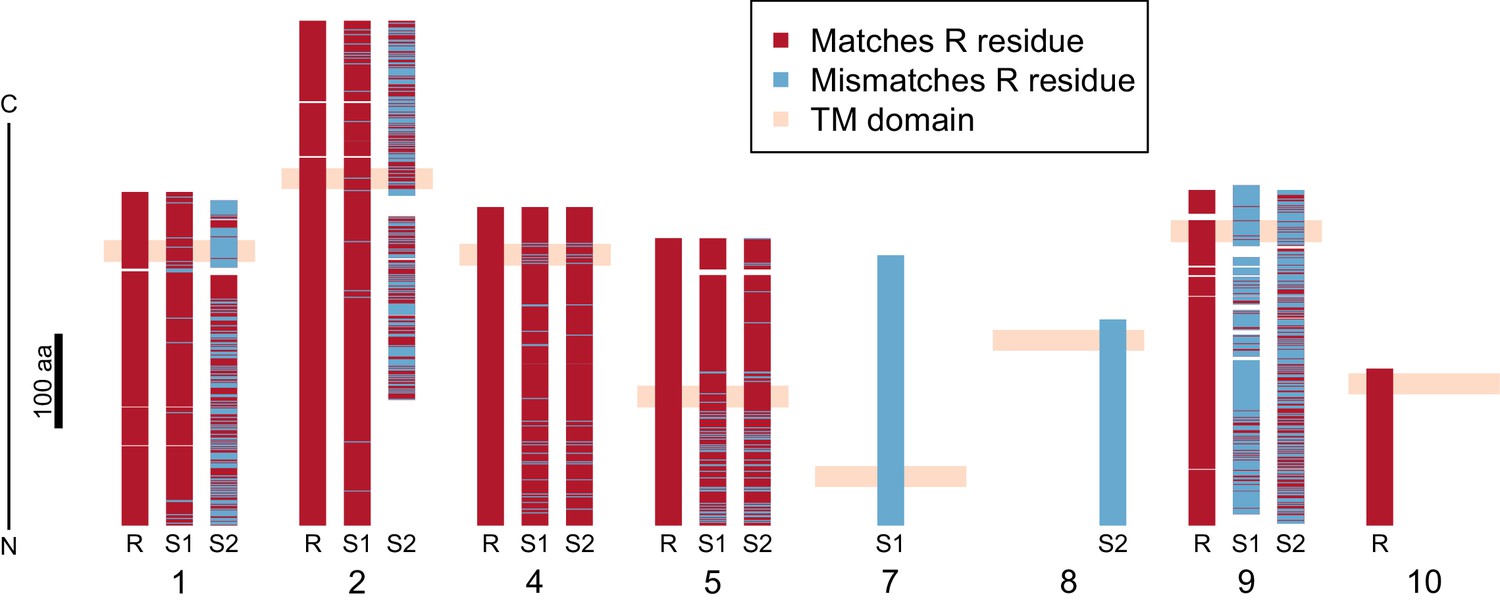
Divergence of PTC2 single-pass transmembrane (TM1) genes.
For each of eight TM1 genes (numbered as in Figure 2; Supplementary file 1D), the protein product is shown, including the transmembrane domain (peach). Orthologous alleles are aligned. Amino acid residues that differ from the R allele are shown in blue. Uncorrected protein sequence divergence between orthologous alleles ranges from 3% to 85%. Three genes (7, 8, and 10) are present only on a single haplotype.
Figure 3—figure supplement 1
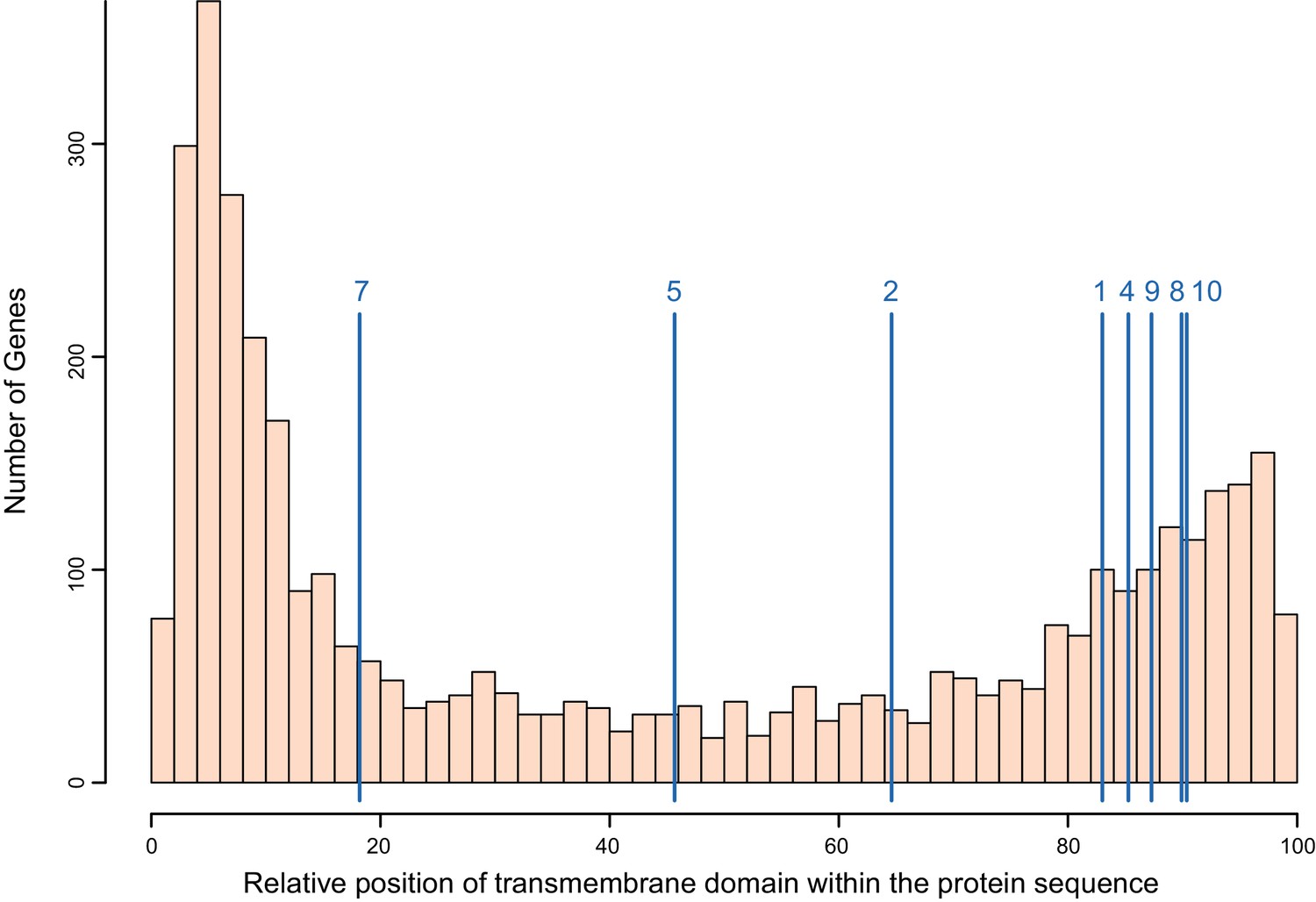
Distribution of the transmembrane domain within the protein sequence for all single-pass transmembrane (TM1) genes in B. glabrata, with positions of the PTC2 genes indicated (blue lines, averaged across alleles; numbered as in Figure 3).
All protein lengths have been normalized to 100. We assessed the distribution of PTC2 genes relative to all TM1 genes with 10,000 replicate trials sampling eight random TM1 proteins each time. The TM1 domains of PTC2 genes are more likely to occur closer to the C-terminus than for other TM1 genes (p<0.05) and tend to be closer to the middle of the protein but not significantly so (p>0.05). This feature is shared with the GRC proteins (Tennessen et al., 2015b) and also Toll-like receptors, suggesting a similar cellular role.
Figure 3—figure supplement 2
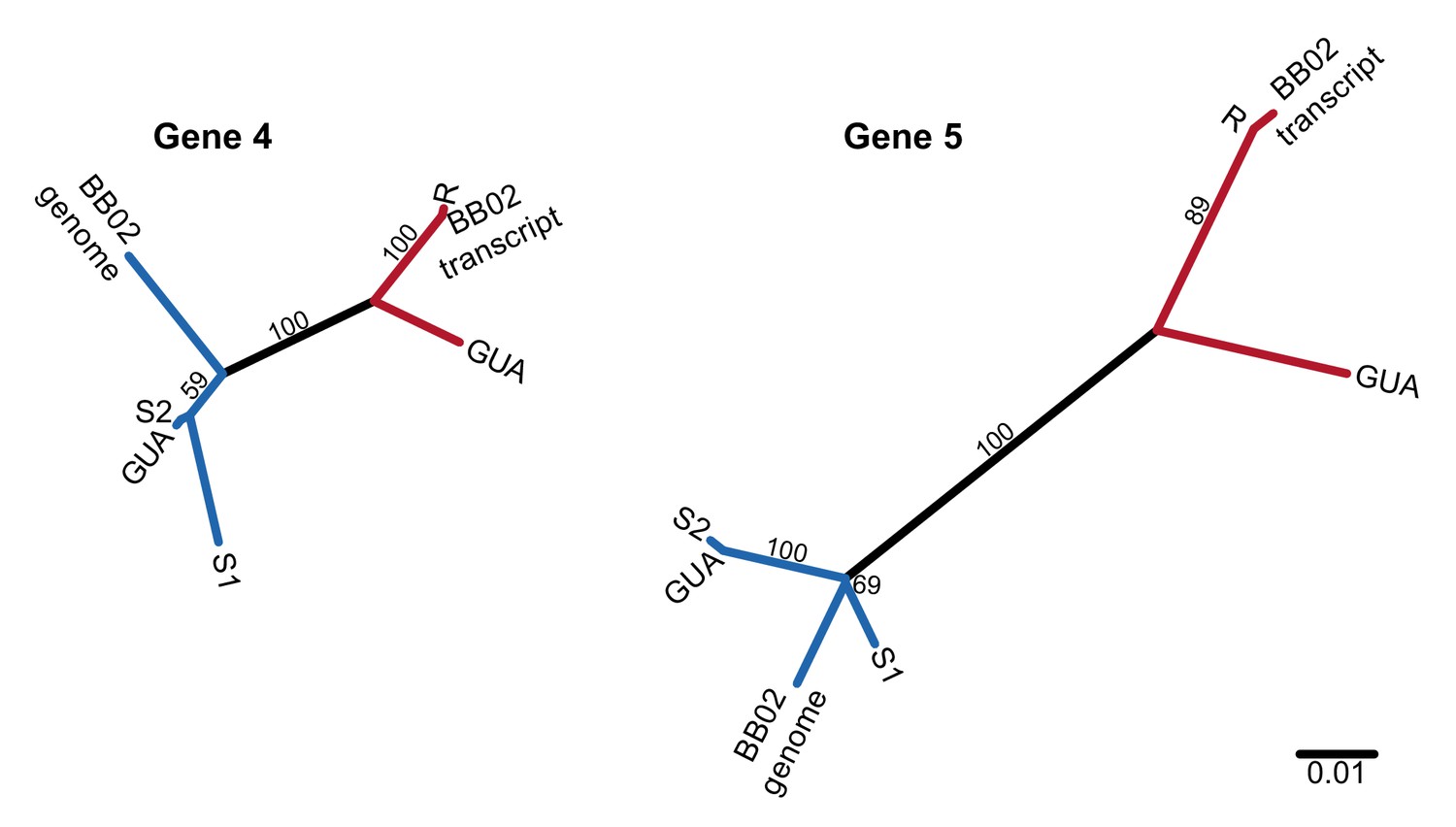
Unrooted nucleotide phylogenies of the coding sequence of the two most conserved PTC2 TM1 genes, including RNA sequence from Caribbean population GUA and genomic and RNA sequence from Brazilian strain BB02.
Numbers on branches indicate bootstrap support. All three populations (13–16-R1, GUA, and BB02) harbor both R-like (red branches) and S-like (blue branches) sequences, which presumably are also allelic in GUA and BB02. Thus, this polymorphism segregates within natural populations throughout the range of B. glabrata and is not an artifact of the admixed history of 13–16-R1, consistent with long-term balancing selection predating the diaspora of B.glabrata across the neotropics.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Biological sample (Biomphalaria glabrata) | 13–16-R1 | PMID:5050093; PMID:7299581 | 5050093:NIH-MH-cc-13-16-1; 7299581:13–16-R1 | Oregon State University population established by C. Bayne |
| Strain, strain background (Schistosoma mansoni) | PR-1 | PMID:5050093; PMID:7299581 | 5050093:NIH-Sm-PR1; 7299581:PR-1 | |
| Sequence-based reagent | PB35_1696 k_F | This paper | PCR primer | GGTTCTCGCTTTTTATTGGCTTTTG |
| Sequence- based reagent | PB35_1696 k_R | This paper | PCR primer | TTAGACGCACCCAAGGATCTC |
| Sequence-based reagent | VB13_859 k_Fb | This paper | PCR primer | ACAAATGGGGCAGTTACACTGTTTAC |
| Sequence-based reagent | VB13_859 k_Rb | This paper | PCR primer | AGCGAAATGTGAGATTGGTTATGTTG |
| Sequence-based reagent | VB13_868 k_Fb | This paper | PCR primer | TCTTTTCACTAAAGCCGCACAAGTT |
| Sequence-based reagent | VB13_868 k_Rb | This paper | PCR primer | CCTACGTTCTCAATATCAACGGGAA |
| Software, algorithm | SimulatePools.pl | https://github.com/jacobtennessen/GOPOPS/ | Perl script | power analysis for pooled sequencing |
| Software, algorithm | MakeFreqTableFromPooledPileup.pl | https://github.com/jacobtennessen/GOPOPS/ | Perl script | estimates allele frequencies |
| Software, algorithm | FstFromJoinedFreqTablesWindow.pl | https://github.com/jacobtennessen/GOPOPS/ | Perl script | calculates mean FST per window |
| Software, algorithm | ChopFastaStaggered.pl | https://github.com/jacobtennessen/MiSCVARS/ | Perl script | subdivides sequence data |
| Software, algorithm | AssessBlatChopped.pl | https://github.com/jacobtennessen/MiSCVARS/ | Perl script | summarizes sequence matches |
Additional files
-
Source code 1
R script for analyzing individual snail genotypes and phenotypes with logistic regression.
- https://cdn.elifesciences.org/articles/59395/elife-59395-code1-v2.r.zip
-
Supplementary file 1
Supplementary tables.
- https://cdn.elifesciences.org/articles/59395/elife-59395-supp1-v2.docx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/59395/elife-59395-transrepform-v2.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Clusters of polymorphic transmembrane genes control resistance to schistosomes in snail vectors
eLife 9:e59395.
https://doi.org/10.7554/eLife.59395




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}