Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry
- Meyer Cancer Center, Department of Medicine, Weill Cornell Medical College, United States
- Department of Microbiology, New York University - Langone Health, United States
- Department of Microbiology and Immunology, Cornell University, United States
- Laboratory of Retrovirology, The Rockefeller University, United States
- Division of Gastroenterology and Hepatology, Department of Medicine, Weill Cornell Medicine, United States
- Department of Physiology, Biophysics and Systems Biology, Weill Cornell Medicine, United States
Abstract
Coagulopathy is a significant aspect of morbidity in COVID-19 patients. The clotting cascade is propagated by a series of proteases, including factor Xa and thrombin. While certain host proteases, including TMPRSS2 and furin, are known to be important for cleavage activation of SARS-CoV-2 spike to promote viral entry in the respiratory tract, other proteases may also contribute. Using biochemical and cell-based assays, we demonstrate that factor Xa and thrombin can also directly cleave SARS-CoV-2 spike, enhancing infection at the stage of viral entry. Coagulation factors increased SARS-CoV-2 infection in human lung organoids. A drug-repurposing screen identified a subset of protease inhibitors that promiscuously inhibited spike cleavage by both transmembrane serine proteases and coagulation factors. The mechanism of the protease inhibitors nafamostat and camostat may extend beyond inhibition of TMPRSS2 to coagulation-induced spike cleavage. Anticoagulation is critical in the management of COVID-19, and early intervention could provide collateral benefit by suppressing SARS-CoV-2 viral entry. We propose a model of positive feedback whereby infection-induced hypercoagulation exacerbates SARS-CoV-2 infectivity.
Editor's evaluation
This study examines the potential role host proteases involved in coagulation may play in proteolytic processing of the SARS CoV-2 virus spike protein, which is required for viral entry. Serine protease inhibitors such as nafamostat and camostat may limit viral entry into host cells and also be useful to treat coagulopathy in patients with SARS CoV-2 infection, particularly if treatment is initiated early.
https://doi.org/10.7554/eLife.77444.sa0Introduction
SARS-CoV-2 emerged into the human population in late 2019 and has evolved into a devastating global health crisis. Despite the recent success of vaccines (Baden et al., 2021; Polack et al., 2020), the limited world-wide vaccine distribution (Kwok et al., 2021; Lin et al., 2020; Nhamo et al., 2021; So and Woo, 2020), the emergence of viral variants (Wang et al., 2021; Weisblum et al., 2020), and the repeated SARS-like zoonotic outbreaks over the last 20 years (Cheng et al., 2007; Ge et al., 2013; Menachery et al., 2015) underscore the urgent need to develop antivirals for coronavirus (Pan et al., 2021).
In addition to attachment to specific receptors on target cells, coronaviruses require proteolytic processing of the spike protein by host cell proteases to facilitate membrane fusion and viral entry (Glowacka et al., 2011; Jaimes et al., 2020c; Walls et al., 2020). In SARS-CoV-2, host cell proteases act on two sites residing at the S1/S2 subunit boundary and at the S2’ region proximal to the fusion peptide (Belouzard et al., 2009; Hoffmann et al., 2020a; Jaimes et al., 2020b; Millet and Whittaker, 2014). S1/S2 site cleavage opens up the spike trimer and exposes the S2’ site, which must be cleaved to allow for the release of the conserved fusion peptide (Benton et al., 2020; Madu et al., 2009). While the prevailing model suggests that furin cleaves the S1/S2 site and TMPRSS2 cleaves the S2’ site (Bestle et al., 2020), it remains unclear to what extent other proteases may be involved (Hoffmann et al., 2021; Ou et al., 2020).
TMPRSS2 is an important host cell factor in proteolytic activation across multiple coronaviruses (Hoffmann et al., 2020a; Jaimes et al., 2019). TMPRSS2 knockout or inhibition reduces infection in mouse models of SARS and MERS (Iwata-Yoshikawa et al., 2019; Zhou et al., 2015). More recently, TMPRSS2 has been highlighted as a drug target for SARS-CoV-2 (Hoffmann et al., 2020a; Hoffmann et al., 2020b).
Furin activity is not essential to produce infectious particles (Tang et al., 2021a) and furin is not necessary for cell fusion (Papa et al., 2021), but deletion of the S1/S2 site attenuates SARS-CoV-2 in vivo (Johnson et al., 2021). Proteolytic activation of envelope proteins presumably coordinates target cell engagement and envelope conformational changes leading to fusion. Furin cleavage during viral biogenesis, before release of viral particles, may render SARS-CoV-2 spike less stable in solution and reduce the likelihood to reach and interact with target cells (Amanat et al., 2021; Berger and Schaffitzel, 2020; Wrobel et al., 2020). Although the S1/S2 site is often referred to as the ‘furin site’ (Johnson et al., 2021), the full spectrum of proteases that catalyze biologically relevant activity in the lung remains incompletely defined.
Proteases also orchestrate the coagulation pathway, via a series of zymogens that are each activated by a chain reaction of proteolytic processing. Coagulopathy and thromboembolic events have emerged as a key component of COVID-19 pathogenesis (McGonagle et al., 2020). Comorbidities associated with severe COVID-19 are also linked to dysregulated blood clotting (Zhou et al., 2020). Patients with a history of stroke prior to infection have nearly twice the risk of in-hospital mortality (Qin et al., 2020). Upon hospital admission, elevated D-dimer levels (an indicator of fibrinolysis and coagulopathy) and low platelet counts (an indicator of consumptive coagulopathy) are predictive biomarkers of severe disease and lethality in COVID-19 patients (Zhou et al., 2020). Systemic activity of clotting factors V, VIII, and X are elevated in severe COVID-19 disease (Stefely et al., 2020). While early phase disease is typically restricted to a local pulmonary hypercoagulable state, late-stage disease may be accompanied by systemic DIC, stroke, and cardio-embolism (Huang et al., 2020; Kipshidze et al., 2020; McGonagle et al., 2020; Tsivgoulis et al., 2020). Ischemic stroke occurred in approximately 1% of hospitalized COVID-19 patients, and strikingly, a fraction of them experienced stroke even prior to onset of respiratory symptoms (Yaghi et al., 2020).
In a drug-repurposing effort to target TMPRSS2, we observed that multiple direct-acting anticoagulants have anti-TMPRSS2 off-target effects. We subsequently investigated overlap in substrate specificity between TMPRSS2, factor Xa, and thrombin. Circulating proteases involved in blood clotting can cleave and activate SARS-CoV-2 spike, enhancing infection, specifically at the stage of viral entry. We propose that the serine protease inhibitor nafamostat may incorporate a combined mechanism in the treatment of COVID-19 through inhibition of TMPRSS2 and coagulation factors.
Results
Serine protease inhibitors suppress SARS-CoV-2 entry via inhibition of TMPRSS2
We developed a fluorescence resonance energy transfer (FRET)-based protease enzymatic assay based on peptides containing either the S1/S2 or S2’ cleavage sites of SARS-CoV-2 spike (Figure 1A, Figure 1—figure supplement 1). Upon cleavage, the liberated 5-FAM emits fluorescent signal proportional to the quantity of product (Figure 1—figure supplement 1A). Camostat and nafamostat resulted in strong inhibition of TMPRSS2 (Figure 1B), as expected (Hoffmann et al., 2020a; Hoffmann et al., 2020b). We also identified that otamixaban and the active form of dabigatran (but not its prodrug dabigatran etexilate) inhibit TMPRSS2 enzymatic activity in vitro (Figure 1B–C).
Figure 1 with 4 supplements see all
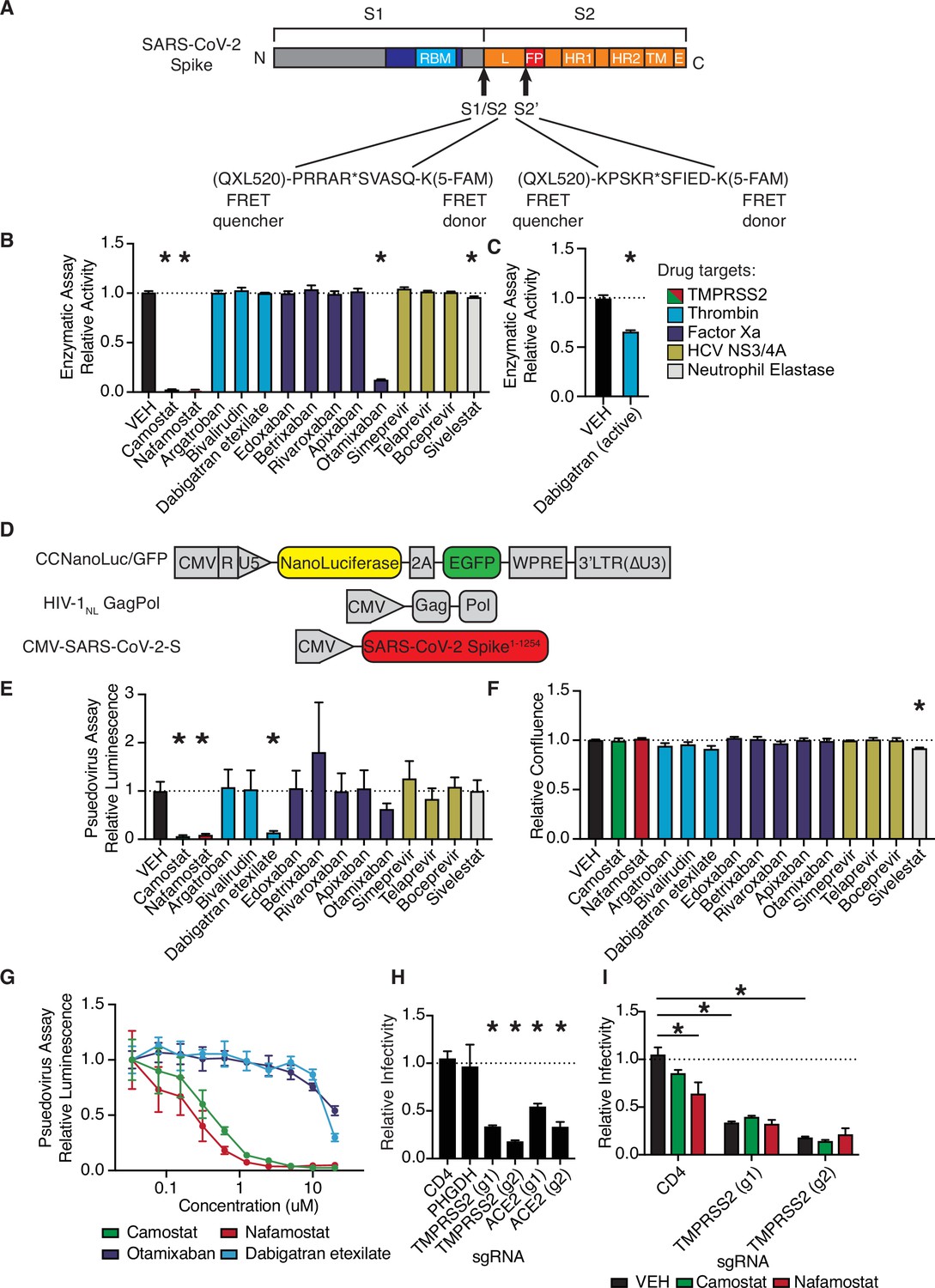
Anticoagulant serine protease inhibitors suppress SARS-CoV-2 entry via inhibition of TMPRSS2.
(A) Peptides derived from two known cleavage sites of SARS-CoV-2 spike were designed with C-terminal fluorophore 5-FAM and N-terminal fluorescence resonance energy transfer (FRET) quencher QXL-520. (B) FDA-approved and investigational serine protease inhibitors were screened by enzymatic assay to inhibit TMPRSS2 cleavage of SARS-CoV-2 S1/S2 peptide substrate. Relative change in fluorescence with respect to DMSO vehicle is shown. Colors indicate the described target of the drugs screened. All drugs screened at 10 µM final concentration. (C) Active form of dabigatran in enzymatic assay for TMPRSS2 inhibition. Relative fluorescence with respect to its corresponding 0.1 N HCl vehicle is shown. (D) Schematic of constructs used to generate SARS-CoV-2 spike-pseudotyped/HIV-1-based particles. (E) Calu3 cells were treated with 10 µM of the indicated drugs for 24 hr prior to infection with HIV-1NL/SARS-CoV-2 pseudovirus. Media was changed at 24 hr post infection and pseudoviral entry was measured by nanoluciferase luminescent signal at 40 hr. (F) Calu3 cells treated with 10 µM of the indicated drugs were monitored for confluence by Incucyte for 40 hr. (G) Pseudoviral entry was measured by nanoluciferase luminescent signal in Calu3 cells treated various concentrations of the indicated drugs for 4 hr prior to infection with SARS-CoV-2 pseudovirus. (H) Caco2 cells were infected with lenti-Cas9-blast and U6-sgRNA-EFS-puro-P2A-tRFP and selected. Neutral controls targeting CD4 (not endogenously expressed) or PHGDH intron 1, two sgRNAs each targeting different regions of ACE2 and TMPRSS2 were included. Cells were subsequently infected with HIV-1NL/SARS-CoV-2 pseudovirus. (I) Caco2 cells co-expressing Cas9 and sgRNAs targeting CD4 (not expressed) or TMPRSS2 were treated with 10 µM camostat, nafamostat, or DMSO vehicle. N = 3, *p < 0.05, two-tailed t-test. Data represented as mean ± SEM.
-
Figure 1—source data 1
Data and summary statistics for enzymatic and pseudovirus assays.
- https://cdn.elifesciences.org/articles/77444/elife-77444-fig1-data1-v2.xlsx
To explore these candidates in a cell-based functional assay of spike protein, SARS-CoV-2 S-pseudotyped HIV-1 particles were employed to infect human lung Calu3 cells (Figure 1D; Schmidt et al., 2020). Consistent with the TMPRSS2 enzymatic assay, camostat, nafamaostat, otamixaban, and dabigatran etexilate suppressed pseudoviral entry, as indicated by nanoluciferase luminescent signal (Figure 1E). No effects on relative cell growth were observed at the same timepoint in Calu3 (Figure 1F) or A549 cells (data not shown), confirming that reduced luminescent signal was not due to cytotoxicity. A dose-response experiment with select protease inhibitors revealed a submicromolar IC50 for camostat and nafamostat and IC50s in the 10–20 µM range for otamixaban and dabigatran in Calu3 cells (Figure 1G).
Using A549 cells with or without ectopic ACE2 expression, we confirmed that HIV-1NL/SARS-CoV-2 pseudovirus infection is dependent on ACE2, while infection with HIV-1NL pseudotyped instead with VSV G envelope protein is not ACE2 dependent (Figure 1—figure supplement 2). Caco2 cells, which endogenously express ACE2 and TMPRSS2, show greater susceptibility to SARS-CoV-2 S-pseudotyped HIV-1NL, but equivalent susceptibility to VSV G-pseudotyped HIV-1NL, when compared to A549/ACE2 cells (Figure 1—figure supplement 2).
To further validate these results in an alternative pseudovirus system, we used recombinant G protein-deficient vesicular stomatitis virus (rVSV∆G) pseudotyped with SARS-CoV-2-S (Figure 1—figure supplement 3A), yielding pseudovirus dependent on spike for cell entry (Figure 1—figure supplement 3B). The antiviral effects of the four candidate protease inhibitors were confirmed in the VSV pseudovirus system in multiple cell lines, and response was associated with TMPRSS2 expression (Figure 1—figure supplement 3C-F).
We aimed to determine whether the effects of camostat and nafamostat are indeed TMPRSS2-dependent, or if other unidentified cellular proteases can compensate for TMPRSS2 suppression. To do so, we knocked out TMPRSS2 in ACE2+ TMPRSS2+ Caco2 cells and found that susceptibility to pseudovirus was significantly reduced, comparable to knockout of ACE2 (Figure 1H, Figure 1—figure supplement 4). Furthermore, both camostat and nafamostat reduce pseudovirus entry into control Caco2 cells harboring control sgRNA, but this trend was lost in cells with two independent TMPRSS2-targeting sgRNAs (Figure 1I). These data indicate that, in the absence of exogenous proteases, TMPRSS2 is a critical host enzyme activating SARS-CoV-2 spike in TMPRSS2+ cells and that TMPRSS2 is the primary target of camostat and nafamostat in these conditions.
Coagulation factors directly cleave SARS-CoV-2 spike
Anticoagulants are highly represented among FDA-approved drugs that target proteases, and among the hits from the screen described above. The off-target effects of anticoagulants on TMPRSS2 imply that these small molecules can interact with the active sites of TMPRSS2 in a similar manner to coagulation factors. This led us to hypothesize that coagulation factors may interact with some of the same substrates as TMPRSS2, including SARS-CoV-2 spike.
To determine the properties of enzyme-substrate relationships, TMPRSS2, factor Xa, and thrombin cleavage of S1/S2 and S2’ peptides were determined over a range of 0–160 µM peptide substrate (Figure 2A–C, Table 1). Surprisingly, factor Xa catalyzed S1/S2 cleavage more than an order of magnitude faster than TMPRSS2 (Figure 2A–B and D), although factor Xa showed lower affinity (higher Km) compared with TMPRSS2 to the S1/S2 peptide (Figure 2A–B and E). Thrombin has greater affinity (lower Km) than TMPRSS2 and factor Xa for the S1/S2 substrate (Figure 2E) and performs S1/S2 cleavage at a rate intermediate between TMPRSS2 and factor Xa (Figure 2D). Unlike factor Xa, thrombin cleaves the S2’ peptide with greater activity than TMPRSS2 (Figure 2D–F).
Figure 2 with 1 supplement see all
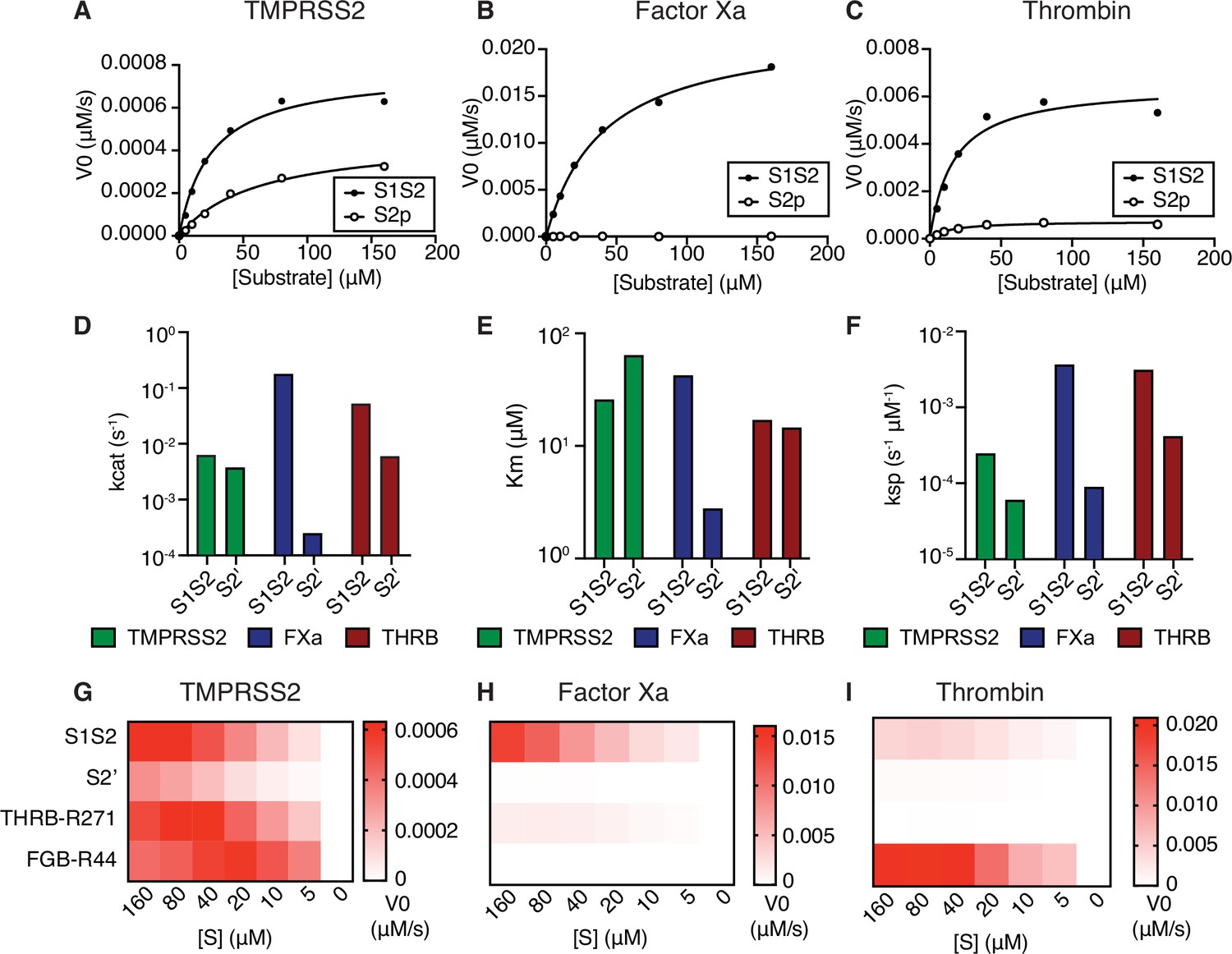
Coagulation factors directly cleave SARS-CoV-2 spike.
Initial velocities for the cleavage of SARS-CoV-2 spike S1/S2 and S2’ peptide substrates by (A) TMPRSS2, (B) factor Xa, and (C) thrombin were measured over a range of 0–160 µM substrate. From initial velocity values, enzyme kinetic constants (D) turnover rate Kcat (s–1), (E) affinity constant Km, and (F) specificity constant (Kcat/Km) were obtained for the indicated enzymes with S1/S2 and S2’ peptides. (G–I) Heatmaps depict the initial velocity V0 of cleavage of the indicated peptide substrates and concentrations by (G) TMPRSS2, (H) factor Xa, and (I) thrombin.
-
Figure 2—source data 1
Data and summary statistics for enzymatic assays.
- https://cdn.elifesciences.org/articles/77444/elife-77444-fig2-data1-v2.xlsx
Table 1
Kinetics of SARS-CoV-2 spike peptide substrate cleavage.
Kinetic constants obtained from initial velocity studies with varying concentrations of SARS-CoV-2 spike S1/S2 and S2’ peptide substrates. Each estimate is based on seven different concentrations of substrate in 1:2 serial dilution (0–160 µM).
| Enzyme | Substrate | Vmax (µM/s) | Kcat (s–1) | Km (µM) | Ksp (s–1 µM–1) |
|---|---|---|---|---|---|
| TMPRSS2 | S1/S2 | 7.71E-04 | 6.17E-03 | 24.71 | 2.50E-04 |
| TMPRSS2 | S2' | 4.60E-04 | 3.68E-03 | 60.94 | 6.04E-05 |
| Factor Xa | S1/S2 | 2.24E-02 | 1.79E-01 | 40.35 | 4.43E-03 |
| Factor Xa | S2' | 3.04E-05 | 2.43E-04 | 2.711 | 8.97E-05 |
| Thrombin | S1/S2 | 6.50E-03 | 5.20E-02 | 16.34 | 3.18E-03 |
| Thrombin | S2' | 7.34E-04 | 5.87E-03 | 13.98 | 4.20E-04 |
We next compared the ability of coagulation factors to cleave SARS-CoV-2 S to their ability to cleave their known substrates. During the physiological process of clotting, factor Xa cleaves prothrombin at R271, which ultimately becomes the activated form α-thrombin (Wood et al., 2011). Thrombin, in turn, cleaves multiple sites of fibrinogen, including the beta chain (FGB) at R44, in a critical step toward aggregation and polymerization of high molecular weight fibrin clots. Fluorogenic peptides corresponding to THRBR271 and FGBR44 were synthesized and assayed with TMPRSS2, factor Xa, and thrombin. TMPRSS2 exhibited relatively broad activity to cleave this collection of substrates (Figure 2G). As expected, factor Xa showed strong selectivity for THRBR271 over FGBR44, while thrombin showed the opposite substrate preference (Figure 2H–I). Remarkably, factor Xa showed ~9-fold greater maximum initial reaction velocity (Vmax) in cleaving the spike S1/S2 peptide compared to cleaving a peptide corresponding to its known substrate, THRBR271 (Figure 2H). The Vmax for thrombin cleavage of the spike S1/S2 peptide was within ~4.5-fold of the Vmax for the benchmark FGBR44 peptide (Figure 2I), indicating that thrombin might also cleave this site when activated during coagulation.
We next assessed the effect of substituting amino acids adjacent to the cleavage site of the S1/S2 peptide on proteolytic cleavage by these proteases. An arginine preceding the cleavage site (P1 position) is a common feature of substrates of many serine proteases. Substitution of the P1 arginine in the S1/S2 substrate with alanine (S1S2-P1A) resulted in a 4-fold reduction in TMPRSS2 cleavage and abolished nearly all cleavage by factor Xa and thrombin (Figure 2—figure supplement 1A-C). Substitutions in the P3 and P4 positions (S1S2-HPN) with features typical of a substrate of type II transmembrane serine proteases (TTSPs), a family which includes TMPRSS2 and hepsin (Damalanka et al., 2019), did not change TMPRSS2 cleavage and greatly reduced factor Xa and thrombin cleavage (Figure 2—figure supplement 1A-C). Although the substrate specificity of TTSPs and coagulation factors are not universally similar, the cleavage sites of SARS-CoV-2 are specifically cleaved by TMPRSS2, factor Xa, and thrombin.
To consider whether the context of protease activity may influence substrate specificity, we repeated the enzymatic assay in the presence of phospholipids. The addition of phospholipid vesicles did not change factor Xa activity or substrate preference in this assay (Figure 2—figure supplement 1E). To ensure the quality of our phospholipid vesicle preparation, we added 0–100 µM PC/PS to a dilute Russell’s viper venom time (dRVVT) clotting assay, where PC/PS drives a significant, concentration-dependent acceleration of clotting of normal pooled human plasma (Figure 2—figure supplement 1F). In summary, the coagulation serine proteases factor Xa and thrombin exhibit proteolytic activity against SARS-CoV-2 spike peptide substrates.
Factor Xa and thrombin facilitate SARS-CoV-2 spike-mediated entry
We next investigated whether coagulation factors could cleave trimeric spike in its native 3D conformation, and whether this activity potentiated spike function in viral entry into cells. To do so, we used replication-defective SARS-CoV-2 spike-pseudotyped VSV or HIV-1 virus (Schmidt et al., 2020). In the VSV pseudovirus system, addition of purified factor Xa or thrombin to the media significantly increased infection in Calu3 cells 16 hr post infection as determined by either quantification of NeonGreen (Figure 3A, Figure 3—figure supplement 1A) or nanoluciferase activity (Figure 3B).
Figure 3 with 2 supplements see all
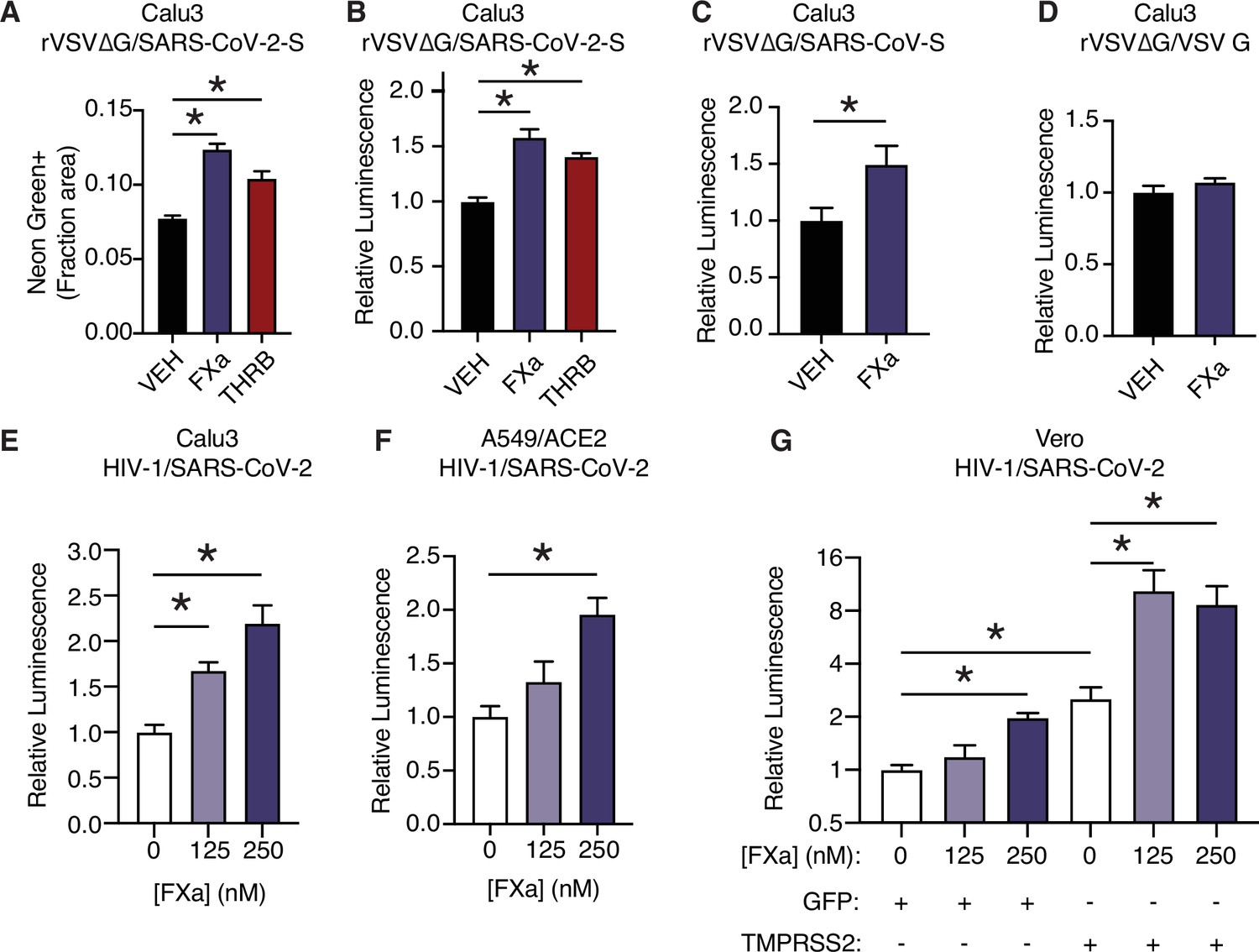
Factor Xa and thrombin facilitate SARS-CoV-2 spike-mediated entry.
(A) Calu3 cells were infected with rVSV∆G/SARS-CoV-2 pseudovirus with concomitant treatment with vehicle, 250 nM factor Xa, or 250 nM thrombin. Quantification of the ratio of green fluorescent area to total confluence (4 fields/replicate well, 4 wells/condition). (B) Nanoluciferase luminescent signal was measured following infection with rVSV∆G/SARS-CoV-2 pseudovirus and the addition of either vehicle, factor Xa, or thrombin. The effect of factor Xa on rVSV∆G complemented with either (C) SARS-CoV spike or (D) VSV-G was measured by luminescent signal. Luminescent signal was measured following HIV-1NL/SARS-CoV-2 pseudovirus infection and concomitant treatment with 125–250 nM factor Xa in (E) Calu3 cells, (F) A549/ACE2, and (G) Vero cells following transduction with lentiviral vectors to express GFP or TMPRSS2. Following selection, cells were infected with HIV-1NL/SARS-CoV-2 pseudovirus and concomitantly treated with 125–250 nM factor Xa. Subsequently, nanoluciferase luminescent signal was determined and plotted relative to vehicle-treated control. *p < 0.05, two-tailed t-test. Data represented as mean ± SEM.
-
Figure 3—source data 1
Data and summary statistics for pseudovirus assays with exogenous proteases.
- https://cdn.elifesciences.org/articles/77444/elife-77444-fig3-data1-v2.xlsx
While factor X and prothrombin levels are variable between individuals in healthy populations (Brummel-Ziedins et al., 2012), the concentration of proteases used in the pseudovirus assay (125–250 nM) are comparable to reference ranges of factor X (Brummel-Ziedins et al., 2012; Tormoen et al., 2013; Williams and Marks, 1994) and prothrombin (Baugh et al., 1998; Brummel-Ziedins et al., 2012; Tormoen et al., 2013). Similar concentrations of active purified proteases were required to normalize in vitro clotting times, where purified factor Xa was used to correct dRVVT of factor X-deficient human plasma (Figure 3A, Figure 3—figure supplement 2A) and purified thrombin was used to correct the prothrombin time of prothrombin-deficient human plasma (Figure 3—figure supplement 2B).
SARS-CoV-2 contains a notable insertion of basic residues at the S1/S2 boundary, distinguishing its sequence from many related betacoronaviruses (Jaimes et al., 2020a). Entry of rVSV∆G was increased when complemented with spike protein from SARS-CoV of the 2002 outbreak (Figure 3C), but not when complemented instead with VSV G (Figure 3D). This indicates that factor Xa spike cleavage could be relevant across multiple coronaviruses, but is not generally associated with VSV entry.
We further validated that factor Xa activated spike-mediated entry the HIV-1-based pseudovirus system (Schmidt et al., 2020). Consistent with the results above, addition of purified factor Xa to the media at the time of infection enhanced entry of HIV-1-based SARS-CoV-2 pseudovirus in Calu3 cells (Figure 3E). Thrombin did not appear to enhance spike-mediated entry by HIV-1/SARS-CoV-2 pseudovirus, unlike rVSV∆G/SARS-CoV-2 pseudovirus (Figure 3—figure supplement 1B-E).
We investigated the functional interplay of TMPRSS2 expression and the effect of exogenous activated coagulation factors. TMPRSS2 is expressed in Calu3 cells and contributes to coronavirus entry (Hoffmann et al., 2020a), whereas A549/ACE2 and Vero cells lack endogenous TMPRSS2 expression. Factor Xa induced a significant dose-dependent effect on pseudovirus entry in both Calu3 and A549/ACE2 (Figure 3E–F). Furthermore, an isogenic pair of Vero cells was generated by expressing TMPRSS2 or GFP control. Pseudovirus infection of both VeroGFP and VeroTMPRSS2 cells were significantly increased by factor Xa, indicating that factor Xa enhancement of infection is not dependent on TMPRSS2 (Figure 3G). This is consistent with the model that FXa cuts the S1/S2 site and TMPRSS2 has functionally important activity at the S2’ site.
Nafamostat broadly inhibits cleavage of spike peptides by both transmembrane serine proteases and coagulation factors
Given that multiple proteases could contribute to SARS-CoV-2 spike cleavage activation, a drug that could inhibit both transmembrane serine proteases and coagulation factors would be potentially valuable as a dual action anticoagulant/antiviral in COVID-19. Accordingly, we next explored the candidate set of inhibitors for cross-reactivity against a broader set of proteases that could facilitate viral entry. Like TMPRSS2, human airway trypsin-like protease (HAT), encoded by TMPRSS11D, is a member of the TTSP family of proteases and could activate SARS-CoV-2 S (Hoffmann et al., 2021). HAT exhibited sensitivity to camostat and nafamostat, similar to TMPRSS2 (Figure 4A–B). Compared to TMPRSS2, HAT was more sensitive to dabigatran and less sensitive to otamixaban (Figure 4A–B). Factor Xa activity against the S1/S2 peptide was most sensitive to otamixaban and moderately sensitive to nafamostat and dabigatran (Figure 4C). Thrombin activity was sensitive to camostat, nafamostat, and dabigatran, and moderately sensitive to otamixaban (Figure 4D).
Figure 4 with 2 supplements see all
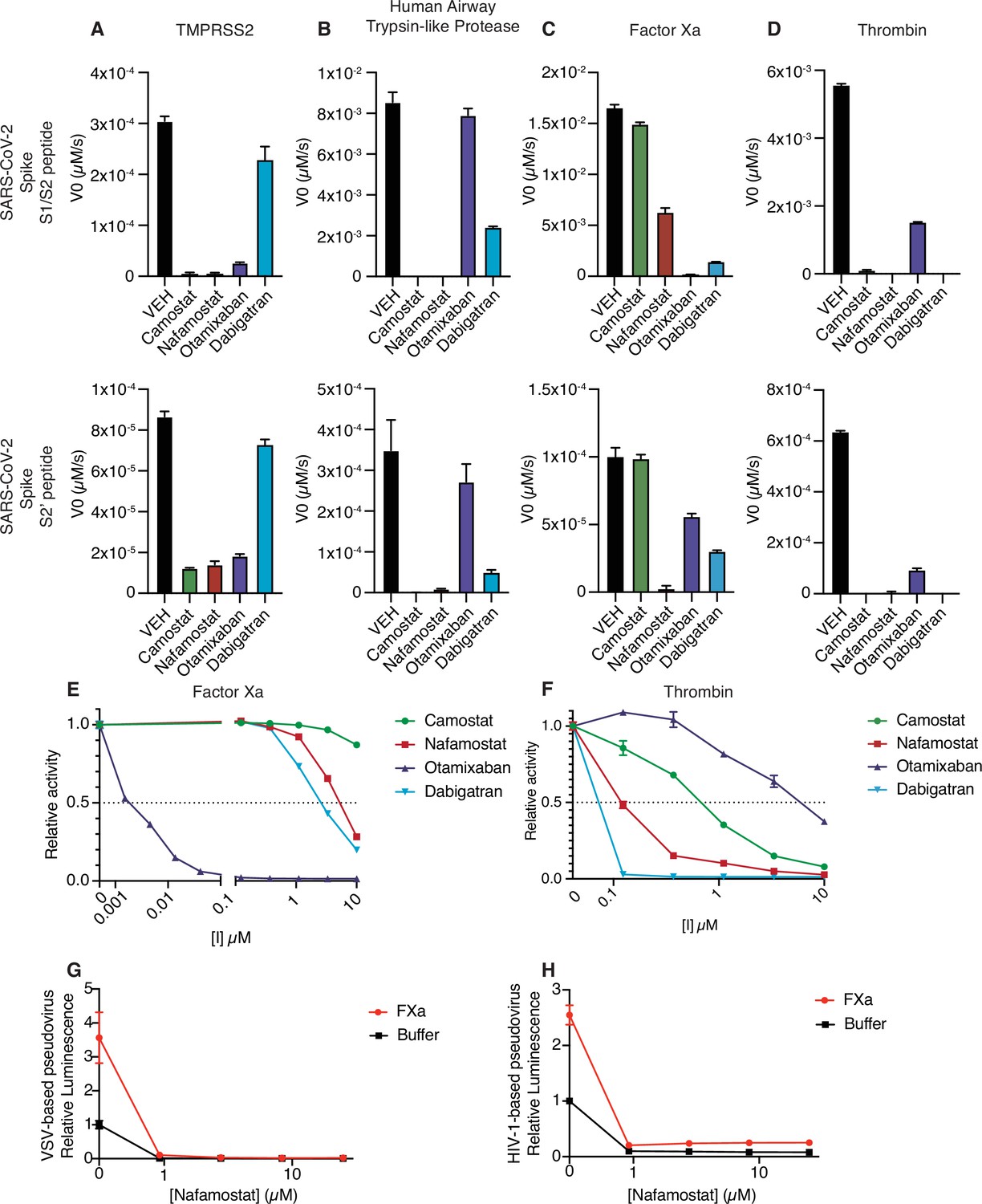
Nafamostat broadly inhibits cleavage of spike peptides by both coagulation factors and transmembrane serine proteases.
Initial velocities for the cleavage of 10 µM SARS-CoV-2 spike S1/S2 (top) and S2’ (bottom) peptide substrates by (A) TMPRSS2, (B) TMPRSS11D/human airway trypsin-like protease (C) factor Xa, and (D) thrombin were measured in the presence of DMSO vehicle, or 10 µM camostat, nafamostat, otamixaban, or dabigatran. The relative activity of (E) factor Xa and (F) thrombin were determined over a range of 0–10 µM of the indicated drugs. Calu3 cells were treated with a range of concentrations of nafamostat with or without addition of 250 nM exogenous factor Xa and infected with (G) rVSV∆G/SARS-CoV-2 pseudovirus or (H) HIV-1NL/SARS-CoV-2 pseudovirus and infectivity was measured by luminescence. N = 3, data represented as mean ± SEM.
-
Figure 4—source data 1
Data and summary statistics for enzymatic assays to determine the effects of protease inhibitors on host proteases.
- https://cdn.elifesciences.org/articles/77444/elife-77444-fig4-data1-v2.xlsx
We performed a dose-response curve of the panel of inhibitors on factor Xa and thrombin S1/S2 cleavage. Otamixaban, a designed factor Xa inhibitor, demonstrated an IC50 at the nanomolar level to factor Xa, while nafamostat and dabigatran demonstrated IC50s in the micromolar range (Figure 4E). Camostat did not potently inhibit factor Xa spike cleavage. Dabigatran, a designed thrombin inhibitor, as well as nafamostat and camostat demonstrated a submicromolar IC50 for thrombin-dependent spike cleavage (Figure 4F). Otamixaban inhibited thrombin spike cleavage in the micromolar range.
Furin showed high activity against the S1/S2 peptide, but not against the S2’ peptide, and was not sensitive to any of the candidate inhibitors (Figure 4—figure supplement 1A-B). While it has been suggested that TMPRSS4 or neutrophil elastase may also cleave SARS-CoV-2 spike, we detected minimal activity against either S1/S2 or S2’ peptide substrates in our enzymatic assay (<1% of furin cleavage of S1/S2) (Figure 4—figure supplement 1A-B).
In the pseudovirus assay, nafamostat effectively suppresses SARS-CoV-2 S-mediated entry with or without the addition of exogenous factor Xa, using either the VSV-based (Figure 4G) or HIV-1-based (Figure 4H) SARS-CoV-2 pseudovirus. To clarify the pleiotropic nature of nafamostat, which inhibits TMPRSS2 and factor Xa, we compared the effect of apixaban, which inhibits factor Xa but not TMPRSS2. Apixaban rescued the effect of exogenous FXa back to the baseline level of infection, but did not affect pseudovirus infection in the absence of exogenous protease (Figure 4—figure supplement 2). Direct oral anticoagulants (DOACs) have the potential to block clotting factor-mediated enhancement of viral entry, but TMPRSS2-mediated cleavage activation would remain unaffected by treatment with DOACs in common usage in North America and Europe. Taken together, nafamostat appears to be a versatile inhibitor of spike activation by a variety of TTSPs and coagulation factors. The multitarget mechanism of nafamostat distinguishes its potential as an antiviral/anticoagulant from currently FDA-approved DOACs.
Factor Xa and thrombin increase SARS-CoV-2 infection in lung organoids
To explore the effect of coagulation factors in a more physiologically relevant setting, we exposed human pluripotent stem cell-derived lung organoids (hPSC-LOs) to SARS-CoV-2 infection under BSL-3 conditions with or without addition of exogenous protease. Stepwise, directed differentiation of human pluripotent stem cells generates lung organoids that form three-dimensional cellular structures and recapitulate functional and molecular characteristics of the lung (Chen et al., 2019; Chen et al., 2017; Han et al., 2021; Huang et al., 2014; Mou et al., 2012). hPSC-LOs express alveolar type II cell markers and were previously shown to be permissive to SARS-CoV-2 infection and replication (Han et al., 2021).
Mature lung organoids were infected with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.1 (Figure 5A–B) or 0.01 (Figure 5C–D) and viral replication was measured by quantitative real-time PCR (qRT-PCR) for SARS-CoV-2 nucleocapsid (N) (Figure 5A and C) and SARS-CoV-2 envelope (E) (Figure 5B and D). The addition of either factor Xa or thrombin increased the levels of viral RNA following infection (Figure 5A–D). This effect was accentuated at 48 hr post infection with respect to 24 hr post infection, and with lower initiating MOI. Factor Xa and thrombin increase SARS-CoV-2 infection in the context of multicycle viral replication in human lung organoids.
Figure 5
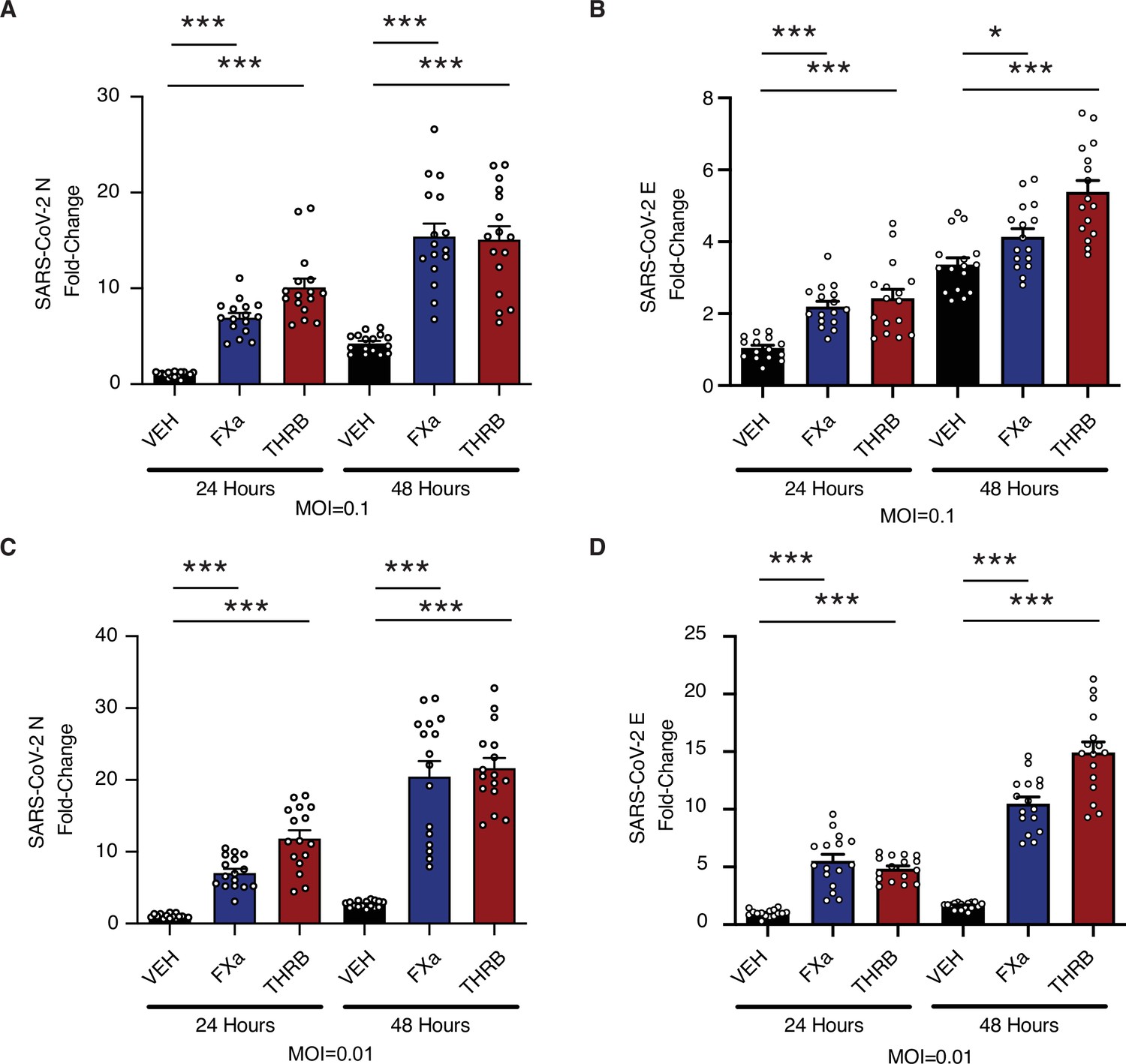
Factor Xa and thrombin increase SARS-CoV-2 infection in lung organoids.
Human pluripotent stem cell (hPSC)-derived lung organoids were infected with SARS-CoV-2. Upon infection, organoids were treated with 170 nM of purified factor Xa or thrombin. (A) Relative level of SARS-CoV-2-N RNA following infection at multiplicity of infection (MOI) = 0.1. (B) Relative level of SARS-CoV-2-E RNA following infection at MOI = 0.1. (C) Relative level of SARS-CoV-2-N RNA following infection at MOI = 0.01. (D) Relative level of SARS-CoV-2-E RNA following infection at MOI = 0.01. N = 16, *p = 0.0144, ***p < 0.0001, two-tailed t-test. Data represented as mean ± SEM.
-
Figure 5—source data 1
Data and summary statistics for infection assays with exogenous proteases.
- https://cdn.elifesciences.org/articles/77444/elife-77444-fig5-data1-v2.xlsx
Discussion
Coagulation factors cleave the SARS-CoV-2 spike protein
Using a FRET-based enzymatic assay, two platforms of pseudovirus assays, and SARS-CoV-2 infection experiments in lung organoids, we demonstrate that coagulation proteases factor Xa and thrombin cleave SARS-CoV-2 spike protein. Coagulation-induced cleavage enhances spike activation and increases viral entry into target cells, potentially instigating a positive feedback loop with infection-induced coagulation. Nafamostat, among currently available drugs, is best suited as a multi-purpose inhibitor against spike cleavage by TTSPs and coagulation factors. These data have numerous implications at the intersection of virology and coagulation.
Viral envelope protein activation by non-target cell proteases
Hijacking of host transmembrane, endosomal, and ER proteases to activate viral envelope proteins has been described in influenza A, human metapneumovirus, HIV, and Sendai virus (Kido et al., 1996; Straus et al., 2020). In the present study, we find an instance where the virus can be primed not by proteases expressed by the target cell, but by host organism proteases derived from the microenvironment of the target cell. Prior studies have described cleavage activation of SARS-CoV by elastase and plasmin, illustrating that microenvironmental host proteases can indeed play an important role in coronavirus spike priming (Belouzard et al., 2010; Kam et al., 2009; Matsuyama et al., 2005). Generally, the scope by which circulating proteases, such as coagulation factors, or proteases expressed by immune cells interact with viral envelope proteins during infection has not been comprehensively explored.
Relatively few studies have examined the interaction of factor Xa or thrombin and viral proteins, and each relies on target cells as the source of coagulation factors. Our results are consistent with a prior study that concluded that factor Xa cleaves and activates SARS-CoV spike (Du et al., 2007). Traditionally, influenza vaccines rely on viral propagation in chicken eggs, where hemagglutinin cleavability by factor Xa is a determinant of the efficiency of strain-specific propagation of influenza A virus in ovo (Gotoh et al., 1990; Straus and Whittaker, 2017). Hepatitis E virus ORF1 polyprotein is processed intracellularly by thrombin and factor Xa in the cytoplasm of hepatocytes, which are the primary cell type responsible for generating and secreting coagulation factors (Kanade et al., 2018).
Activation of coagulation has the potential to exacerbate SARS-CoV-2 infectivity in both TMPRSS2+ and TMPRSS2- host cells. Reliance on extracellular proteolytic activity could expand the field of susceptible cell types and regions of the airway. Extrapulmonary infection has been described, particularly in small intestinal enterocytes (Xiao et al., 2020; Zang et al., 2020) and, in some cases, the central nervous system (Song et al., 2021). It warrants investigation whether hypercoagulation is linked to extrapulmonary infection.
Evolutionary perspective on viral-host interaction
Proteolytic cleavage of the spike forms a barrier to zoonotic crossover independent of receptor binding (Menachery et al., 2020). Hemostasis is of central importance in mammals and represents a major vulnerability of mammals to predators and pathogens, either through hyperactivation of coagulation or uncontrolled bleeding. The dysregulation of hemostasis is a convergent mechanism of toxins of snakes, bees, and bats (Ma et al., 2013; Markland and Swenson, 2013; Prado et al., 2010) and a driver of virulence in Ebola and dengue virus infection (Geisbert et al., 2003; Rathore et al., 2019). Acute lung injury from viral cytopathic effects, the induction of a COVID-19-associated cytokine storm, complement activation, and anti-phospholipid autoantibodies have all been suggested to instigate the coagulation cascade (Merrill et al., 2020; Zuo et al., 2020). Furthermore, one model posits that COVID-19 coagulopathy is platelet-driven and an Arg-Gly-Asp (RGD) motif on SARS-CoV-2 spike directly interacts with GPIIb/GPIIIa integrins on the surface of platelets (Cox, 2021), consistent with in silico predictions of integrin binding (Mészáros et al., 2021). Perhaps, SARS-CoV-2 has undergone selection to exploit an environment locally enriched in coagulation proteases for enhanced entry. As infection spreads, more clotting is induced, instigating a positive feedback loop to promote entry into additional host cells.
Clinical relevance of potential antiviral activity of anticoagulants
Effective anticoagulation is a critical area of investigation to improve outcomes in coronavirus infection. Vitamin K antagonists, including heparin, are commonly used for preventing venous thromboembolism in COVID-19, although no strong evidence yet supports any specific anticoagulant (Cuker et al., 2021). Three large randomized clinical trials to determine the benefit of therapeutic intensity vs. prophylactic intensity heparin in critically ill COVID-19 patients were suspended at interim analysis for futility (NCT02735707, NCT04505774, and NCT04372589). There has been interest in the use of direct-acting oral anticoagulants (DOACs) to manage COVID-19-related coagulopathy, but optimal protocols for managing coagulopathy in COVID-19 patients have not yet been developed (Capell et al., 2021; Lopes et al., 2021). The most prominent risk of anticoagulants is bleeding, and notably DOACs, as well as nafamostat, have a reduced risk of intracranial hemorrhage and other bleeding events compared to vitamin K antagonists (Chen et al., 2020; Hellenbart et al., 2017; Makino et al., 2016).
In our studies, anticoagulant serine protease inhibitors, otamixaban and dabigatran, exhibited off-target activity against TMPRSS2 and other TTSPs, but likely require concentrations higher than those safely reached in vivo (Paccaly et al., 2006; Stangier and Clemens, 2009). On the other hand, our data suggest that nafamostat and camostat may offer three distinct therapeutic mechanisms against SARS-CoV-2 infection; these compounds have the potential to block spike cleavage mediated by TMPRSS2 and other TTSPs, to block spike cleavage by coagulation factors, and to serve as an anticoagulant. It is also plausible that nafamostat, like related protease inhibitor pentamidine, could also interfere with platelet GPIIb/IIIa, platelet aggregation, and thrombus formation (Cox, 2021; Cox et al., 1996). Nafamostat (Fujii and Hitomi, 1981; Keck et al., 2001; Takeda et al., 1996) and Camostat (Ramsey et al., 2019) have been in clinical use in Asia for many years for the treatment of pancreatitis. Nafamostat has also been used as an anticoagulant during hemodialysis (Akizawa et al., 1993) and extracorporeal membrane oxygenation (Park et al., 2015), and to manage disseminated intravascular coagulopathy (Kobayashi et al., 2001). As of this writing, there have been eight clinical trials initiated (reported on https://www.clinicaltrials.gov/) to investigate the use of nafamostat in COVID-19, while 24 active clinical trials of camostat for COVID-19 were identified.
Inhibition of coagulation factor-induced spike cleavage may contribute to the molecular mechanism of these agents, if treatment is given sufficiently early. Many COVID-19-associated complications leading to hospitalization occur as immune hyperactivation waxes and peak viral titer wanes (Griffin et al., 2021). To take advantage of the potential antiviral effect of anticoagulants, early intervention in an outpatient setting may be beneficial.
Limitations
The experiments of this study, like prior studies using similar techniques, have some limitations. Protease enzymatic assays on peptide substrates allow for detailed biochemical characterization of a specific site, but peptide substrates may not have the equivalent three-dimensional conformation or post-translational modifications of the full-length protein produced in appropriate cells. For instance, SARS-CoV-2 S is extensively glycosylated (Watanabe et al., 2020). The possibility of additional spike cleavage sites and potential pro- and anti-viral consequence of proteases acting on cell surface proteins including ACE2 cannot be excluded. The amount, density, and accessibility of spike protein could be different between pseudovirus assays and wild type (WT) SARS-CoV-2 infection. However, antibody neutralization is highly correlated between authentic virus and corresponding pseudotyped viruses, suggesting similar conformation (Schmidt et al., 2020). We confirmed our results using WT SARS-CoV-2 infection, which alone does not easily allow for precise definition of which stage of the viral replication cycle is being affected, but pseudovirus assays confirm a cell-entry mechanism. We have attempted to mitigate the risk of artifact by using multiple orthogonal platforms.
Conclusion
Collectively, our data provide rationale for the investigation of early intervention with judiciously selected anticoagulant treatment, which may have collateral benefit in limiting progressive spread of SARS-CoV-2 infection throughout the lung in infected individuals. Preparedness to mitigate future coronavirus outbreaks is critical to pursue through the understanding of coronavirus-host interactions.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Chemical compound, drug | Camostat | Selleck | Cat# S2874 | |
| Chemical compound, drug | Nafamostat | Selleck | Cat# S1386 | |
| Chemical compound, drug | Apixaban | Medchem Express | Cat# HY-50667 | |
| Chemical compound, drug | Betrixaban | Medchem Express | Cat# HY-10268 | |
| Chemical compound, drug | Bivalirudin (TFA) | Medchem Express | Cat# HY-15664 | |
| Chemical compound, drug | Boceprevir | Medchem Express | Cat# HY-10237 | |
| Chemical compound, drug | Dabigatran etexilate | Medchem Express | Cat# HY-10274 | |
| Chemical compound, drug | Edoxaban | Medchem Express | Cat# HY-10264 | |
| Chemical compound, drug | Otamixaban | Medchem Express | Cat# HY-70035 | |
| Chemical compound, drug | Rivaroxaban | Medchem Express | Cat# HY-50903 | |
| Chemical compound, drug | Simeprevir | Medchem Express | Cat# HY-10241 | |
| Chemical compound, drug | Sivelestat | Medchem Express | Cat# HY-17443 | |
| Chemical compound, drug | Telaprevir | Medchem Express | Cat# HY-10235 | |
| Chemical compound, drug | Dabigatran | Medchem Express | Cat# HY-10163 | |
| Peptide, recombinant protein | Thrombin | Millipore Sigma | Cat# 605195 | |
| Peptide, recombinant protein | Factor Xa | Millipore Sigma | Cat# 69036 | |
| Peptide, recombinant protein | TMPRSS2 | LSBio | Cat# LS-G57269 | |
| Peptide, recombinant protein | TMPRSS4 | Aviva System Biology | Cat# OPCA0240 | |
| Peptide, recombinant protein | Furin | Thermo Fisher Scientific | Cat# 1503SE010 | |
| Peptide, recombinant protein | Neutrophil elastase | Thermo Fisher Scientific | Cat# 9167SE020 | |
| Peptide, recombinant protein | S1/S2 | Anaspec | QXL520-PRRARSVASQ-K(5-FAM)-NH2 | |
| Peptide, recombinant protein | S2’ | Anaspec | QXL520-KPSKRSFIED-K(5-FAM)-NH2 | |
| Peptide, recombinant protein | THRB-R271 | Anaspec | QXL520-AIEGRTATSE-K(5-FAM)-NH2 | |
| Peptide, recombinant protein | FGB-R44 | Anaspec | QXL520-FFSARGHRPL-K(5-FAM)-NH2 | |
| Peptide, recombinant protein | S1/S2-P1A | Anaspec | QXL520-PRRAASVASQ-K(5-FAM)-NH2 | |
| Peptide, recombinant protein | S1/S2-HPN | Anaspec | QXL520-PSQARSVASQ-K(5-FAM)-NH2 | |
| Chemical compound, drug | Phosphatidylcholine | Avanti Polar Lipids | Cat# 850375C | 1,2-Dioleoyl-sn-glycero-3-phosphocholine |
| Chemical compound, drug | phosphatidylserine | Avanti Polar Lipids | Cat# 840035C | 1,2-Dioleoyl-sn-glycero-3-phospho-L-serine |
| Cell line (Homo sapiens) | Calu3 | ATCC | Cat# HTB-55; RRID:CVCL_0609 | |
| Cell line (Homo sapiens) | A549 | ATCC | Cat# CCL-185; RRID:CVCL_0023 | |
| Cell line (Homo sapiens) | Caco2 | ATCC | Cat# HTB-37; RRID:CVCL_0025 | |
| Cell line (Chlorocebus sabaeus) | Vero | Laboratory of Benjamin tenOever | RRID:CVCL_0059 | |
| Cell line (Homo sapiens) | HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 | |
| Recombinant DNA reagent | pEGPN | This paper | ||
| Recombinant DNA reagent | pEGPN-ACE2 | This paper | ||
| Recombinant DNA reagent | pEGPN-TMPRSS2 | This paper | ||
| Recombinant DNA reagent | Lenti-Cas9-blast | Addgene | Cat# 52962 | |
| Recombinant DNA reagent | ipUSEPR | Francisco Sanchez-Rivera & Scott Lowe | ||
| Recombinant DNA reagent | CMV-SARS-CoV-2-S | Schmidt et al., 2020 | ||
| Recombinant DNA reagent | CCNanoLuc/GFP | Schmidt et al., 2020 | ||
| Recombinant DNA reagent | HIV-1NL GagPol | Schmidt et al., 2020 | ||
| Commercial assay or kit | NEBuilder master mix | New England Biolabs | Cat# E2621 | |
| Chemical compound, drug | XtremeGene9 | Millipore Sigma | Cat# 6365787001 | |
| Chemical compound, drug | Polybrene | Santa Cruz Biotechnology | Cat# SC-134220 | |
| Chemical compound, drug | Lenti-X | Takara Bio | Cat# 631232 | |
| Chemical compound, drug | G418 | Sigma-Aldrich | Cat# # G8168 | |
| Chemical compound, drug | Blasticidin | Invivogen | Cat# ANT-BL-1 | |
| Chemical compound, drug | Puromycin | Thermo Fisher Scientific | Cat# A1113803 | |
| Commercial assay or kit | Cell Lysis Buffer | Promega | Cat# E1531 | |
| Commercial assay or kit | NanoGlo Luciferase Assay | Promega | Cat# N1130 | |
| Biological sample (Homo sapiens) | Normal human plasma | Pacific Hemostasis | Cat# 95059–698 | |
| Biological sample (Homo sapiens) | Factor X-deficient plasma | Haematologic Technologies | Cat# FX-ID | |
| Biological sample (Homo sapiens) | Prothrombin-deficient plasma | Haematologic Technologies | Cat# FII-ID | |
| Biological sample (Vipera russelli) | Russell’s Viper Venom | Sigma-Aldrich | Cat# V2501 | |
| Strain, strain background (Indiana vesiculovirus) | rVSVdG/NG-NanoLuc | Schmidt et al., 2020 | ||
| Strain, strain background (SARS-CoV-2) | SARS-CoV-2, isolate USA-WA1/2020 | BEI Resources, NIAID, NIH | Cat# NR-52281 | |
| Sequence-based reagent | sgRNA: CD4 | This study | sgRNA | GGTGCAATGTAGGAGTCCAA |
| Sequence-based reagent | sgRNA: PHGDH intron1 | This study | sgRNA | GGGCGAGAGAGAGAAAATTG |
| Sequence-based reagent | sgRNA: ACE2 g1 | This study | sgRNA | CACCGCAAAGGCGAGAGATAGTTG |
| Sequence-based reagent | sgRNA: ACE2 g2 | This study | sgRNA | CACCGACATCTTCATGCCTATGTG |
| Sequence-based reagent | sgRNA: TMPRSS2 g1 | This study | sgRNA | CACCGCTGGAACGAGAACTACGGG |
| Sequence-based reagent | sgRNA: TMPRSS2 g2 | This study | sgRNA | CACCGGGGACGGGTAGTACTGAGC |
| Sequence-based reagent | Primer: CD4-Forward | This study | PCR primers | GATAATGGAGAGATGTTGTTGGTTT |
| Sequence-based reagent | Primer: CD4- Reverse | This study | PCR primers | ATGTCCAGGTGCCACTATCCT |
| Sequence-based reagent | Primer: PHGDH intron 1 – Forward | This study | PCR primers | AAAGCAGAACCTTAGCAAAGAGG |
| Sequence-based reagent | Primer: PHGDH intron 1 – Reverse | This study | PCR primers | GAACTAATTGATACGGGGTGCAT |
| Sequence-based reagent | Primer: ACE2-g1- Forward | This study | PCR primers | TCCCTACTTTTTGTCGTTATTAGCA |
| Sequence-based reagent | Primer: ACE2-g1- Reverse | This study | PCR primers | GGTGATCCACAGCTAATGTATTGTT |
| Sequence-based reagent | Primer: ACE2-g2- Forward | This study | PCR primers | TCAAAATGCGATTTCTACAATGTTA |
| Sequence-based reagent | Primer: ACE2-g2- Reverse | This study | PCR primers | TGGGCTTTTCAGATTAAACCATTAT |
| Sequence-based reagent | Primer: TMPRSS2-g1-Forward | This study | PCR primers | ACAAATTCCACCTGCTGGTTATAG |
| Sequence-based reagent | Primer: TMPRSS2-g1- Reverse | This study | PCR primers | ACTTCATCCTTCAGGTGTACTCATC |
| Sequence-based reagent | Primer: TMPRSS2-g2- Forward | This study | PCR primers | CAGGAAATAAACACAAAGAGAATCC |
| Sequence-based reagent | Primer: TMPRSS2-g2-Reverse | This study | PCR primers | ACTATGAAAACCATGGATACCAACC |
| Sequence-based reagent | SARS-CoV-2-N-F | PCR primers | TAATCAGACAAGGAACTGATTA | |
| Sequence-based reagent | SARS-CoV-2-N-R | PCR primers | CGAAGGTGTGACTTCCATG | |
| Sequence-based reagent | SARS-CoV-2-E-F | PCR primers | ACAGGTACGTTAATAGTTAATAGCGT | |
| Sequence-based reagent | SARS-CoV-2-E-R | PCR primers | ATATTGCAGCAGTACGCACACA | |
| Sequence-based reagent | Human 18S-F | PCR primers | GGCCCTGTAATTGGAATGAGTC | |
| Sequence-based reagent | Human 18S-R | PCR primers | CCAAGATCCAACTACGAGCTT | |
| Software, algorithm | Prism 9 | GraphPad Software |
Enzymatic assay
Request a detailed protocolThrombin (605195, human, activated by factor Xa, factor Va, and phospholipid) and factor Xa (69036, bovine, activated by Russell’s Viper Venom) were obtained from Millipore Sigma. Recombinant TMPRSS2, purified from yeast, was obtained from LSBio (LS-G57269). TMPRSS4 was obtained from Aviva System Biology (OPCA0240), furin was obtained from Thermo Fisher Scientific (1503SE010), neutrophil elastase was obtained from Thermo Fisher Scientific (9167SE020). FRET peptides were obtained from Anaspec, and a peptide sequences are listed in Figure 2—figure supplement 1D. Protease assay buffer was composed of 50 mM Tris-HCl, 150 mM NaCl, pH 8. Enzyme dilution/storage buffer was 20 mM Tris-HCl, 500 mM NaCl, 2 mM CaCl2, 50% glycerol, pH 8. Peptides were reconstituted and diluted in DMSO. Enzyme kinetics were assayed in black 96-well plates with clear bottom and measured using a BMG Labtech FLUOstar Omega plate reader, reading fluorescence (excitation 485 nm, emission 520 nm) every minute for 20 cycles, followed by every 5 min for an additional eight cycles. A standard curve of 5-FAM from 0 to 10 µM (1:2 serial dilutions) was used to convert RFU to µM of cleaved FRET peptide product. Calculation of enzyme constants was performed with GraphPad Prism software (version 9.0). Camostat and nafamostat were obtained from Selleck Chemicals and all other inhibitors were obtained from MedChem Express.
Phospholipid vesicles
Request a detailed protocolPhosphatidylcholine (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids #850375C) and phosphatidylserine (1,2-dioleoyl-sn-glycero-3-phospho-L-serine, Avanti Polar Lipids #840035C) were mixed in a 3:1 w/w ratio in chloroform solvent in a screw top vial and the chloroform solvent was evaporated under a nitrogen stream. Unilamellar vesicles were isolated by extrusion using 0.1 µm pore filters and diluted in buffer AB2 (50 mM Tris-HCl, 150 mM NaCl, pH 8).
Cell culture
Request a detailed protocolCalu3, A549, Caco2, and Vero cells were tested for mycoplasma (Lonza MycoAlert detection kit) and human cell line identity was authenticated by ATCC. A549 and Vero cells were grown in DMEM, supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. Calu3 and Caco2 cells were grown in MEM, supplemented with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 1% MEM NEAA, and 1 mM sodium pyruvate.
Plasmids and lentivirus infection
View detailed protocolOverexpression constructs pEGPN-GFP, pEGPN-ACE2, and pEGPN-TMPRSS2 were constructed by Gibson cloning using NEBuilder master mix (New England Biolabs, E2621) with overlapping PCR generated inserts for promoter EF1α, the gene of interest, promoter PGK, and neomycin/resistance gene. Lentiviral vectors were co-transduced with MD2G and PAX2 in 293T cells (5 million cells/10 cm plate) with 25 µl of XtremeGene9 (Millipore Sigma, #6365787001) and supernatant was harvested at 48 and 72 hr post transfection. Target cells were transduced with the addition of 4 µg/ml polybrene (Santa Cruz, sc-134220). Infected cells were selected and maintained in 500 µg/ml G418 (Life Technologies, #10131027). lentiCas9-Blast was a gift from Feng Zhang (Sanjana et al., 2014) (Addgene plasmid #52962). ipUSEPR was a gift from Francisco Sanchez-Rivera and Scott Lowe. sgRNAs were selected from the Brunello CRISPR database (Doench et al., 2016). Four guides per gene were tested in Caco2 cells and the most efficient two sgRNAs/gene were used in subsequent experiments (Figure 4—figure supplement 1). Knockout efficiency was determined by next-generation amplicon sequencing (Genewiz).
Pseudovirus
Request a detailed protocolRecombinant VSV-based and HIV-1-based SARS-CoV-2 pseudovirus was generated as described previously (Schmidt et al., 2020). To generate rVSVΔG/SARS-CoV-2 pseudovirus, 293T cells (12 million cells/15 cm plate) were transfected with 12.5 µg pSARS-CoV-2Δ19, and 24 hr post transfection, were infected with VSV-G-complemented rVSVΔG virus at an MOI of 1. Supernatant was collected 16 hr post infection, centrifuged at 350 g × 10 min, filtered through a 0.45 µm filter, and concentrated using Lenti-X-Concentrator (Takara Bio). Prior to infection of target cells, the viral stock was incubated with anti-VSV-G antibody (3 µg/ml) for 1 hr at 37°C to neutralize contaminating rVSVΔG/NG/NanoLuc/VSV-G particles.
To generate HIV-1NL/SARS-CoV-2 pseudovirus, 293T cells (12 million cells/15 cm plate) were co-transfected with 15.75 µg CCNanoLuc/GFP, 15.75 µg HIV-1NL GagPol, and 5.625 µg CMV-SARS-CoV-2-S, using 50 µl per 15 cm plate X-tremeGENE 9 (Sigma-Aldrich, 8724121001). Media was changed at 24 hr post transfection, and supernatant was collected at 48 and 72 hr. Centrifuged and filtered pseudovirus was concentrated with Lenti-X-Concentrator or with 40% (w/v) PEG-8000, 1.2 M NaCl, pH 7.2.
Incucyte
Request a detailed protocolCells were imaged and analyzed using an Incucyte ZOOM (Essen BioScience). Four fields of view per well were averaged and 3–6 wells/condition were assayed in each experiment. Confluence was calculated from brightfield images, GFP/NeonGreen object confluence was calculated from green fluorescent images taken with 400 ms exposure time, and GFP+ fractional area is the ratio of these variables.
Luciferase assay
Request a detailed protocolFollowing pseudovirus infection, cells were washed twice with PBS, which was subsequently aspirated. Lysis buffer (Promega, E1531) was added (50 µl/well) and incubated rotating for 15 min at room temperature. NanoGlo Substrate (Promega, N1130) was diluted 1:50 in assay buffer and 25 µl/well was added and incubated for an additional 15 min. Samples were transferred to a white, opaque-bottom 96-well plate and luminescence was read using a BMG Labtech FLUOstar Omega plate reader.
Clotting assays
Request a detailed protocolPooled normal human plasma was obtained from Pacific Hemostasis (VWR #95059–698). Factor X and prothrombin-deficient plasma were obtained from Haematologic Technologies (#FX-ID and #FII-ID). Russell’s viper venom test was performed with 10 µg/ml snake venom from Vipera russelli (RVV, Sigma-Aldrich #V2501) diluted in Tris buffer (20 mM Tris-HCl, 150 mM NaCl, 14 mM CaCl2, pH 7.5). Pre-warmed plasma was mixed with pre-warmed dilute venom (100 µl each) and monitored for visible clotting at 37°C. Prothrombin time was determined by mixing 100 µl pre-warmed plasma with 200 µl pre-warmed thromboplastin (VWR #95059–802) and monitoring for visible clotting at 37°C.
hPSC lung organoids
Request a detailed protocolThe hPSC-derived lung organoids were differentiated and cultured as described previously (Han et al., 2021). Briefly, hPSCs were differentiated into endoderm in serum-free differentiation (SFD) medium (DMEM/F12 (3:1) (Life Technologies) supplemented with 1 × N2 (Life Technologies), 1 × B27 (Life Technologies), 50 μg/ml ascorbic acid, 2 mM Glutamax (Gibco), 0.4 μM monothioglycerol and 0.05% BSA) in a 5% O2 incubator; followed by 10 μM Y-27632, 0.5 ng/ml human BMP4 (R&D Systems), 2.5 ng/ml human bFGF and 100 ng/ml human activin A (R&D Systems) for 3 days; and subsequently single cells were plated on fibronectin-coated plates. Differentiation to anterior foregut endoderm was performed with SFD with 1.5 μM dorsomorphin dihydrochloride (R&D Systems) and 10 μM SB431542 (R&D Systems) for 3 days, and then 10 μM SB431542 and 1 μM IWP2 (R&D Systems) treatment for 3 days. Differentiation to early stage lung progenitor cells was accomplished by treatment with 3 μM CHIR99021 (CHIR, Stem-RD), 10 ng/ml human FGF10, 10 ng/ml human KGF, 10 ng/ml human BMP4, and 50–60 nM all-trans retinoic acid (ATRA) in a 5% CO2/air incubator. Differentiation to late-stage lung progenitor cells, cells were treated with SFD containing 3 μM CHIR99021, 10 ng/ml human FGF10, 10 ng/ml human FGF7, 10 ng/ml human BMP4 and 50 nM ATRA on fibronectin-coated plates, and subsequently maintained for 1 week in SFD medium containing 3 μM CHIR99021, 10 ng/ml human FGF10 and 10 ng/ml human KGF, in a 5% CO2/air incubator. Mature lung organoids were generated by growing late-stage lung progenitor cells in 90% Matrigel in SFD medium containing 3 μM CHIR99021, 10 ng/ml human FGF10, 10 ng/ml human KGF, 50 nM dexamethasone, 0.1 mM 8-bromo-cAMP (Sigma-Aldrich), and 0.1 mM IBMX (3,7-dihydro-1-methyl-3-(2-methylpropyl)-1H-purine-2,6-dione; Sigma-Aldrich) for ~20 days.
SARS-CoV-2 virus infection
Request a detailed protocolSARS-CoV-2 was maintained and infections were performed as described previously (Tang et al., 2021b). SARS-CoV-2, isolate USA-WA1/2020 (NR-52281), was deposited by the Center for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH. SARS-CoV-2 was propagated in Vero E6 cells in DMEM supplemented with 2% FBS, 4.5 g/l D-glucose, 4 mM L-glutamine, 10 mM non-essential amino acids, 1 mM sodium pyruvate, and 10 mM HEPES. MOI of SARS-CoV-2 was determined by plaque assay in Vero E6 cells in Minimum Essential Media supplemented with 2% FBS, 4 mM L-glutamine, 0.2% BSA, 10 mM HEPES and 0.12% NaHCO3, and 0.7% agar.
All work involving live SARS-CoV-2 was performed in the CDC/USDA-approved BSL-3 facility of the Global Health and Emerging Pathogens Institute at the Icahn School of Medicine at Mount Sinai in accordance with institutional biosafety requirements.
At 24 or 48 hpi, RNA was extracted with TRIzol and Direct-zol RNA Miniprep Plus kit (Zymo Research). SARS-CoV-2-N and SARS-CoV-2-E transcripts were quantified by two-step qRT-PCR using LunaScript RT SuperMix Kit (E3010L) for c-DNA synthesis and Luna Universal qPCR Master Mix (NEB #M3003) for RT-qPCR. qRT-PCRs were performed on CFX384 Touch Real-Time PCR Detection System (Bio-Rad). Primers specific for the N gene (SARS-CoV-2-N-F: TAATCAGACAAGGAACTGATTA, SARS-CoV-2-N-R: CGAAGGTGTGACTTCCATG), for the E gene (SARS-CoV-2-E-F: ACAGGTACGTTAATAGTTAATAGCGT, SARS-CoV-2-E-R: ATATTGCAGCAGTACGCACACA), as well as internal control human 18S (Forward: GGCCCTGTAATTGGAATGAGTC, Reverse: CCAAGATCCAACTACGAGCTT) were used. The delta-delta-cycle threshold (ΔΔCT) was determined relative to 18S and vehicle-treated samples.
Data availability
All new plasmids will be made available through Addgene.
References
-
Efficacy and Safety of the mRNA-1273 SARS-CoV-2 VaccineNew England Journal of Medicine 384:403–416.https://doi.org/10.1056/NEJMoa2035389
-
Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitorThe Journal of Biological Chemistry 273:4378–4386.https://doi.org/10.1074/jbc.273.8.4378
-
Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domainThe Journal of Biological Chemistry 285:22758–22763.https://doi.org/10.1074/jbc.M110.103275
-
Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cellsThe Journal of Experimental Medicine 216:674–687.https://doi.org/10.1084/jem.20181155
-
Direct Oral Anticoagulant Use: A Practical Guide to Common Clinical ChallengesJournal of the American Heart Association 9:e017559.https://doi.org/10.1161/JAHA.120.017559
-
Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infectionClinical Microbiology Reviews 20:660–694.https://doi.org/10.1128/CMR.00023-07
-
Pentamidine is a specific, non-peptide, GPIIb/IIIa antagonistThrombosis and Haemostasis 75:503–509.
-
Targeting SARS-CoV-2-Platelet Interactions in COVID-19 and Vaccine-Related ThrombosisFrontiers in Pharmacology 12:708665.https://doi.org/10.3389/fphar.2021.708665
-
Discovery of Selective Matriptase and Hepsin Serine Protease Inhibitors: Useful Chemical Tools for Cancer Cell BiologyJournal of Medicinal Chemistry 62:480–490.https://doi.org/10.1021/acs.jmedchem.8b01536
-
Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9Nature Biotechnology 34:184–191.https://doi.org/10.1038/nbt.3437
-
Cleavage of spike protein of SARS coronavirus by protease factor Xa is associated with viral infectivityBiochemical and Biophysical Research Communications 359:174–179.https://doi.org/10.1016/j.bbrc.2007.05.092
-
New synthetic inhibitors of C1r, C1 esterase, thrombin, plasmin, kallikrein and trypsinBiochimica et Biophysica Acta (BBA) - Enzymology 661:342–345.https://doi.org/10.1016/0005-2744(81)90023-1
-
Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key eventThe Journal of Infectious Diseases 188:1618–1629.https://doi.org/10.1086/379724
-
Evaluation of bleeding in patients receiving direct oral anticoagulantsVascular Health and Risk Management 13:325–342.https://doi.org/10.2147/VHRM.S121661
-
Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19Antimicrobial Agents and Chemotherapy 64:e00754-20.https://doi.org/10.1128/AAC.00754-20
-
Clinical features of patients infected with 2019 novel coronavirus in Wuhan, ChinaLancet (London, England) 395:497–506.https://doi.org/10.1016/S0140-6736(20)30183-5
-
A Fluorogenic Peptide Cleavage Assay to Screen for Proteolytic Activity: Applications for coronavirus spike protein activationJournal of Visualized Experiments 10:3791.https://doi.org/10.3791/58892
-
Viral Coagulopathy in Patients With COVID-19: Treatment and CareClinical and Applied Thrombosis/Hemostasis 26:1076029620936776.https://doi.org/10.1177/1076029620936776
-
Diagnosis and management of acute obstetrical DICSeminars in Thrombosis and Hemostasis 27:161–167.https://doi.org/10.1055/s-2001-14076
-
Influenza vaccine uptake, COVID-19 vaccination intention and vaccine hesitancy among nurses: A surveyInternational Journal of Nursing Studies 114:103854.https://doi.org/10.1016/j.ijnurstu.2020.103854
-
Comparison of nafamostat mesilate and unfractionated heparin as anticoagulants during continuous renal replacement therapyThe International Journal of Artificial Organs 39:16–21.https://doi.org/10.5301/ijao.5000465
-
Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumoniaThe Lancet. Rheumatology 2:e437–e445.https://doi.org/10.1016/S2665-9913(20)30121-1
-
Trypsin Treatment Unlocks Barrier for Zoonotic Bat Coronavirus InfectionJournal of Virology 94:e01774-19.https://doi.org/10.1128/JVI.01774-19
-
Emerging evidence of a COVID-19 thrombotic syndrome has treatment implicationsNature Reviews. Rheumatology 16:581–589.https://doi.org/10.1038/s41584-020-0474-5
-
Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial ResultsThe New England Journal of Medicine 384:497–511.https://doi.org/10.1056/NEJMoa2023184
-
Nafamostat mesilate as a regional anticoagulant in patients with bleeding complications during extracorporeal membrane oxygenationThe International Journal of Artificial Organs 38:595–599.https://doi.org/10.5301/ijao.5000451
-
Safety and Efficacy of the BNT162b2 mRNA Covid-19 VaccineNew England Journal of Medicine 383:2603–2615.https://doi.org/10.1056/NEJMoa2034577
-
Dengue virus-elicited tryptase induces endothelial permeability and shockThe Journal of Clinical Investigation 129:4180–4193.https://doi.org/10.1172/JCI128426
-
Improved vectors and genome-wide libraries for CRISPR screeningNature Methods 11:783–784.https://doi.org/10.1038/nmeth.3047
-
Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric virusesThe Journal of Experimental Medicine 217:e20201181.https://doi.org/10.1084/jem.20201181
-
Reserving coronavirus disease 2019 vaccines for global access: cross sectional analysisBMJ (Clinical Research Ed.) 371:m4750.https://doi.org/10.1136/bmj.m4750
-
Neuroinvasion of SARS-CoV-2 in human and mouse brainThe Journal of Experimental Medicine 218:e20202135.https://doi.org/10.1084/jem.20202135
-
Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitorClinical and Applied Thrombosis/Hemostasis 15:9S–16S.https://doi.org/10.1177/1076029609343004
-
Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolismAmerican Journal of Hematology 95:1522–1530.https://doi.org/10.1002/ajh.25979
-
Continuous regional arterial infusion of protease inhibitor and antibiotics in acute necrotizing pancreatitisAmerican Journal of Surgery 171:394–398.https://doi.org/10.1016/S0002-9610(97)89617-1
-
SARS-CoV-2 infection induces beta cell transdifferentiationCell Metabolism 33:1577–1591.https://doi.org/10.1016/j.cmet.2021.05.015
-
COVID-19 and cerebrovascular diseases: a comprehensive overviewTherapeutic Advances in Neurological Disorders 13:1756286420978004.https://doi.org/10.1177/1756286420978004
-
BookScientific Foundations of Biochemistry in Clinical PracticeButterworth Heinemann.
-
SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effectsNature Structural & Molecular Biology 27:763–767.https://doi.org/10.1038/s41594-020-0468-7
-
Evidence for Gastrointestinal Infection of SARS-CoV-2Gastroenterology 158:1831–1833.https://doi.org/10.1053/j.gastro.2020.02.055
-
Protease inhibitors targeting coronavirus and filovirus entryAntiviral Research 116:76–84.https://doi.org/10.1016/j.antiviral.2015.01.011
-
Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19Science Translational Medicine 12:eabd3876.https://doi.org/10.1126/scitranslmed.abd3876
Article and author information
Author details
Edward R Kastenhuber

Funding
National Institutes of Health (R01AI35270)
- Gary R Whittaker
National Cancer Institute (R35CA197588)
- Lewis Cantley
- Lewis Cantley
Pershing Square Foundation
- Lewis Cantley
- Lewis Cantley
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors would like to thank Pilar Mendoza (Rockefeller University), John Blenis (WCMC), Elena Piskounova (WCMC), Tim McGraw (WCMC), Marco Straus (Cornell University), Tomer Yaron (WCMC), and all members of the Cantley Lab for insightful discussion and helpful comments. We thank Paul Bieniasz (Rockefeller University) and Theodora Hatziioannou (Rockefeller University) for providing reagents and helping to establish the pseudovirus platform in our laboratory. We thank Danielle Bulaon (Weill Cornell) for provisioning inhibitors, Benjamin tenOever (Mount Sinai) for providing Vero cells, and Francisco Sanchez-Rivera and Scott Lowe (MSKCC) for the ipUSEPR plasmid. This work was funded in part by the National Institute of Health research grants R01AI35270 (to GW) and R35 CA197588 (to LCC) and the Pershing Square Foundation (LCC).
Copyright
© 2022, Kastenhuber et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,965
- views
-
- 477
- downloads
-
- 40
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry
eLife 11:e77444.
https://doi.org/10.7554/eLife.77444
Further reading
-
- Biochemistry and Chemical Biology
- Cell Biology
Stem cell differentiation involves a global increase in protein synthesis to meet the demands of specialized cell types. However, the molecular mechanisms underlying this translational burst and the involvement of initiation factors remains largely unknown. Here, we investigate the role of eukaryotic initiation factor 3 (eIF3) in early differentiation of human pluripotent stem cell (hPSC)-derived neural progenitor cells (NPCs). Using Quick-irCLIP and alternative polyadenylation (APA) Seq, we show eIF3 crosslinks predominantly with 3’ untranslated region (3’-UTR) termini of multiple mRNA isoforms, adjacent to the poly(A) tail. Furthermore, we find that eIF3 engagement at 3’-UTR ends is dependent on polyadenylation. High eIF3 crosslinking at 3’-UTR termini of mRNAs correlates with high translational activity, as determined by ribosome profiling, but not with translational efficiency. The results presented here show that eIF3 engages with 3’-UTR termini of highly translated mRNAs, likely reflecting a general rather than specific regulatory function of eIF3, and supporting a role of mRNA circularization in the mechanisms governing mRNA translation.
-
- Biochemistry and Chemical Biology
- Microbiology and Infectious Disease
Mycofactocin is a redox cofactor essential for the alcohol metabolism of mycobacteria. While the biosynthesis of mycofactocin is well established, the gene mftG, which encodes an oxidoreductase of the glucose-methanol-choline superfamily, remained functionally uncharacterized. Here, we show that MftG enzymes are almost exclusively found in genomes containing mycofactocin biosynthetic genes and are present in 75% of organisms harboring these genes. Gene deletion experiments in Mycolicibacterium smegmatis demonstrated a growth defect of the ∆mftG mutant on ethanol as a carbon source, accompanied by an arrest of cell division reminiscent of mild starvation. Investigation of carbon and cofactor metabolism implied a defect in mycofactocin reoxidation. Cell-free enzyme assays and respirometry using isolated cell membranes indicated that MftG acts as a mycofactocin dehydrogenase shuttling electrons toward the respiratory chain. Transcriptomics studies also indicated remodeling of redox metabolism to compensate for a shortage of redox equivalents. In conclusion, this work closes an important knowledge gap concerning the mycofactocin system and adds a new pathway to the intricate web of redox reactions governing the metabolism of mycobacteria.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}