Genome-wide detection of imprinted differentially methylated regions using nanopore sequencing
- Canada's Michael Smith Genome Sciences Centre, BC Cancer Agency, Canada
- Department of Medical Genetics, University of British Columbia, Canada
- Department of Microbiology and Immunology, Michael Smith Laboratories, University of British Columbia, Canada
Abstract
Imprinting is a critical part of normal embryonic development in mammals, controlled by defined parent-of-origin (PofO) differentially methylated regions (DMRs) known as imprinting control regions. Direct nanopore sequencing of DNA provides a means to detect allelic methylation and to overcome the drawbacks of methylation array and short-read technologies. Here, we used publicly available nanopore sequencing data for 12 standard B-lymphocyte cell lines to acquire the genome-wide mapping of imprinted intervals in humans. Using the sequencing data, we were able to phase 95% of the human methylome and detect 94% of the previously well-characterized, imprinted DMRs. In addition, we found 42 novel imprinted DMRs (16 germline and 26 somatic), which were confirmed using whole-genome bisulfite sequencing (WGBS) data. Analysis of WGBS data in mouse (Mus musculus), rhesus monkey (Macaca mulatta), and chimpanzee (Pan troglodytes) suggested that 17 of these imprinted DMRs are conserved. Some of the novel imprinted intervals are within or close to imprinted genes without a known DMR. We also detected subtle parental methylation bias, spanning several kilobases at seven known imprinted clusters. At these blocks, hypermethylation occurs at the gene body of expressed allele(s) with mutually exclusive H3K36me3 and H3K27me3 allelic histone marks. These results expand upon our current knowledge of imprinting and the potential of nanopore sequencing to identify imprinting regions using only parent-offspring trios, as opposed to the large multi-generational pedigrees that have previously been required.
Editor's evaluation
This work uses nanowire sequencing to detect genome-Wide imprinted differentially methylated regions. It will be of broad interest to DNA methylation researchers.
https://doi.org/10.7554/eLife.77898.sa0Introduction
The addition of a methyl group to the fifth carbon of cytidine is the most prevalent and stable epigenetic modification of human DNA (Laurent et al., 2010). DNA methylation is involved in gene regulation and influences a vast array of biological mechanisms, including embryonic development and cell fate, genome imprinting, X-chromosome inactivation, and transposon silencing (Moore et al., 2013; Smith and Meissner, 2013). In mammals, there are two copies or alleles of a gene, one inherited from each parent. Most gene transcripts are expressed from both alleles. However, a subset of genes are only expressed from a single allele; this allele can be selected either randomly, as seen in X-chromosome inactivation in females, or based upon the parent-of-origin (PofO), referred to as imprinting (Chess, 2013; Khamlichi and Feil, 2018).
In imprinting, mono-allelic expression of a gene or cluster of genes is controlled by a cis-acting imprinting control region (ICR) (Bartolomei and Ferguson-Smith, 2011). The main mechanism by which this occurs is PofO-defined differential methylation at ICRs, also known as imprinted differentially methylated regions (DMRs) (Bartolomei and Ferguson-Smith, 2011; Maupetit-Méhouas et al., 2016). Imprinted DMRs are classified as germline (primary) or somatic (secondary), hereinafter referred to as gDMR and sDMR. gDMRs are established during the first wave of methylation reprogramming in germ cell development and escape the second methylation reprogramming after fertilization (Zink et al., 2018). sDMRs are established de novo after fertilization during somatic development, usually under the control of a nearby gDMR (Zink et al., 2018). Imprinted clusters of genes may span up to ~4 Mb, by acting through a CCCTC-binding factor (CTCF)-binding site or by allelic expression of a long non-coding RNA (Bartolomei and Ferguson-Smith, 2011; da Rocha and Gendrel, 2019). By contrast, individually imprinted genes are typically regulated by PofO-derived differential methylation at the gene promoter (Bartolomei and Ferguson-Smith, 2011).
Imprinting is implicated in various genetic disorders, either from aberrations in imprinted methylation or from deleterious variants affecting the ICR and imprinted genes. Aberrant imprinted methylation is also detected in several human cancers (Goovaerts et al., 2018; Jelinic and Shaw, 2007; Tomizawa and Sasaki, 2012). Thus, accurate mapping and characterization of imprinting in humans is key to the treatment and actionability of genetic disorders, and to personalized oncogenomonics.
To detect imprinted methylation, accurate assignment of methylation data to paternal and maternal alleles is required. Achieving this with traditional bisulfite sequencing or arrays is challenging. Several studies have used samples with large karyotypic abnormalities, such as uniparental disomies, teratomas, and hydatidiform moles, to infer regions of imprinting (Court et al., 2014; Hernandez Mora et al., 2018; Joshi et al., 2016). This approach relies not only on rare structural variants, but also on the assumption that both normal methylation and the imprinted state remain intact in spite of substantial genomic aberrations. A study by Zink et al., 2018, leveraged a genotyped, multi-generation pedigree spanning nearly half the population of Iceland (n=150,000), in combination with whole-genome oxidative bisulfite sequencing, to phase methylation and infer PofO (Zink et al., 2018). However, despite being able to phase nearly every SNP in that cohort, they were only able to phase 84% of the human autosomal methylome in over 200 samples due to the short length of reads. Furthermore, the study was based on a single, genetically isolated population, which may not be representative of the wider human population. A comprehensive mapping of imprinted methylation using a technology more suited to phasing reads, based on individuals more representative of the human population, could greatly advance our understanding of imprinting, with direct benefits for human health.
Previously, we have shown that nanopore sequencing can detect allelic methylation in a single sample and accurately determine PofO using only trio data. We also previously developed the software NanoMethPhase for this purpose (Akbari et al., 2021). Here, we applied NanoMethPhase to public nanopore data from a diverse set of 12 lymphoblastoid cell lines (LCLs) from the 1000 Genomes Project (1KGP) and Genome in a Bottle (GIAB) to investigate genome-wide allele-specific methylation (ASM) and detect novel imprinted DMRs (Figure 1A; Auton et al., 2015; De Coster et al., 2019; Jain et al., 2018; Shafin et al., 2020; Zook et al., 2016; Zook et al., 2019). Using trio data from GIAB and 1KGP for these cell lines, we phased nanopore long reads to their PofO and inferred allelic methylation (Akbari et al., 2021; Auton et al., 2015; Zook et al., 2019). We were able to detect haplotype and methylation status for 26.5 million autosomal CpGs comprising 95% of the human autosomal methylome (GRCh38 main chromosomes). We further used public whole-genome bisulfite sequencing (WGBS) data to confirm the presence of the detected DMRs in other tissues and to classify the novel DMRs as germline or somatic. We captured 94% of the well-characterized DMRs and detected 42 novel DMRs (16 gDMRs and 26 sDMRs). Of these novel DMRs, 40.5% show evidence of conservation. We also detected seven blocks of PofO methylation bias at seven imprinted clusters with mutual exclusive allelic H3K36me3 and H3K27me3 histone marks. Collectively, our results extend the set of known imprinted intervals in humans and demonstrate a major contribution in our ability to characterize imprinting by ASM, brought about by the capabilities of long-read nanopore sequencing.
Figure 1 with 1 supplement see all
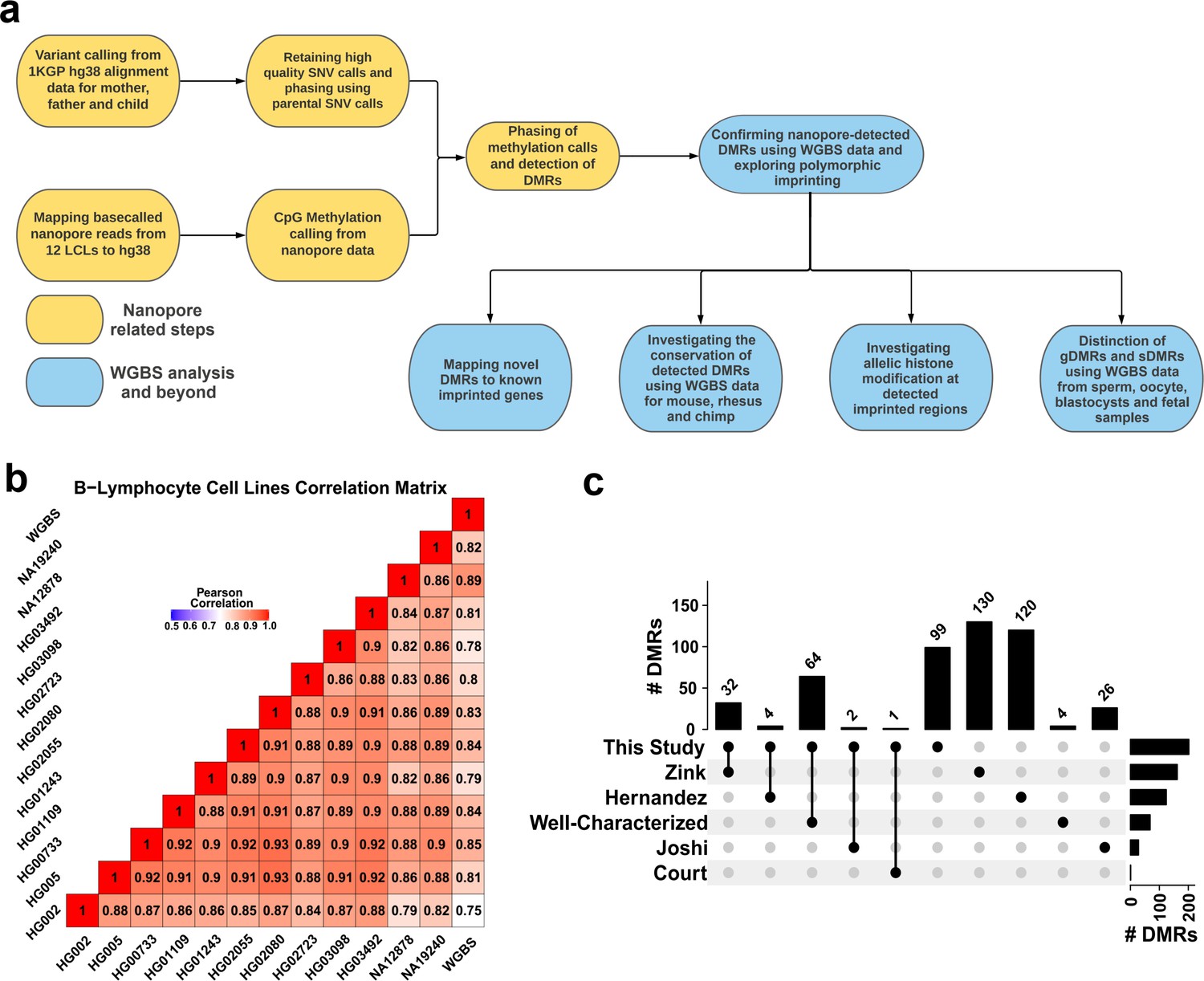
Detection of allelic methylation using nanopore sequencing.
(a) Flowchart of the study representing all the analysis steps. (b) Pearson correlation matrix of the nanopore CpG methylation frequencies for the 12 cell lines and NA12878 whole-genome bisulfite sequencing (WGBS) from ENCODE (ENCFF835NTC). (c) Upset plot of the number of differentially methylated regions (DMRs) detected in our study and previous studies, including overlaps.
Results
Assessing the effectiveness of nanopore methylation calling and detection of known imprinted DMRs
Using the set of 12 LCLs for which we called methylation data, we conducted correlation analysis among nanopore-called methylation data and another WGBS dataset for NA12878 cell line (ENCFF835NTC) to confirm the reliability of methylation calling (Figure 1B). We observed high correlation across cell lines (r=0.75–0.93), as expected as they were the same cell type. NA12878 nanopore-called methylation also showed the highest correlation (r=0.89) with NA12878 WGBS, as expected (Figure 1B). To assess the use of nanopore long reads in detecting known DMRs, we identified previously reported DMRs, including 383 imprinted intervals (Court et al., 2014; Hernandez Mora et al., 2018; Joshi et al., 2016; Zink et al., 2018). Of these 383, we classified 68 as ‘well-characterized’ as they were reported by at least two genome-wide mapping studies or were previously known to be imprinted (Supplementary file 1). Subsequently, we haplotyped the methylome in each cell line, and performed differential methylation analysis (DMA) between alleles across cell lines; 95% (26.5M) of the human autosomal CpGs could be assigned to a haplotype. We detected 200 allelic DMRs (p-value <0.001, |methylation difference|>0.20, and detected in at least four cell lines in each haplotype) (Supplementary file 2). Out of the 200 detected DMRs, 101 overlapped with 103 previously reported DMRs with consistent PofO (Supplementary file 3), while the remaining 99 were novel (Figure 1C). Of the well-characterized DMRs, 64/68 (94%) were detected in our study (Figure 1C; Supplementary file 3).
Similarly, we assessed methylation haplotyping and detection of imprinted DMRs within a single sample. On average, 90% (M ± SD = 25 M ± 1.61 M) of the human methylome could be assigned to a parental haplotype in each cell line (Figure 1—figure supplement 1). Among the well-characterized DMRs, ~73% (M ± SD = 49.5 ± 4.5) could be detected in a single cell line. An additional 33 DMRs (SD = 9.6) reported by only one previous study were detected in each cell line (Figure 1—figure supplement 1).
Confirmation of novel imprinted DMRs
We detected 99 imprinted DMRs that did not overlap with previously reported imprinted DMRs (Court et al., 2014; Hernandez Mora et al., 2018; Joshi et al., 2016; Zink et al., 2018). In order to confirm these DMRs in human tissues and detect potential novel imprinted regions, we investigated WGBS datasets for partial methylation at nanopore-detected DMRs (Materials and methods). We used 60 WGBS datasets from 29 tissue types and 119 blood samples from 87 individuals (Bernstein et al., 2010; ENCODE Project Consortium, 2012; Stunnenberg et al., 2016). We first examined the 68 well-characterized DMRs, 91% of them demonstrated partial methylation (more than 60% of the CpGs at the DMR having between 0.35 and 0.65 methylation) in at least one tissue and individual blood samples (Figure 2A and B). As controls, we used 100 randomly selected 1, 2, 3 kb bins, and CpG islands (CGIs) in 100 resampling iterations. Of these, 0.65%, 0.74%, 2.28%, and 4.83% of the randomly selected 3, 2, 1 kb, and CGIs, respectively, demonstrated partial methylation (Figure 2—figure supplement 1). Applying this approach to the 99 previously unreported DMRs, the WGBS data supported 42 of the novel DMRs (Figure 2, Table 1). In agreement with previous studies reporting a higher number of maternally methylated intervals (Court et al., 2014; Hernandez Mora et al., 2018; Joshi et al., 2016), 74% of the novel DMRs were maternally methylated. Overall, we detected 143 imprinted DMRs of which 101 were found to overlap with previously reported DMRs while 42 were novel DMRs detected by nanopore and confirmed using WGBS data (Figure 2C, Supplementary file 4).
Figure 2 with 1 supplement see all

Confirmation of nanopore-detected differentially methylated regions (DMRs) using whole-genome bisulfite sequencing (WGBS) data.
(a) and (b) Violin plots representing the average methylation of each DMR in WGBS tissue and blood samples. (c) Idiogram of the 101 DMRs overlapping to reported intervals and 42 novel DMRs which were confirmed by WGBS. Paternally methylated DMRs are labelled on the left side of each chromosome while maternally methylated DMRs are on the right. Red labels represent germline DMRs while blue labels represent somatic DMRs. Novel DMRs are boxed and named based on their nearest gene (Ensembl Gene 103 GRCh38.p13).
-
Figure 2—source data 1
The average methylation of each DMR in human WGBS tissue samples.
- https://cdn.elifesciences.org/articles/77898/elife-77898-fig2-data1-v1.txt
-
Figure 2—source data 2
The average methylation of each DMR in human WGBS blood samples.
- https://cdn.elifesciences.org/articles/77898/elife-77898-fig2-data2-v1.txt
Table 1
Forty-two detected imprinted differentially methylated regions (DMRs) from nanopore data and confirmed using whole-genome bisulfite sequencing (WGBS) data.
DMRs are named after the nearest gene (EnsemblGene 103 GRCh38.p13).
| ID | DMR name | Origin | Type | Distance to nearest imprinted gene | % Individuals with partial methylation | % Tissues with partial methylation |
|---|---|---|---|---|---|---|
| 22 | AC024940.1 | Maternal | Germline | 0 | 15.9 | 3.8 |
| 35 | DDA1 | Maternal | Germline | 0 | 7.3 | 15.4 |
| 38 | ACTL10;NECAB3 | Maternal | Germline | 0 | 3.7 | 8.7 |
| 42 | SYCE1 | Maternal | Germline | 3.2 kb | 4 | 9.1 |
| 12 | FAM83H | Maternal | Germline | 149 kb | 48.8 | 12 |
| 20 | OPCML | Maternal | Germline | 744.1 kb | 45.1 | 25 |
| 19 | YBX2P1 | Maternal | Germline | >2 Mb | 3.7 | 7.7 |
| 26 | NALCN-AS1 | Maternal | Germline | >2 Mb | 30.5 | 10 |
| 28 | PTGDR | Maternal | Germline | >2 Mb | 8.4 | 3.4 |
| 32 | AATK | Maternal | Germline | >2 Mb | 23.2 | 9.1 |
| 34 | MIR7-3HG | Maternal | Germline | >2 Mb | 8.1 | 3.6 |
| 2 | AC007391.3 | Maternal | Germline | >2 Mb | 37.2 | 60.7 |
| 5 | C4orf50 | Maternal | Germline | >2 Mb | 14.5 | 22.2 |
| 7 | CPLANE1 | Maternal | Germline | >2 Mb | 2.3 | 7.1 |
| 14 | SPTLC1 | Maternal | Germline | >2 Mb | 5.8 | 48.1 |
| 1 | BMP8A | Maternal | Somatic | 0 | 4.5 | 26.1 |
| 24 | LPAR6;RB1 | Maternal | Somatic | 0 | 2.3 | 10.3 |
| 36 | ZNF714 | Maternal | Somatic | 0 | 43.9 | 29.6 |
| 17 | BTBD7P1 | Maternal | Somatic | >2 Mb | 73.5 | 55.6 |
| 18 | ANKRD2 | Maternal | Somatic | >2 Mb | 34.1 | 3.8 |
| 23 | GJA3 | Maternal | Somatic | >2 Mb | 25.6 | 21.4 |
| 27 | RANBP20P | Maternal | Somatic | >2 Mb | 28 | 32 |
| 33 | NFE2L3P1 | Maternal | Somatic | >2 Mb | 29.3 | 44.4 |
| 39 | AL096828.3 | Maternal | Somatic | >2 Mb | 50 | 7.7 |
| 41 | SLC5A1 | Maternal | Somatic | >2 Mb | 56.1 | 25.9 |
| 8 | TMEM232 | Maternal | Somatic | >2 Mb | 25.6 | 37.9 |
| 9 | NME5 | Maternal | Somatic | >2 Mb | 22.1 | 10.7 |
| 11 | AC092634.8 | Maternal | Somatic | >2 Mb | 6.1 | 3.8 |
| 13 | CDRT15P6 | Maternal | Somatic | >2 Mb | 12.7 | 5.6 |
| 15 | TMOD1 | Maternal | Somatic | >2 Mb | 35.4 | 14.8 |
| 16 | LHX6 | Maternal | Somatic | >2 Mb | 44.6 | 25 |
| 30 | MIR6085 | Paternal | Germline | >2 Mb | 27.1 | 25.9 |
| 3 | PAX8;PAX8-AS1 | Paternal | Somatic | 0 | 24.4 | 32.1 |
| 4 | ZDBF2 | Paternal | Somatic | 0 | 53.6 | 58.6 |
| 29 | SNHG14 | Paternal | Somatic | 3 kb | 4.7 | 37.9 |
| 37 | AC092296.3 | Paternal | Somatic | 90 kb | 1.2 | 7.7 |
| 40 | RIMBP3 | Paternal | Somatic | 296 kb | 17.4 | 11.1 |
| 10 | RNU6-293P | Paternal | Somatic | 1.03 Mb | 65.1 | 37 |
| 21 | PARP11 | Paternal | Somatic | >2 Mb | 10.8 | 22.2 |
| 25 | UBAC2 | Paternal | Somatic | >2 Mb | 1.2 | 6.9 |
| 31 | CFAP161 | Paternal | Somatic | >2 Mb | 22.4 | 6.9 |
| 6 | MIR4456 | Paternal | Somatic | >2 Mb | 11.3 | 22.2 |
Novel imprinted DMRs display inter-individual variation
Although imprinted methylation is generally regarded as consistent between individuals and resistant to environmental factors, there are examples of polymorphic imprinting where imprinted methylation is not consistently observed across individuals. In order to assess the inter-individual variation of the novel imprinted DMRs, we examined partial methylation in the 119 blood samples from 87 individuals. Some imprinted DMRs such as VTRNA2-1, IGF2, RB1, PARD6G, CHRNE, and IGF2R are known to be polymorphic (Joshi et al., 2016; Zink et al., 2018). The detected DMRs that mapped to these imprinted regions displayed partial methylation in 2–65% of the individuals in our analysis (M ± SD = 40% ± 22%; Supplementary file 5). ZNF331 DMR is known to be consistently imprinted across individuals (Zink et al., 2018). In our analysis, the DMR that mapped to ZNF331 reported interval displayed partial methylation in 99% of the individuals (Supplementary file 5). We then examined inter-individual variation across the 42 novel DMRs. Imprinted methylation at all the novel DMRs demonstrated variation ranging from 1.2% to 73.5% of the individuals (M ± SD = 23.6% ± 19.2%; Table 1). Among the novel DMRs, maternal sDMR near BTBD7P1 is the most consistent with partial methylation in 73.5% of the individuals (Table 1). On the other hand, the novel paternal sDMRs within AC092296.3 and UBAC2 are the most variable with partial methylation in 1.2% of the individuals (Table 1). Among the individuals, four displayed hypermethylation at several of the well-characterized and novel DMRs (Figure 2B), in line with a previous study that identified rare individuals with consistent hyper- or hypomethylation at dozens of imprinted loci, indicative of a generalized imprinting disruption (Joshi et al., 2016).
As demonstrated in Figure 1C, a considerable number of imprinted DMRs detected in different studies are not overlapping between studies. Different studies used different samples and individuals, therefore, we examined inter-individual variation at DMRs detected in two or more studies (including the current work) and those that detected in one study (Supplementary file 5). The DMRs that detected in at least two studies demonstrated more consistency across individuals (M ± SD = 41.2% ± 33%) while DMRs detected in a single study showed more variability (M ± SD = 10.6% ± 15.4%) (Supplementary file 5). These results suggest that polymorphic imprinting can explain this non-overlapping DMRs across studies.
Determination of germline versus somatic status of novel imprinted DMRs
We investigated the methylation status of the detected novel DMRs in sperm and oocyte to determine if they are germline or somatic imprinted intervals. Maternally methylated gDMRs must display high methylation in oocyte and very low or no methylation in sperm with partial methylated after fertilization. Paternally methylated gDMRs must show high methylation in sperm and very low or no methylation in oocyte with partial methylated after fertilization. For the novel DMRs, 16 were detected as germline (more than 70% methylation in oocyte and less than 20% in sperm and vice versa) from which 15 were maternally methylated and one was paternally methylated (Figure 3A and B). Moreover, novel candidate gDMRs showed partial methylation in the blastocyst and fetal samples, indicating the gDMRs escaped de-methylation after fertilization. Meanwhile, sDMRs displayed partial methylation in fetal tissues, indicating their establishment during somatic development (Figure 3A and B). Overall, 16 of the novel DMRs were found to be germline while 26 were sDMRs.
Figure 3
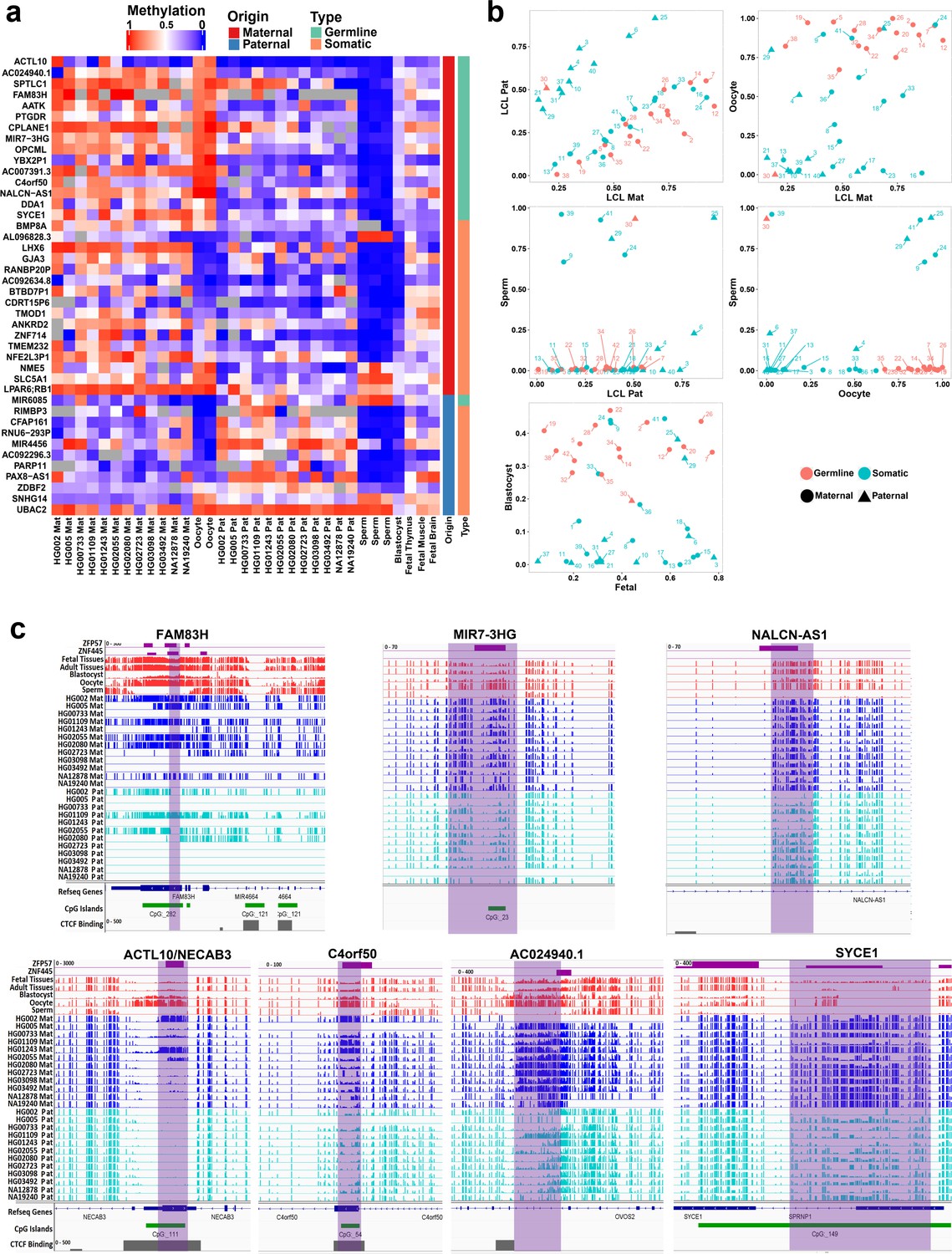
Detection of novel germline and somatic differentially methylated regions (DMRs).
(a) Heatmap displaying average methylation of the 42 nanopore-detected DMRs confirmed by whole-genome bisulfite sequencing (WGBS). DMRs are named based on their nearest gene (Ensembl Gene 103 GRCh38.p13). (b) Dot plots representing the methylation of novel germline and somatic DMRs in each sample with respect to other samples. (c) IGV screenshots from six novel germline DMRs overlapping with ZNF445 and/or ZFP57 chromatin immunoprecipitation sequencing (ChIP-seq) peaks. The range for all methylation tracks is 0–1.
During germ cell development, gDMRs are bound by proteins critical for their methylation maintenance during post-fertilization reprogramming. ZFP57 and ZNF445 have been identified as imprinting maintenance proteins (Takahashi et al., 2019). Using ZFP57 and ZNF445 ChIP-seq peak calling information from human embryonic stem cells and the HEK 293T cell line (Imbeault et al., 2017; Takahashi et al., 2019), 44% of the novel gDMRs and 49% of the reported gDMRs were bound by ZFP57 and/or ZNF445 (Figure 3C; Supplementary file 4). Of these gDMRs, 89% had a ZFP57 peak and 45% had a ZNF445 peak. This highlights the importance of ZFP57 as an important factor for the maintenance of imprinted methylation at gDMRs. 5′-TGC(5mC)GC-3′ is the canonical binding motif for ZFP57 (Quenneville et al., 2011). Eighty-eight percent of the gDMRs with a ZFP57 peak had at least one 5′-TGCCGC-3′ motif, while 40% of the gDMRs without ZFP57 peak had at least one 5′-TGCCGC-3′ motif in the human genome (GRCh38; Supplementary file 4). Moreover, at gDMRs the number of 5′-TGCCGC-3′ motifs demonstrated a significant positive correlation with the number of individuals demonstrating partial methylation (Pearson = 0.54, p-value = 3.6e−07; Appendix 1—figure 1). This suggests that a greater number of motifs provide more functional binding opportunities for ZFP57 and also less likelihood that all ZFP57 motifs could be perturbed through polymorphism or DNA sequence variation resulting in the imprinted methylation being less polymorphic.
Allelic H3K4me3 histone mark at detected DMRs
The H3K4me3 histone mark is protective against DNA methylation. At imprinted DMRs, the unmethylated allele is usually enriched for this histone modification (Court et al., 2014; John and Lefebvre, 2011). We used H3K4me3 chromatin immunoprecipitation sequencing (ChIP-seq) data for six LCLs and their heterozygous single-nucleotide variant (SNV) calls from 1KGP. Fifty of the DMRs mapped to reported intervals and 19 of the novel DMRs could be examined. Of these, 47 previously reported and 16 novel DMRs showed a significant allelic count in ChIP-seq data (Fisher’s combined p-value binomial <0.05 with at least 80% of the reads on one allele) (Figure 4a; Supplementary file 6). We also examined if the allelic H3K4me3 and methylation are in opposite alleles in NA12878 and NA19240. Forty of the previously reported DMRs and 10 of the novel DMRs with significant allelic H3K4me3 could be examined in NA12878 and/or NA19240. Thirty-seven previously reported and seven novel DMRs showed opposite allelic states between H3K4me3 and methylation (Figure 4b; Supplementary file 6).
Figure 4
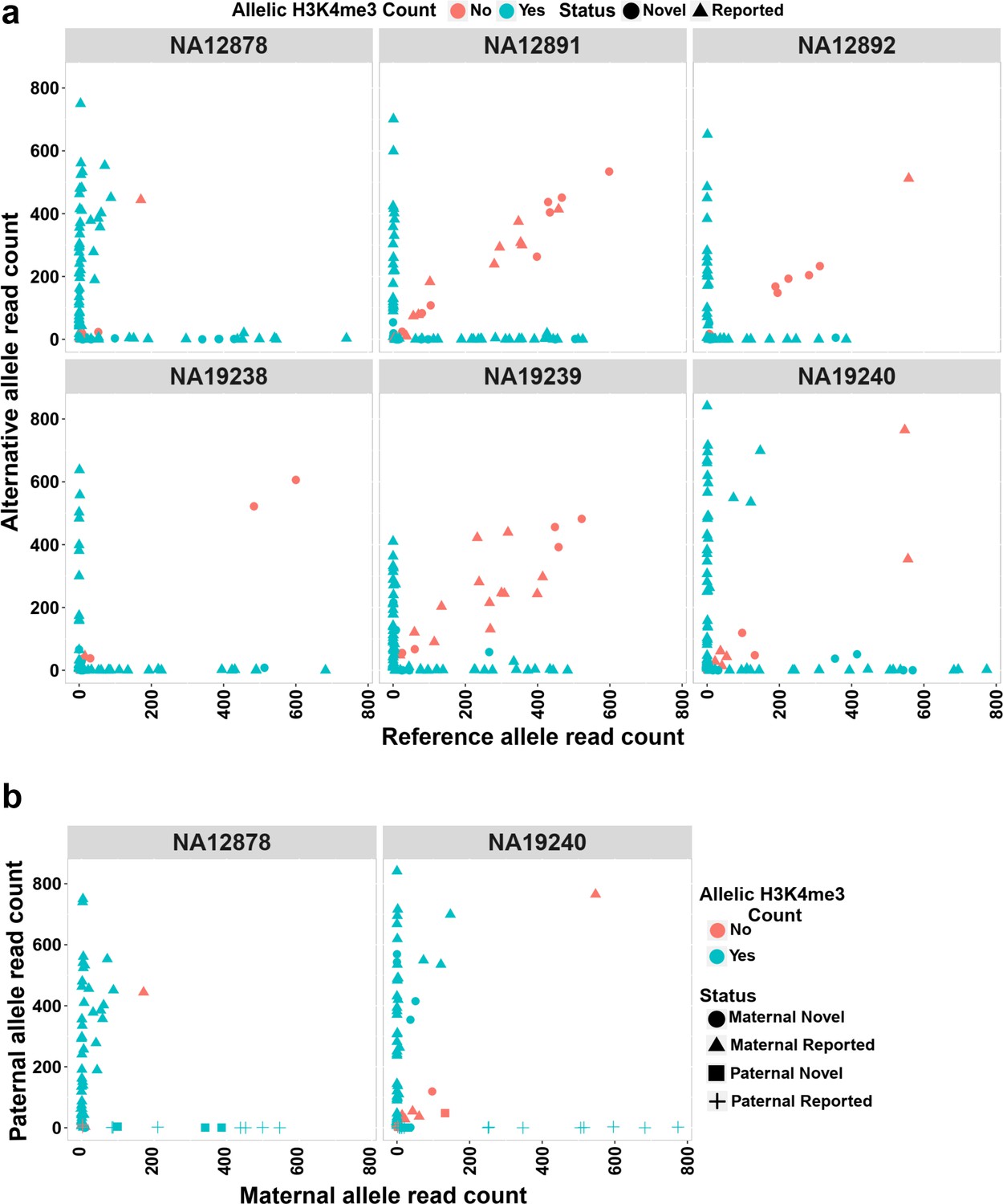
Allelic H3K4me3 histone mark at detected imprinted differentially methylated regions (DMRs).
(a) The plots representing reference and alternative alleles H3K4me3 read counts for the heterozygous single-nucleotide variants (SNVs) mapped to the detected DMRs for the six examined samples. Each point represents an SNV. Blue color displays SNVs with Fisher’s combined p-value binomial <0.05 and at least 80% of the reads on one allele and red color represent those SNVs that did not satisfy either of these thresholds. (b) The plots representing paternal and maternal H3K4me3 read counts for the heterozygous SNVs at DMRs in NA12878 and NA19240. Each point represents an SNV. The ‘Status’ indicates the methylation origin of the DMR and if the DMR is novel or reported.
-
Figure 4—source data 1
H3K4me3 allelic read counts for the heterozygous single-nucleotide variants (SNVs) mapped to the detected DMRs.
- https://cdn.elifesciences.org/articles/77898/elife-77898-fig4-data1-v1.zip
-
Figure 4—source data 2
H3K4me3 allelic read counts for the paternal and maternal heterozygous single-nucleotide variants (SNVs) mapped to the detected DMRs.
- https://cdn.elifesciences.org/articles/77898/elife-77898-fig4-data2-v1.zip
Overall, gDMRs were enriched more with the H3K4me3 mark. Sixty-three percent of the gDMRs and 48% of the sDMRs with at least one heterozygous SNV demonstrated an allelic H3K4me3 mark (Supplementary file 4). This is consistent with previous studies demonstrating the protective role of H3K4me3 against DNA methylation, specifically at germline ICRs in the second round of re-methylation during implantation and somatic development (Chen and Zhang, 2020; Hanna and Kelsey, 2014).
Conservation of detected imprinted DMRs across mammals
To investigate the conservation of detected DMRs and determine if any of the novel DMRs are conserved in mammals, we used WGBS data from mouse (Mus musculus), rhesus macaque (Macaca mulatta), and chimpanzee (Pan troglodytes) (Hon et al., 2013; Jeong et al., 2021; Tung et al., 2012). In determining whether any of the orthologous regions in these mammals displayed partial methylation, we found that 81 of the detected intervals which overlapped with previously reported DMRs and 17 of the novel imprinted DMRs displayed partial methylation in at least one tissue sample in one or more mammals (Figure 5A; Supplementary file 4). In the mouse, orthologs of the 33 detected DMRs were partially methylated, 20 of these were previously reported to be imprinted in mice (Gigante et al., 2019; Xie et al., 2012). Most (88%) of the partially methylated DMRs in the mouse were also partially methylated in rhesus macaque and/or chimpanzee suggesting conservation across species. These shared DMRs mapped to well-known imprinted clusters including KCNQ1, H19, GNAS, SNURF/SNRPN, PLAGL, SGCE, BLCAP, PEG3, PEG10, PEG13, GRB10, BLCAP, NAP1L5, INPP5F, and MEG3 where their allelic PofO expression has already been reported in mouse and other mammals (Geneimprint, 2021; Morison et al., 2001).
Figure 5

Conservation of detected differentially methylated regions (DMRs).
(a) Upset plot representing the number of previously reported and novel DMRs with evidence of conservation (partial methylation) in each of the mammals. (b) Heatmap representing human DMRs (DMR names on the left) and average methylation of their orthologous intervals in mouse and macaque in different tissues and also in sperm, oocyte, and embryonic samples. Gray regions represent NA values that either did not have an ortholog or enough CpG in the whole-genome bisulfite sequencing (WGBS) data.
Sperm, oocyte, and embryo WGBS data for mouse and rhesus macaque were used to investigate if DMRs classified as germline or somatic in humans were still germline or somatic in other mammals and vice versa (Figure 5B; Dahlet et al., 2020; Gao et al., 2017; Jung et al., 2017; Saenz-de-Juano et al., 2019). Overall, imprinted DMRs preserved their identity as germline or somatic in the two other mammals examined (Figure 5B). However, in a few cases, the type of imprinted DMR was not consistent between humans and other mammals (Figure 5B). This finding is supported by an earlier study indicated that imprinting is largely conserved in mammals while the identity of ICR at the germline stage is not completely conserved (Cheong et al., 2015).
Novel DMRs within known imprinted gene domains
To examine the vicinity of novel DMRs to known imprinted genes, we assembled a list of 259 imprinted genes identified in previous studies (Supplementary file 7; Babak et al., 2015; Baran et al., 2015; Geneimprint, 2021; Jadhav et al., 2019; Morison et al., 2001; Zink et al., 2018). Fifteen of the novel DMRs (six germline and nine somatic) identified in our study could be mapped nearby (<1.03 Mb) to known imprinted genes (Table 1; Supplementary file 4).
Novel sDMRs close to known imprinted genes were mostly paternal of origin. Five of them mapped within known imprinted genes including ZDBF2, PAX8/PAX8-AS1, LPAR6/RB1, BMP8A, and ZNF714 while four mapped close to imprinted genes including PWAR1, LINC00665, DGCR6, and IGF2R (Figure 6; Figure 6—figure supplements 1–7). For ZNF714 and PAX8/PAX8-AS1, there are no reported imprinted DMRs within the gene or very close to them that explain their imprinted expression. Two of the novel sDMRs mapped to the promoters of these genes with a reverse relation between origin of methylation and expression (Figure 6), suggesting these DMRs could directly suppress paternal and maternal alleles in PAX8-AS1 and ZNF714, respectively.
Figure 6 with 7 supplements see all
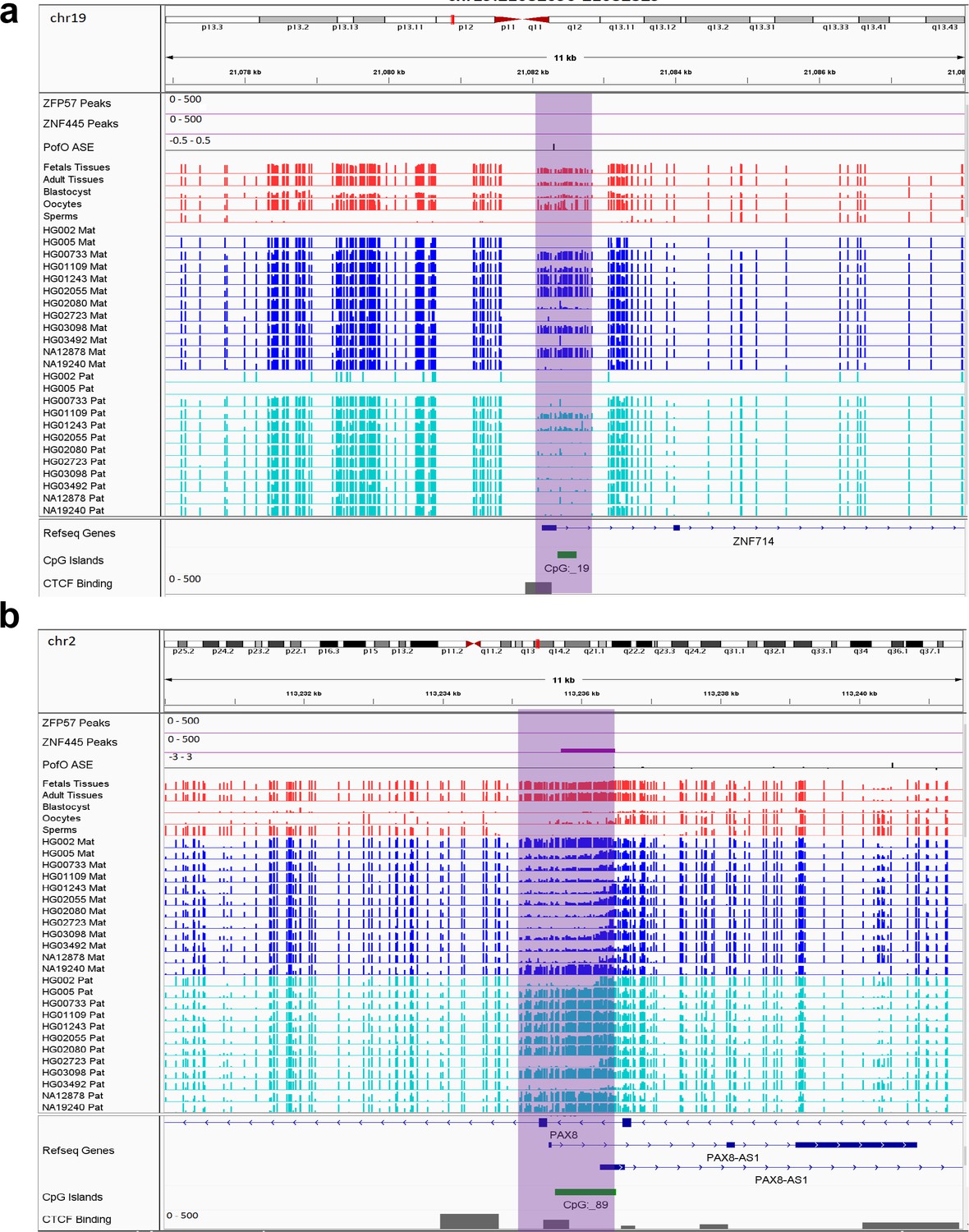
IGV screenshots of two novel somatic differentially methylated regions (DMRs).
(a) Novel maternally methylated somatic DMR overlapping with the promoter of paternally expressed ZNF714 gene. (b) Novel paternally methylated somatic DMR overlapping with the promoter of maternally expressed PAX8-AS1gene. Highlighted box regions represent the DMRs. Parent-of-origin (PofO) allele-specific expression (ASE) track is created using publicly available ASE data from Zink et al., 2018 (see Materials and methods). Positive vertical bars (upward) represent paternal expression and negative bars (downward) represent maternal expression. The range for all methylation tracks is 0–1.
All novel gDMRs close to imprinted genes were maternal of origin. Three of them mapped within known imprinted genes including ACTL10/NECAB3, DDA1, and AC024940.1 while three of them mapped close to imprinted genes including SYCE1, NAPRT, and NTM (Figure 7; Figure 7—figure supplements 1–4). Three of the germline DMRs mapped within or very close to three known imprinted genes without reported ICR including AC024940.1 (OVOS2), ACTL10/NECAB3, and SYCE1. A novel maternal gDMR mapped to the promoter of the paternally expressed ACTL10 (Zink et al., 2018; Figure 7A). In a previous study, a CpG site located ~130 bp away from the DMR we detected was demonstrated to be a cis-methylation quantitative trait loci with PofO association (Cuellar Partida et al., 2018). Thus, the novel gDMR might be the ICR of this gene and directly suppress the maternal allele. Another novel maternal gDMR mapped to the promoter of SYCE1, which demonstrates paternal expression bias in the allele-specific expression (ASE) track (Zink et al., 2018; Figure 7B). Nakabayashi et al., 2011, also observed two array probes consistent with an imprinted DMR at this region, but were unable to validate them because of the difficulty in designing bisulfite PCR primers (Nakabayashi et al., 2011). The novel maternal gDMR at the promoter of SYCE1 could be the ICR for this gene and directly suppress the maternal allele.
Figure 7 with 4 supplements see all
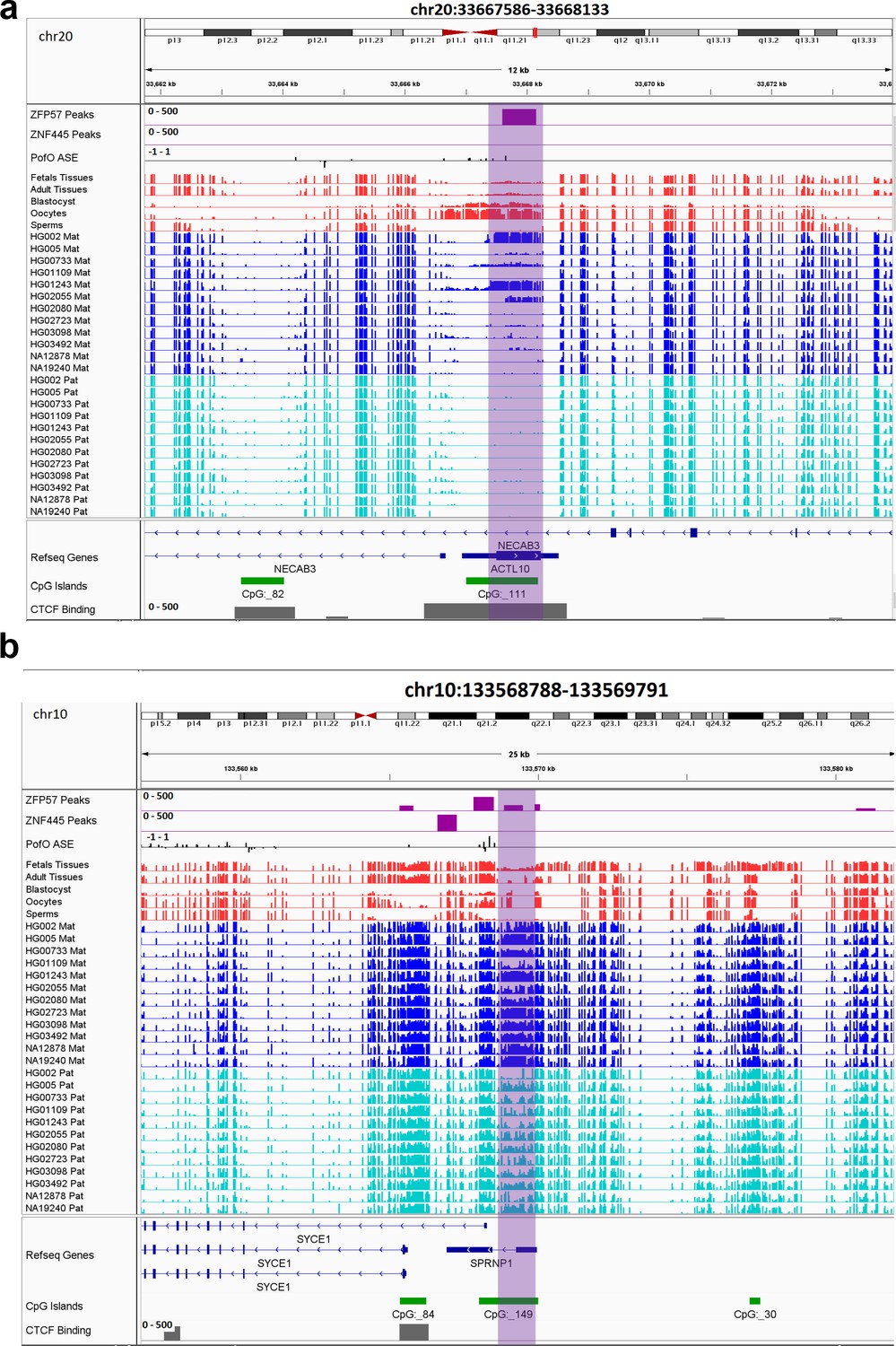
IGV screenshots of two novel maternal germline differentially methylated regions (DMRs).
(a) Novel maternally methylated germline DMR overlapping with the promoter of the paternally expressed ACTL10 gene. (b) Novel maternally methylated germline DMR overlapping with the promoter of the SYCE1 gene, which demonstrates paternal expression bias from parent-of-origin (PofO) allele-specific expression (ASE) track. Highlighted box regions represent the DMRs. PofO ASE track is created using publicly available ASE data from Zink et al., 2018 (see Materials and methods). Positive vertical bars (upward) represent paternal expression and negative bars (downward) represent maternal expression. The range for all methylation tracks is 0–1.
Contiguous blocks of parental methylation bias
Previous studies demonstrated two paradigms of imprinting at the PWS/AS imprinted cluster, either PofO methylation confined to particular regulatory regions such as CGIs or subtle paternal bias across this cluster with spikes of maternal methylation (Court et al., 2014; Joshi et al., 2016; Sharp et al., 2010; Zink et al., 2018). Probes with paternal methylation bias at the SNORD116 cluster have been reported, spanning about a 95 kb region, and paternal deletion of this cluster results in PWS phenotypes (Hernandez Mora et al., 2018; Joshi et al., 2016; Matsubara et al., 2019). Slight hypomethylation of SNORD116 cluster in cases with PWS phenotype and hypermethylation in the cases with AS phenotype have been reported (Matsubara et al., 2019). We did not observe paternal methylation bias across the whole PWS/AS cluster; however, we detected a paternal methylation block spanning ~200 kb, immediately downstream of the known, maternally methylated PWS SNURF/SNRPN ICR (Figure 8). This block encompasses the SNORD116 cluster genes and several other genes such as PWAR1, 5 and 6, PWARSN and IPW. In addition to the PWS/AS block, we detected six other PofO methylation bias blocks ranging from 35 to 65 kb in size, were located within ZNF331, KCNQ1OT1, GNAS/GNAS-AS1, L3MBTL1, ZNF597/NAA60, and GPR1-AS/ZDBF2 imprinted clusters (Figure 8—figure supplements 1–6).
Figure 8 with 6 supplements see all
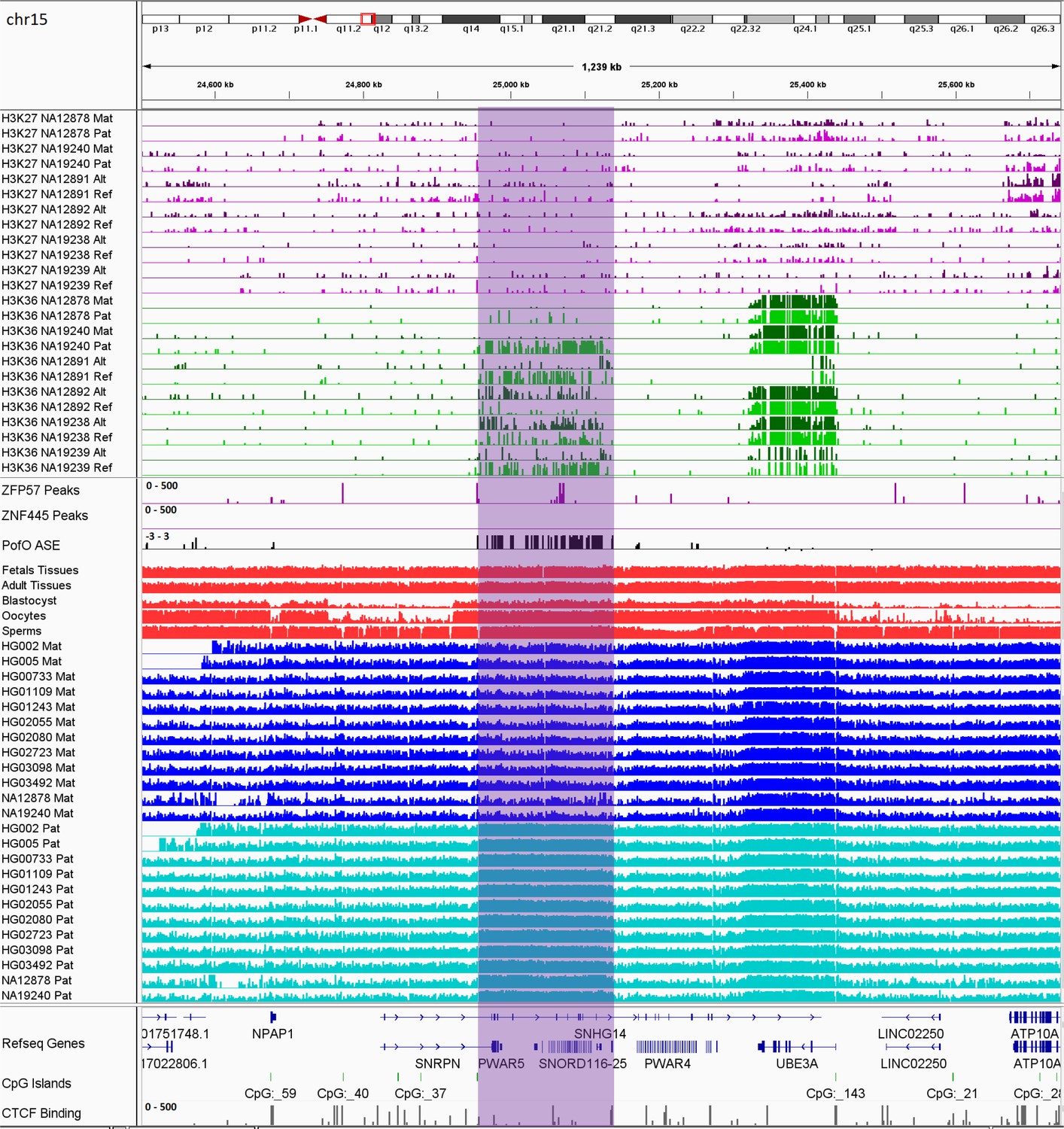
IGV screenshot of 200 kb paternally methylated biased methylation block in the PWS/AS imprinted cluster.
The range for all methylation tracks is 0–1. The histone mark tracks represent allelic read counts for H3K36me3 and H3K27me3 modifications. The range for all histone mark tracks is 0–20. In H3K27 and H3K36 tracks, for NA12878 and NA19240 the parent-of-origin could be determined and specified by maternal (Mat) and paternal (Pat) alleles. While the other samples are specified by reference (Ref) and alternative (Alt) alleles.
As mentioned in the ‘Confirmation of novel imprinted DMRs’ section, only 42 out of 99 detected novel DMRs in the nanopore data could be confirmed in the WGBS data as partially methylated. Forty of the novel nanopore-detected DMRs that did not show partial methylation in the WGBS data mapped to the seven PofO-biased blocks. At imprinted intervals one allele is methylated and the other one is not. Therefore, at these intervals aggregated methylation from both alleles demonstrate partial methylation (~50% methylation) in WGBS data. However, in the subtle PofO bias blocks both alleles are methylated with a subtle hypomethylation on one of the alleles. Therefore, in contrast to imprinted intervals, aggregated methylation at these blocks usually do not show partial methylation in WGBS data. The weaker or subtle differential methylation can therefore explain why several novel DMRs detected in the nanopore data did not show partial methylation in the WGBS data and demonstrates the utility of nanopore sequencing in detecting subtle ASM differences.
Enriched allelic H3K36me3 and H3K27me3 histone marks at contiguous blocks
RNA polymerase II recruits SETD2 during elongation which results in the deposition of the H3K36me3 mark in the gene body. In turn, H3K36me3 recruits de novo DNA methyltransferases through their PWWP domain which results in DNA methylation in the gene body (Wagner and Carpenter, 2012).
Within the seven PofO methylation-biased blocks, parentally expressed or active allele demonstrated hypermethylation suggesting that subtle methylation is linked to parental ASE. Except ZNF597/NAA60, all the blocks demonstrated hypermethylation and ASE on the paternal allele. ZNF597/NAA60 demonstrated hypermethylation and ASE on the maternal allele. Therefore, to assess allelic H3K36me3, we used ChIP-seq data from six LCLs (Kasowski et al., 2013). H3K36me3 and H3K27me3 histone marks are mutually exclusive (Yuan et al., 2011). Moreover, DNA methylation and H3K27me3 shown to be mutually exclusive at CGIs (Brinkman et al., 2012). Thus, we also examined allelic H3K27me3 in the same cell line samples (Kasowski et al., 2013).
To analyze allelic histone modifications and detect blocks of allelic histone marks at large blocks of PofO bias, we binned the genome into 10 kb intervals and performed a binomial test with Fisher’s combined p-value test to determine the significance of allelic read counts at 10 kb intervals with >3 informative heterozygous SNVs (having at least five mapped reads) within each block in each sample. A 10 kb bin considered as significant for allelic histone mark if it had an adjusted p-value <0.001 and if at least 70% of the SNVs within the 10 kb bin having ≥80% of the reads mapped to one allele. In total, 174 bins for H3K36me3 and 132 bins for H3K27me3 could be examined. Of these, 147 bins for H3K36me3 and 51 bins for H3K27me3 were significant. Thirty-eight bins were significant for both histone marks in the same sample. All the seven blocks demonstrated multiple significant bins for H3K36me3 at almost all the samples. L3MBTL1, GPR1-AS/ZDBF2, GNAS/GNAS-AS1, and ZNF597/NAA60 demonstrated multiple significant H3K27me3 bins in majority of the samples and KCNQ1OT1, PWS/AS, and ZNF331 had significant H3K27me3 bins at 3, 2, and 1 of the samples, respectively. H3K36me3 and H3K27me3 demonstrated mutual exclusive pattern and H3K36me3 appeared on the hypermethylated allele while H3K27me3 on the hypomethylated allele (Figure 8; Figure 8—figure supplements 1–6; Figure 9; Supplementary file 8).
Figure 9 with 8 supplements see all

Mutual exclusive allelic H3K36me3 and H3K27me3 histone marks at seven parent-of-origin (PofO) methylation-biased blocks.
All blocks demonstrate allelic H3K36me3 on hypermethylated allele and H3K27me3 on hypomethylated allele. For NA12878 and NA19240, allele1 is the paternal and allele2 is maternal. For sake of visualization in other four cell lines without parental information, allele1 for H3K36me3 mark demonstrates the allele with more mapped reads at all blocks except ZNF597/NAA60. Therefore, for H3K36me3 we swapped the reference allele read count with the alternative allele read count if the reference allele count was less than the alternative allele count. At ZNF597/NAA60, we swapped the reference allele read count with the alternative allele read count if the reference had higher read count. We also swapped the reference and the alternative allele counts for the same SNVs for H3K27me3. Each point represents a heterozygous SNV. Lines are connecting SNVs that have mapped reads for both histone modifications.
-
Figure 9—source data 1
H3K36me3 and H3K27me3 allelic read counts for the heterozygous single-nucleotide variants (SNVs) mapped to the detected PofO-biased methylation blocks.
- https://cdn.elifesciences.org/articles/77898/elife-77898-fig9-data1-v1.zip
To determine if allelic histone marks are unique to the PofO methylation-biased blocks, we examined allelic histone marks on several other imprinted clusters with strong ASE which did not display PofO bias methylation. For this, we examined PPIEL, MEG3, MEST, DIRAS3, IGF2, MTRNR2L4, and ADNP2/PARD6G-AS1 clusters. Eighty-three bins for H3K36me3 and 138 bins for H3K27me3 could be examined at the seven test blocks. Of these, only five bins for H3K36me3 and seven bins for H3K27me3 were significant and none of the bins were significant for both histone marks (Figure 9—figure supplements 1–8; Supplementary file 9). These results suggest that the blocks of PofO methylation bias in the gene body of active alleles are mediated by transcription and histone marks at their gene bodies.
Discussion
Here, we describe the first genome-wide map of human ASM intended to detect novel imprinted intervals using nanopore sequencing. Leveraging long reads and parental SNVs allowed us to phase methylation for ~26.5 million autosomal CpGs representing 95% of the CpGs in the human autosomal genome (GRCh38) across 12 LCLs. This effort achieves a much higher resolution than previous studies aimed at capturing allelic methylation using bisulfite sequencing or methylation arrays (Court et al., 2014; Hernandez Mora et al., 2018; Joshi et al., 2016; Zink et al., 2018). Fourteen of our novel DMRs did not have any phased CpG from previous WGBS or array studies (Supplementary file 4), illustrating the utility of longer reads for imprinted methylation calling. DMRs that are detected in only a single study displayed higher variations across individuals compared to those detected by at least two studies. Therefore, lack of phasing at some novel DMRs in previous studies and higher variation in imprinted methylation at novel DMRs could explain the reason they were not detected previously. We also demonstrated that germline DMRs with a greater number of ZFP57 motif tend to be more consistently imprinted across individuals suggesting motifs redundancy increases ZFP57 recruitment and tolerance to any DNA sequence variation. However, due to the availability of DNA sequence in a limited number of samples, we were not able to examine sequence variation at the DMRs and the ZFP57-binding motifs for any possible association with polymorphic imprinted methylation which will require further study.
Even though we detected methylation for all the CpGs in the human genome (GRCh38), we were not able to phase 5% of the human methylome (Kent et al., 2002). We used SNVs detected from short-reads data in the 1KGP and GIAB databases for phasing (Auton et al., 2015; Zook et al., 2019). Seventy-five percent of the unphased CpGs mapped to the ENCODE blacklist, regions with low mappability, indicative of lack of SNVs to phase reads (Amemiya et al., 2019). Improvement in base calling and variant calling from nanopore reads could enable the phasing of a complete genome-wide methylome using nanopore-detected SNVs.
We detected 16 novel gDMRs and 26 novel sDMRs. These novel DMRs were supported by several lines of evidence in our analyses. (1) They displayed significant PofO methylation bias in nanopore-sequenced cell line samples. (2) They were partially methylated in WGBS data. (3) gDMRs demonstrated establishment of methylation in sperm or oocyte and escape from the second de-methylation step. (4) Eighty-four percent of those for which H3K4me3 ChIP-seq data could be phased and examined showed significant allelic H3K4me3. (5) Forty percent showed evidence of conservation. (6) Eighty-three percent mapped to at least one regulatory region including CGI, CTCF-binding site, and enhancer (Supplementary file 4). These novel DMRs represent a substantial and well-confirmed expansion of known regions of imprinting, which may aid future research and diagnosis in the fields of genetic medicine and oncology.
We detected seven blocks of allelic methylation bias (Figure 8; Figure 8—figure supplements 1–6). All of the blocks represented several common features. (1) They were detected in imprinted genes that appeared in a cluster. (2) There was at least one well-characterized and conserved gDMR in each block (except ZNF597/NAA60 block with a conserved sDMR). (3) The well-characterized DMRs in these blocks displayed significant allelic H3K4me3 (except the DMR in the L3MBTL1 block, which could not be examined due to the lack of an SNV). (4) The well-characterized DMRs in these blocks overlapped with the promoters of genes with subtle PofO methylation bias at the gene body and DMR itself displayed opposite PofO methylation (except for GPR1-AS/ZDBF2 block where DMR did not map to the promoter and had the same PofO with the gene body). (5) All the blocks were accompanied by a strong allelic expression and H3K36me3 histone mark on the subtle hypermethylated allele and H3K27me3 on the hypomethylated allele. This represents a novel facet of imprinting biology and suggests a link between allelic expression and histone modifications with biased PofO methylation at these blocks. However, the mechanism regulating such blocks and the rule of these PofO-biased methylation remain to be determined. One possible explanation could be that the subtle parental methylation bias is used by cells to express important genes (genes that can regulate other genes in the cluster or have regulatory roles) in an imprinted cluster with higher fidelity through its gene body methylation on the active allele. For example, at the KCNQ1OT1 and GNAS/GNAS-AS1 clusters, the methylation blocks overlap with KCNQ1OT1 and GNAS-AS1 gene bodies, both of which encode antisense RNA transcripts that regulate other genes in the imprinted cluster (Chiesa et al., 2012; Turan and Bastepe, 2013).
Orthologous regions of ~40% of the detected DMRs demonstrated partial methylation in one or more of the three mammals including chimpanzee, rhesus macaque, and mouse, suggesting their conservation. There were a considerably higher number of orthologous sites and partially methylated orthologous DMRs in chimpanzee and rhesus macaque, in agreement with more similarities and less distance to these primates compared to the mouse in human evolution. Previously, Court et al., 2014, detected 14 novel DMRs, and did not detect any imprinted orthologs of their novel DMRs in mice (Court et al., 2014). All 14 DMRs also overlapped with our detected DMRs and six of them had orthologous regions in mm10 using the UCSC liftover file (Kent et al., 2002). Two of the orthologs displayed partial methylation in mouse; the first is MEG8 human DMR with its orthologous Rian gene in the mouse, which was not examined by Court et al., 2014, and the other is found in the Htr5a gene, which was previously reported as not conserved in mouse (Court et al., 2014). After reviewing their analysis, Court et al., 2014, seem to have examined different orthologous region (Appendix 2—figure 1). For Htr5a, they examined the CGI (CpG:_102) ~50 kb away from the gene, while we examined the region spanning the first or second exon (two transcripts) of Htr5a which was partially methylated while CpG:_102 was also unmethylated in our study.
Using reported imprinted genes, 36% of the novel DMRs mapped close to known imprinted genes (Babak et al., 2015; Baran et al., 2015; Geneimprint, 2021; Jadhav et al., 2019; Morison et al., 2001; Zink et al., 2018). Five of our novel DMRs could be potential ICRs for reported imprinted genes. Specifically, imprinted DMRs overlapping the promoters of ZNF714, PAX8-AS1, ACTL10, and SYCE1 genes (Figures 6 and 7). ZNF714 is a member of the zinc finger family of proteins which have several imprinted genes with developmental roles (Babak et al., 2015; Baran et al., 2015; Camargo et al., 2012; Jadhav et al., 2019; Zink et al., 2018). ZNF714 has been reported to be associated with non-syndromic cleft lip (Camargo et al., 2012). Aberrant methylation of multiple CpGs overlapping with the novel DMR at PAX8-AS1 has been implicated in thyroid disorders (Candler et al., 2021). SYCE1 and ACTL10 are also implicated in human diseases (Bak et al., 2016; Maor-Sagie et al., 2015). Thus, these imprinted DMRs could be of potential clinical value.
In addition to the aforementioned novel DMRs, two of the reported DMRs in PTCHD3 and FANCC are also interesting. Paternal expression of PTCHD3 and maternal expression for FANCC were previously detected by Zink et al., 2018, though they could not detect any associated DMR due to the lack of phased CpG (Zink et al., 2018). Hernandez Mora et al., 2018, detected three maternally methylated probes at the promoter of PTCHD3 and one maternally methylated probe in intron 1 of FANCC, but were unable to examine the parental expression (Hernandez Mora et al., 2018). We also detected two maternally methylated gDMRs overlapping with the promoter of PCTHD3 and intron 1 of FANCC (Appendix 3—figure 1; Appendix 3—figure 2). Therefore, these gDMRs could be the ICRs for these genes. The maternal gDMR at the PTCHD3 promoter can directly suppress the maternal allele and results in paternal expression. FANCC gDMR overlaps with a CGI and CTCF-binding site. CTCF is a methylation-sensitive DNA-binding protein and CpG methylation can inhibit CTCF binding (Hashimoto et al., 2017; Renda et al., 2007). Moreover, CTCF binding to the first intron of major immediate early (MIE) gene of the human cytomegalovirus (HCMV) in HCMV-infected cells resulted in repression of this gene (Puerta et al., 2014). Therefore, the maternally methylated DMR in intron 1 of maternally expressed FANCC suggests a mechanism through which the paternal allele is suppressed by CTCF binding at DMR while DNA methylation inhibits CTCF binding at the maternal allele.
Overall, our study demonstrates a near-complete genome-wide map of human ASM by leveraging long-read nanopore technology. The use of nanopore technology allowed us to expand the set of known imprinted DMRs using 12 LCLs with parental SNPs. Moreover, we detected seven large PofO bias methylation blocks with enriched allelic expression and histone modifications. We showed that nanopore sequencing has the ability to achieve a higher resolution of phased CpGs using a small sample size and allows for the calling of imprinted methylation in a single sample, potentially reducing the cost by reducing the sample size.
Materials and methods
Nanopore sequencing data and detection of ASM
Request a detailed protocolWe used publicly available nanopore sequencing data for 12 LCLs with trio data available. Raw and base-called nanopore data for HG002, HG005, HG00733, HG01109, HG01243, HG02055, HG02080, HG02723, HG03098, and HG03492 were obtained from the Human Pangenomics and GIAB (Shafin et al., 2020; Zook et al., 2016). NA19240 data (ERR3046934 and ERR3046935 raw nanopore and their base-called reads ERR3219853 and ERR3219854) were obtained from De Coster et al., 2019. Raw and base-called nanopore data for NA12878 were obtained from rel6 nanopore WGS consortium (Jain et al., 2018). Reads were mapped to the GRCh38 human reference genome using Minimap2 with the setting minimap2 –ax map-ont (Kent et al., 2002; Li, 2018). For all the cell lines and their parents, except HG002 and HG005, high-quality SNVs were called using Strelka2 with default parameters from alignment files in the 1KGP GRCh38 (Auton et al., 2015; Kim et al., 2018). High-quality SNVs for HG002 and HG005 and their parents were obtained from GIAB v.3.3.2 high confidence variant calls (Zook et al., 2019). CpG methylations were called from nanopore data using nanopolish with default parameters (Simpson et al., 2017). Methylation calls for each sample were preprocessed using the NanoMethPhase methyl_call_processor default setting (Akbari et al., 2021). Subsequently, haplotyping and PofO methylation detection were performed using NanoMethPhase and trio (mother, father, and child) variant call data with the setting nanomethphase phase –mbq 0. Finally, DMRs between haplotypes were called using the default setting of NanoMethPhase dma module that uses Dispersion Shrinkage for Sequencing data R package for DMA (Park and Wu, 2016). To avoid the confounding effects of X-chromosome inactivation, and because previous studies demonstrated no evidence of imprinting at sex chromosomes, we only examined autosomal chromosomes (Court et al., 2014; Joshi et al., 2016; Zink et al., 2018).
WGBS data and detection of novel DMRs
Request a detailed protocolTo confirm allelic methylation in other tissues and also detect potential novel imprinted DMRs, we used 60 public WGBS data records for 29 tissue type samples from the Epigenomics Roadmap and ENCODE projects (Supplementary file 10) and 119 blood WGBS datasets for 87 individuals from the Blueprint project (Bernstein et al., 2010; ENCODE Project Consortium, 2012; Stunnenberg et al., 2016; Supplementary file 10). CpGs with at least five mapped reads were used for further analysis. At imprinted DMRs, only one allele is methylated and we expect to observe partial methylation (~50%) at such regions. Therefore, we investigated the partial methylation of nanopore-detected DMRs in WGBS data (code is available on https://github.com/vahidAK/NanoMethPhase/tree/master/scripts (Akbari, 2022): PartialMethylation_AtDMR.sh). As controls, we examined 100 randomly selected CGIs: 1, 2, and 3 kb intervals with more than 15 CpGs each resampled 100 times.
Detection of gDMRs and sDMRs
Request a detailed protocolTo discriminate gDMRs from sDMRs, we used publicly available WGBS data for three sperms, two oocytes, and one blastocyst first published by Okae et al., 2014, and three fetal tissue libraries (GSM1172595 thymus, GSM1172596 muscle, and GSM941747 brain) from the Roadmap project (Bernstein et al., 2010; Okae et al., 2014).
Allelic H3K4me3, H3K36me3, and H3K27me3 analysis
Request a detailed protocolH3K4me3, H3K36me3, and H3K27me3 ChIP-seq fastq files were obtained for NA12878, NA12891, NA12892, NA19238, NA19239, and NA19240 (SRP030041) (Kasowski et al., 2013). ChIP-seq data were aligned to the GRCh38 reference genome using the bwa-mem default setting (Kent et al., 2002; Li and Durbin, 2009). High-quality SNVs were called for these samples from 1KGP GRCh38 alignment files using strelka2 (Auton et al., 2015; Kim et al., 2018). We then counted the number of reads with a minimum mapping quality of 20 and base quality of 10 at each heterozygous SNV and kept those with at least five mapped reads. The reference allelic counts and total counts at each heterozygous SNV were used to detect significant allelic bias using a two-sided binomial test under the default probability of p=0.5 in python SciPy package (codes are available on GitHub https://github.com/vahidAK/NanoMethPhase/tree/master/scripts: CountReadsAtSNV.py & Binomial_test.py) (Virtanen et al., 2020).
ASE track
Request a detailed protocolASE data from Zink et al., 2018 (PofO_ASE.tsv; https://doi.org/10.6084/m9.figshare.6816917) were used to create ASE track for IGV. In PofO_ASE.tsv file from Zink et al., they have calculated lor_paternal_maternal across individuals which is (lor_ref_alt_pref - lor_ref_alt_palt)/2. lor_ref_alt_pref is log(#reads with ref allele/#reads with alt allele) when paternal homologue has ref allele and lor_ref_alt_palt is log(#reads with ref allele/#reads with alt allele) when paternal homologue has alt allele. For visualization in IGV, we converted the PofO_ASE.tsv file from Zink et al., to a bigwig format file using the UCSC tool bedGraphToBigWig version 4 and we kept lor_paternal_maternal as ASE value (Kent et al., 2010).
Mammalian conservation of DMRs
Request a detailed protocolWe used 16 WGBS datasets for mouse (GSM1051150-60 and GSM1051162-66), 34 WGBS datasets for rhesus macaque (GSE34128 and GSE151768), and 22 WGBS datasets for chimpanzee (GSE151768) to examine partial methylation in orthologous intervals (Hon et al., 2013; Jeong et al., 2021; Tung et al., 2012). Mouse, macaque, and chimpanzee coordinates lifted over to mm10, RheMac8, and PanTro5 coordinates using CrossMap and the appropriate liftover file from the UCSC genome browser. The list of detected human DMRs were also converted to the orthologous regions for each mammal using CrossMap and the appropriate liftover file (Kent et al., 2002; Zhao et al., 2014). Since many coordinates in the human splitted to several orthologs in other mammals, we merged orthologs that were ≤200 bp apart.
To examine the somatic and germline ortholog DMRs, we used embryo (GSM3752614, GSM4558210), sperm (GSE79226), and oocyte (GSM3681773, GSM3681774, GSM3681775) WGBS libraries from mouse; and embryo (GSM1466814), sperm (GSM1466810), and oocyte (GSM1466811) WGBS libraries from rhesus macaque (Dahlet et al., 2020; Gao et al., 2017; Jung et al., 2017; Saenz-de-Juano et al., 2019).
Appendix 1
Appendix 1—figure 1
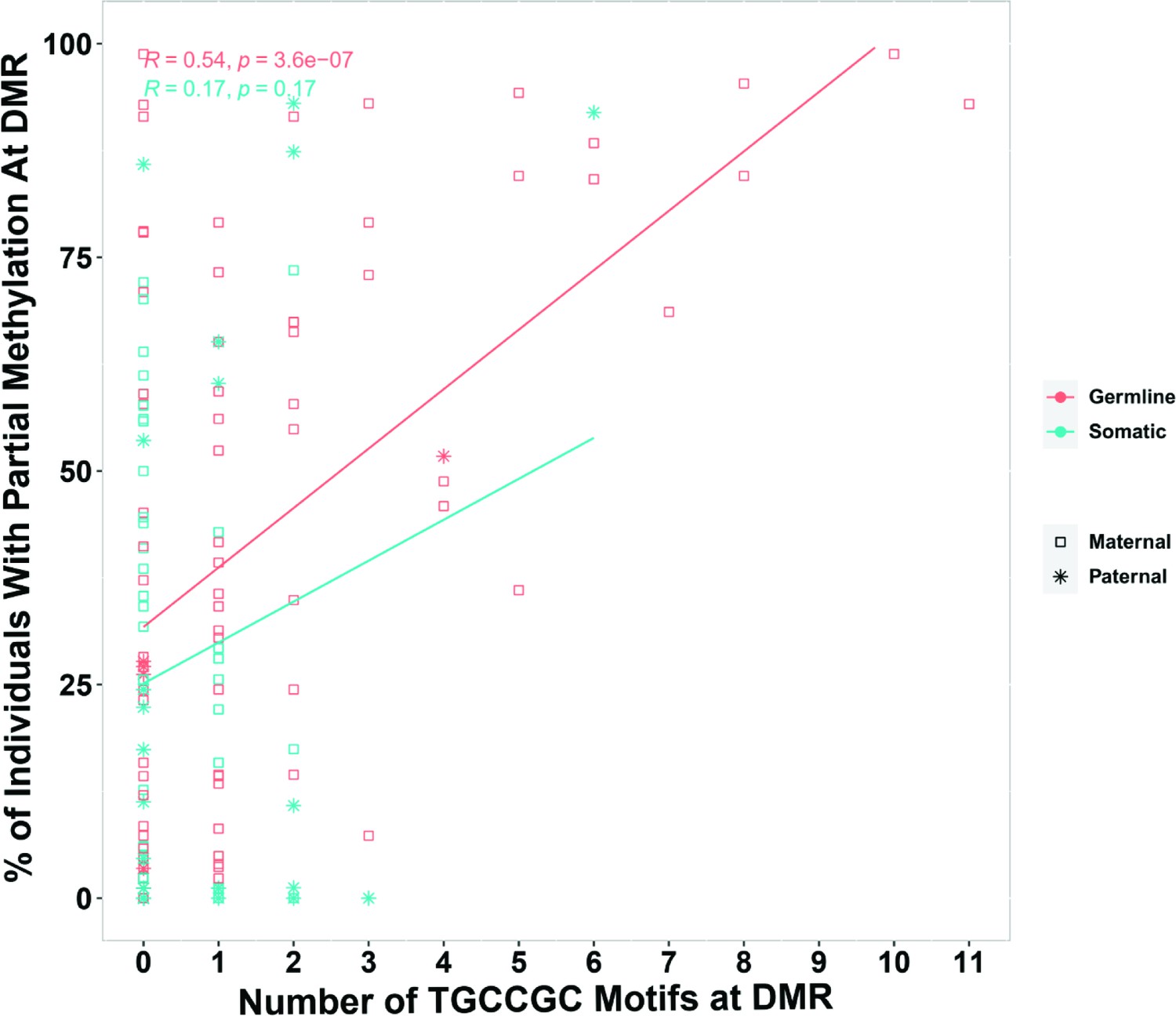
Pearson correlation for the number of ZFP57-binding motif (TGCCGC) at differentially methylated regions (DMRs) and percent of individuals that demonstrated partial methylation in their whole-genome bisulfite sequencing data at the DMRs.
-
Appendix 1—figure 1—source data 1
Number of ZFP57-binding motif (TGCCGC) at differentially methylated regions (DMRs).
- https://cdn.elifesciences.org/articles/77898/elife-77898-app1-fig1-data1-v1.zip
Appendix 2
Appendix 2—figure 1
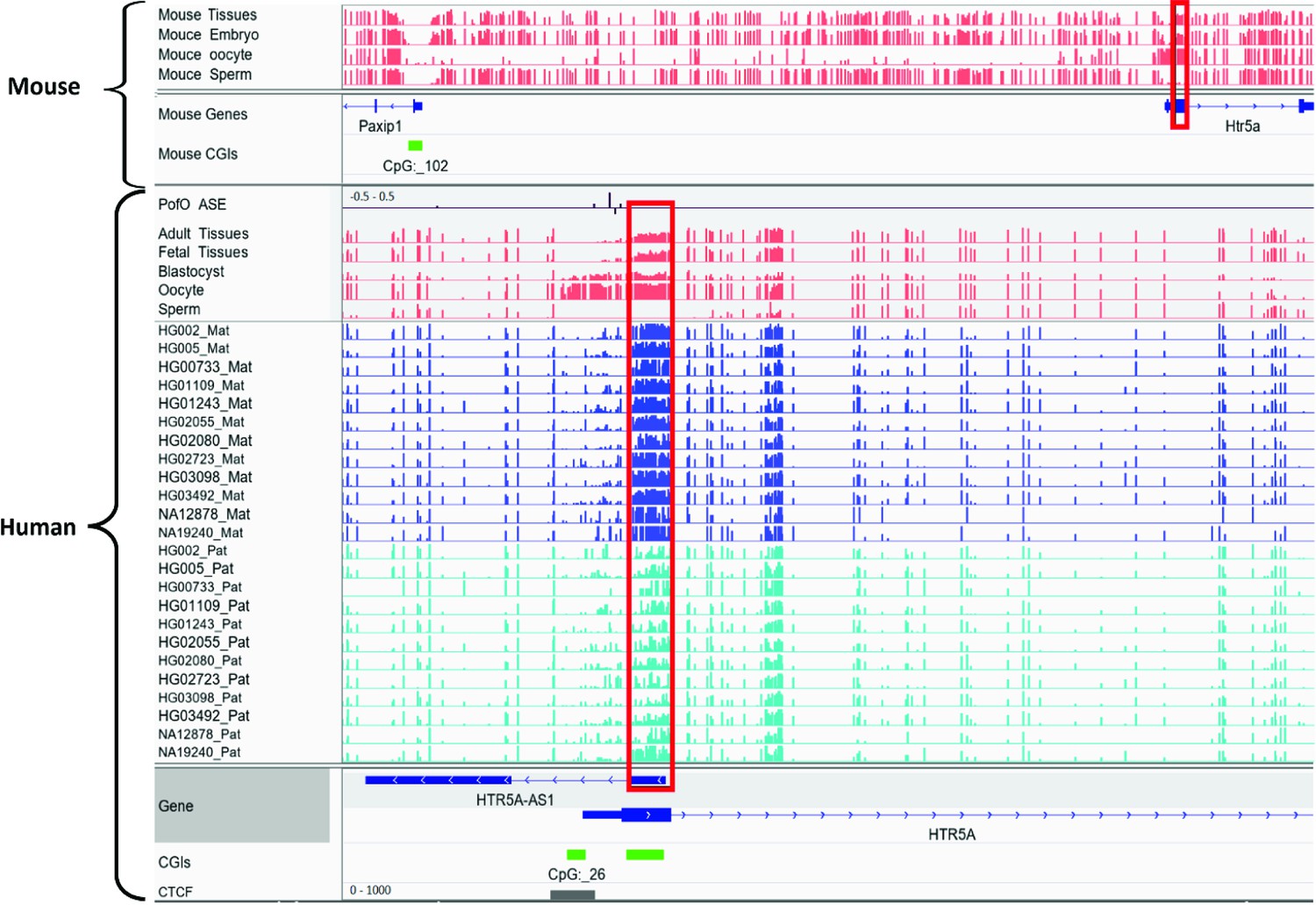
Conserved differentially methylated region (DMR) at HTR5A in human and mouse.
The known DMR at HTR5A reported to be not conserved in mouse by Court et al. (PMID: 24402520; see supplementary figure S6 from Court et al., 2014), however we detected it as conserved due to a different orthologous examined in our study. Court et al. examined CGI 102 which is also not imprinted in our analysis, however the ortholog we examined spans beginning of the HTR5A and is partially methylated which suggests the region is imprinted. Red boxes are showing germline DMRs. The range for all methylation tracks is 0–1. In parent-of-origin (PofO) ASE track, positive or upward bars represent paternal expression bias and negative or downward bars represent maternal expression bias.
Appendix 3
Appendix 3—figure 1
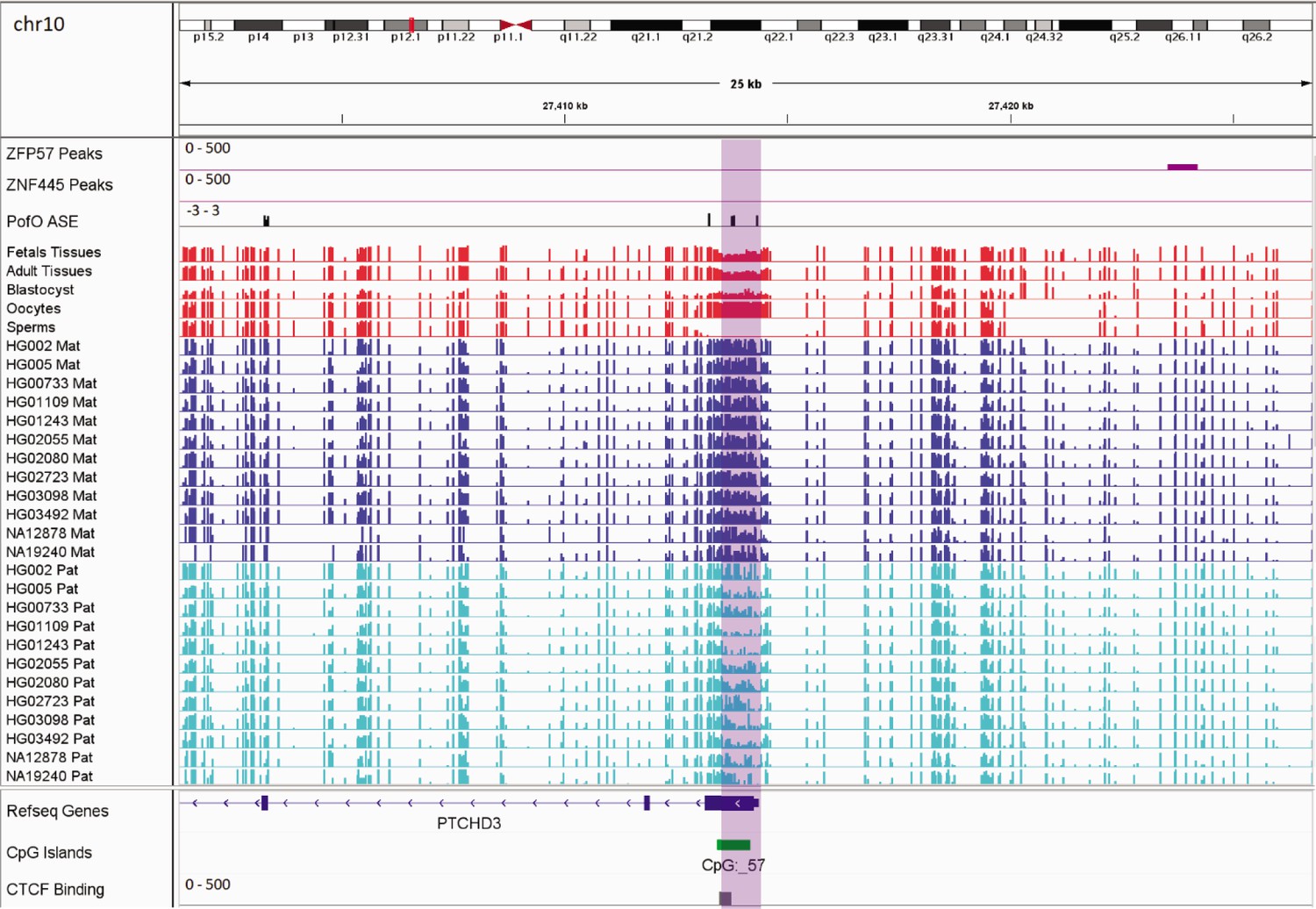
Germline maternally methylated differentially methylated region (DMR) in the promoter of paternally expressed PTCHD3 gene.
The range for all methylation tracks is 0–1. In parent-of-origin (PofO) ASE track, positive or upward bars represent paternal expression bias and negative or downward bars represent maternal expression bias.
Appendix 3—figure 2
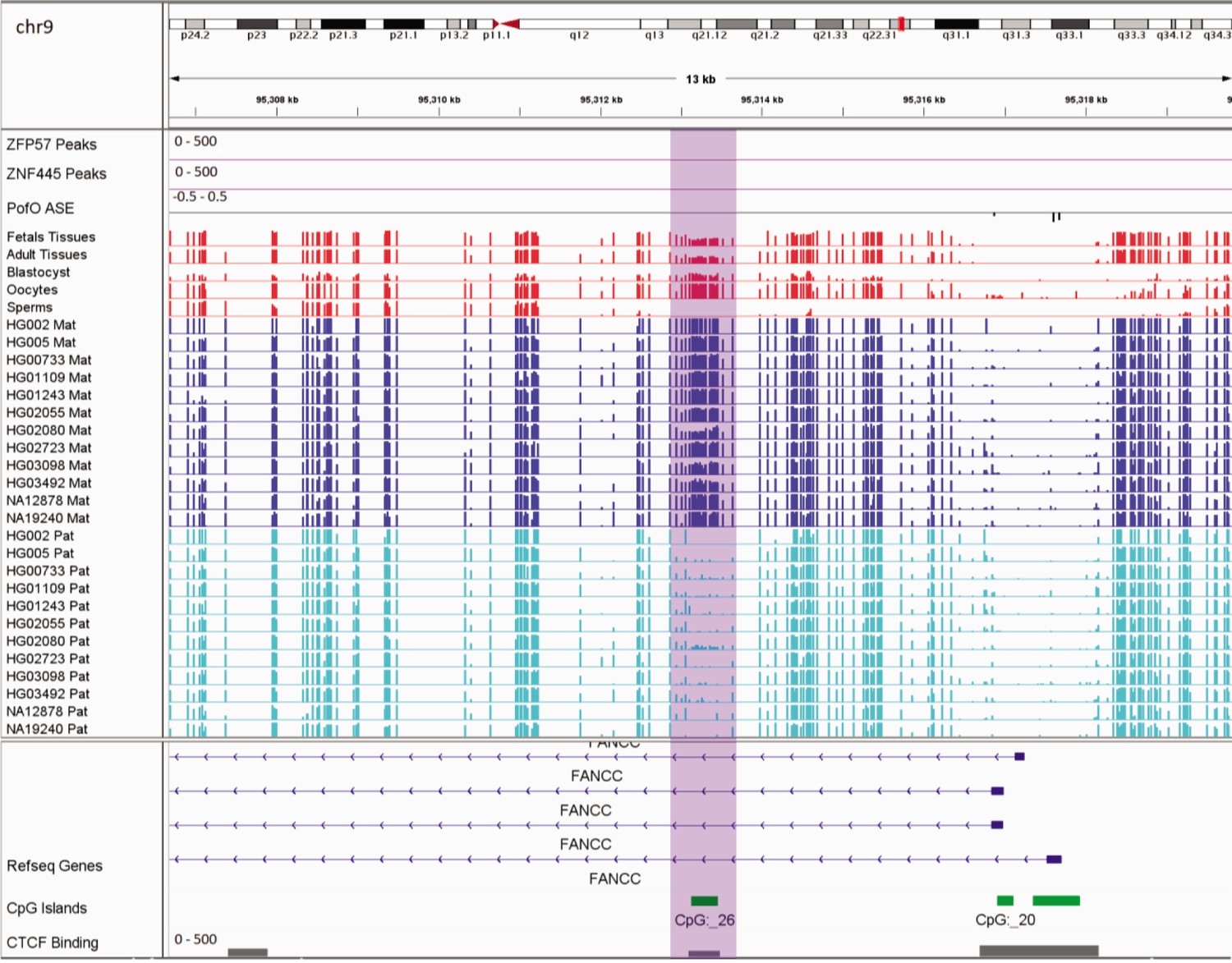
Germline maternally methylated differentially methylated region (DMR) in the intron 1 of maternally expressed FANCC gene.
The range for all methylation tracks is 0–1. In parent-of-origin (PofO) ASE track, positive or upward bars represent paternal expression bias and negative or downward bars represent maternal expression bias.
Data availability
The current manuscript is a computational study, so no new datasets have been generated for this manuscript. The source of each dataset is provided under the ‘“Materials and methods’” section under the appropriate subsection. Genomic tracks generated in this study including DNA methylation and histone modification tracks are deposited in the Mendeley data repository (https://doi.org/10.17632/f4k2gytbh5.1). Codes are uploaded to GitHub https://github.com/vahidAK/NanoMethPhase/tree/master/scripts (copy archived at swh:1:rev:1657f7aed60604aa7c7f3e77d992d76bee6bf6d3): PartialMethylation_AtDMR.sh, CountReadsAtSNV.py and Binomial_test.py.
-
Mendeley DataGenome-Wide Detection of Imprinted Differentially Methylated Regions Using Nanopore Sequencing_Akbari-etal.https://doi.org/10.17632/f4k2gytbh5.1
-
Human Pangenome Reference ConsortiumID hpgp-data. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes.
-
Nanopore WGS ConsortiumID NA12878. Nanopore sequencing and assembly of a human genome with ultra-long reads.
-
European Nucleotide ArchiveID PRJEB26791. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome.
-
NCBI Genome in a Bottle FTPID FTP. Extensive sequencing of seven human genomes to characterize benchmark reference materials.
-
The International Genome Sample ResourceID 30x-grch38. A global reference for human genetic variation.
-
Blueprint EpigenomeID blueprint. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery.
-
NCBI Gene Expression OmnibusID epigenomics. The NIH Roadmap Epigenomics Mapping Consortium.
-
ENCODEID encodeproject. An Integrated Encyclopedia of DNA Elements in the Human Genome.
-
DNA Data Bank of JapanID DRA003802. Genome-wide analysis of DNA methylation dynamics during early human development.
-
NCBI Sequence Read ArchiveID SRP030041. Extensive Variation in Chromatin States Across Humans.
-
NCBI Gene Expression OmnibusID GSE42836. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues.
-
NCBI Gene Expression OmnibusID GSE34128. Social environment is associated with gene regulatory variation in the rhesus macaque immune system.
-
NCBI Gene Expression OmnibusID GSE151768. Evolution of DNA methylation in the human brain.
-
NCBI Gene Expression OmnibusID GSE130735. Genome-wide analysis in the mouse embryo reveals the importance of DNA methylation for transcription integrity.
-
NCBI Gene Expression OmnibusID GSE79226. Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes.
-
NCBI Gene Expression OmnibusID GSE128656. Genome-wide assessment of DNA methylation in mouse oocytes reveals effects associated with in vitro growth, superovulation, and sexual maturity.
-
NCBI Gene Expression OmnibusID GSE60166. De novo DNA methylation during monkey pre-implantation embryogenesis.
-
figshareMethylation and expression data for whole-genome human imprinting study.https://doi.org/10.6084/m9.figshare.6816917
-
NCBI Gene Expression OmnibusID GSE78099. ChIP-exo of human KRAB-ZNFs transduced in HEK 293T cells and KAP1 in hES H1 cells.
-
NCBI Gene Expression OmnibusID GSE115387. ZNF445 is a primary regulator of genomic imprinting.
References
-
SoftwareNanoMethPhase, version swh:1:rev:1657f7aed60604aa7c7f3e77d992d76bee6bf6d3Software Heritage.
-
Mammalian genomic imprintingCold Spring Harbor Perspectives in Biology 3:a002592.https://doi.org/10.1101/cshperspect.a002592
-
The NIH Roadmap Epigenomics Mapping ConsortiumNature Biotechnology 28:1045–1048.https://doi.org/10.1038/nbt1010-1045
-
GWAS reveals new recessive loci associated with non-syndromic facial cleftingEuropean Journal of Medical Genetics 55:510–514.https://doi.org/10.1016/j.ejmg.2012.06.005
-
Maternal H3K27me3-dependent autosomal and X chromosome imprintingNature Reviews. Genetics 21:555–571.https://doi.org/10.1038/s41576-020-0245-9
-
Random and non-random monoallelic expressionNeuropsychopharmacology 38:55–61.https://doi.org/10.1038/npp.2012.85
-
Genome-wide survey of parent-of-origin effects on DNA methylation identifies candidate imprinted loci in humansHuman Molecular Genetics 27:2927–2939.https://doi.org/10.1093/hmg/ddy206
-
The influence of DNA methylation on monoallelic expressionEssays in Biochemistry 63:663–676.https://doi.org/10.1042/EBC20190034
-
Using long-read sequencing to detect imprinted DNA methylationNucleic Acids Research 47:e46.https://doi.org/10.1093/nar/gkz107
-
Nanopore sequencing and assembly of a human genome with ultra-long readsNature Biotechnology 36:338–345.https://doi.org/10.1038/nbt.4060
-
Loss of imprinting and cancerThe Journal of Pathology 211:261–268.https://doi.org/10.1002/path.2116
-
Evolution of DNA methylation in the human brainNature Communications 12:2021.https://doi.org/10.1038/s41467-021-21917-7
-
Developmental regulation of somatic imprintsDifferentiation 81:270–280.https://doi.org/10.1016/j.diff.2011.01.007
-
DNA Methylation Profiling of Uniparental Disomy Subjects Provides a Map of Parental Epigenetic Bias in the Human GenomeAmerican Journal of Human Genetics 99:555–566.https://doi.org/10.1016/j.ajhg.2016.06.032
-
Extensive variation in chromatin states across humansScience (New York, N.Y.) 342:750–752.https://doi.org/10.1126/science.1242510
-
BigWig and BigBed: enabling browsing of large distributed datasetsBioinformatics (Oxford, England) 26:2204–2207.https://doi.org/10.1093/bioinformatics/btq351
-
Parallels between Mammalian Mechanisms of Monoallelic Gene ExpressionTrends in Genetics 34:954–971.https://doi.org/10.1016/j.tig.2018.08.005
-
Dynamic changes in the human methylome during differentiationGenome Research 20:320–331.https://doi.org/10.1101/gr.101907.109
-
Fast and accurate short read alignment with Burrows-Wheeler transformBioinformatics (Oxford, England) 25:1754–1760.https://doi.org/10.1093/bioinformatics/btp324
-
Minimap2: pairwise alignment for nucleotide sequencesBioinformatics (Oxford, England) 34:3094–3100.https://doi.org/10.1093/bioinformatics/bty191
-
Deleterious mutation in SYCE1 is associated with non-obstructive azoospermiaJournal of Assisted Reproduction and Genetics 32:887–891.https://doi.org/10.1007/s10815-015-0445-y
-
DNA methylation and its basic functionNeuropsychopharmacology 38:23–38.https://doi.org/10.1038/npp.2012.112
-
The imprinted gene and parent-of-origin effect databaseNucleic Acids Research 29:275–276.https://doi.org/10.1093/nar/29.1.275
-
Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genesHuman Molecular Genetics 20:3188–3197.https://doi.org/10.1093/hmg/ddr224
-
Differential methylation analysis for BS-seq data under general experimental designBioinformatics (Oxford, England) 32:1446–1453.https://doi.org/10.1093/bioinformatics/btw026
-
CTCF Binding to the First Intron of the Major Immediate Early (MIE) Gene of Human Cytomegalovirus (HCMVNegatively Regulates MIE Gene Expression and HCMV Replication. Journal of Virology 88:7389–7401.https://doi.org/10.1128/JVI.00845-14
-
Critical DNA Binding Interactions of the Insulator Protein CTCFJournal OF Biological Chemistry 282:33336–33345.https://doi.org/10.1074/jbc.M706213200
-
Detecting DNA cytosine methylation using nanopore sequencingNature Methods 14:407–410.https://doi.org/10.1038/nmeth.4184
-
DNA methylation: roles in mammalian developmentNature Reviews. Genetics 14:204–220.https://doi.org/10.1038/nrg3354
-
ZNF445 is a primary regulator of genomic imprintingGenes & Development 33:49–54.https://doi.org/10.1101/gad.320069.118
-
The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted geneHormone Research in Paediatrics 80:229–241.https://doi.org/10.1159/000355384
-
Understanding the language of Lys36 methylation at histone H3Nature Reviews. Molecular Cell Biology 13:115–126.https://doi.org/10.1038/nrm3274
-
H3K36 methylation antagonizes PRC2-mediated H3K27 methylationThe Journal of Biological Chemistry 286:7983–7989.https://doi.org/10.1074/jbc.M110.194027
-
CrossMap: a versatile tool for coordinate conversion between genome assembliesBioinformatics (Oxford, England) 30:1006–1007.https://doi.org/10.1093/bioinformatics/btt730
-
An open resource for accurately benchmarking small variant and reference callsNature Biotechnology 37:561–566.https://doi.org/10.1038/s41587-019-0074-6
Article and author information
Author details
Funding
The University of British Columbia, 4-Year Doctoral Fellowship
- Vahid Akbari
Canada Research Chairs
- Marco A Marra
- Steven JM Jones
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
SJMJ and MAM acknowledge funding from the Canada Research Chairs program and the Canadian Foundation for Innovation. VA acknowledges funding from the University of British Columbia with a Four-Year Doctoral Fellowship.
Copyright
© 2022, Akbari et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,591
- views
-
- 598
- downloads
-
- 36
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Genome-wide detection of imprinted differentially methylated regions using nanopore sequencing
eLife 11:e77898.
https://doi.org/10.7554/eLife.77898
Further reading
-
- Computational and Systems Biology
- Genetics and Genomics
Obesity is a major risk factor for type 2 diabetes, dyslipidemia, cardiovascular disease, and hypertension. Intriguingly, there is a subset of metabolically healthy obese (MHO) individuals who are seemingly able to maintain a healthy metabolic profile free of metabolic syndrome. The molecular underpinnings of MHO, however, are not well understood. Here, we report that CTRP10/C1QL2-deficient mice represent a unique female model of MHO. CTRP10 modulates weight gain in a striking and sexually dimorphic manner. Female, but not male, mice lacking CTRP10 develop obesity with age on a low-fat diet while maintaining an otherwise healthy metabolic profile. When fed an obesogenic diet, female Ctrp10 knockout (KO) mice show rapid weight gain. Despite pronounced obesity, Ctrp10 KO female mice do not develop steatosis, dyslipidemia, glucose intolerance, insulin resistance, oxidative stress, or low-grade inflammation. Obesity is largely uncoupled from metabolic dysregulation in female KO mice. Multi-tissue transcriptomic analyses highlighted gene expression changes and pathways associated with insulin-sensitive obesity. Transcriptional correlation of the differentially expressed gene (DEG) orthologs in humans also shows sex differences in gene connectivity within and across metabolic tissues, underscoring the conserved sex-dependent function of CTRP10. Collectively, our findings suggest that CTRP10 negatively regulates body weight in females, and that loss of CTRP10 results in benign obesity with largely preserved insulin sensitivity and metabolic health. This female MHO mouse model is valuable for understanding sex-biased mechanisms that uncouple obesity from metabolic dysfunction.
-
- Computational and Systems Biology
Mass spectrometry imaging (MSI) is a powerful technology used to define the spatial distribution and relative abundance of metabolites across tissue cryosections. While software packages exist for pixel-by-pixel individual metabolite and limited target pairs of ratio imaging, the research community lacks an easy computing and application tool that images any metabolite abundance ratio pairs. Importantly, recognition of correlated metabolite pairs may contribute to the discovery of unanticipated molecules in shared metabolic pathways. Here, we describe the development and implementation of an untargeted R package workflow for pixel-by-pixel ratio imaging of all metabolites detected in an MSI experiment. Considering untargeted MSI studies of murine brain and embryogenesis, we demonstrate that ratio imaging minimizes systematic data variation introduced by sample handling, markedly enhances spatial image contrast, and reveals previously unrecognized metabotype-distinct tissue regions. Furthermore, ratio imaging facilitates identification of novel regional biomarkers and provides anatomical information regarding spatial distribution of metabolite-linked biochemical pathways. The algorithm described herein is applicable to any MSI dataset containing spatial information for metabolites, peptides or proteins, offering a potent hypothesis generation tool to enhance knowledge obtained from current spatial metabolite profiling technologies.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}