Machine Learning: Lighting up protein design
Using a neural network to predict how green fluorescent proteins respond to genetic mutations illuminates properties that could help design new proteins.
- MRC Human Genetics Unit, The University of Edinburgh, United Kingdom
Protein engineering is a growing area of research in which scientists use a variety of methods to design new proteins that can perform certain functions. For instance, enzymes that can biodegrade plastics, materials inspired by spider silk, or antibodies to neutralize viruses (Lu et al., 2022; Shan et al., 2022).
In the past, protein engineering has commonly relied on directed evolution, a laboratory procedure that mimics natural selection. This involves randomly mutating the genetic sequence of a naturally occurring protein to create multiple variants with slightly different amino acids. Various selection pressures are then applied to identify the ‘fittest’ variants that best carry out the desired role (Chen et al., 2018). Alternatively, researchers can use a rational design approach, in which new proteins are built using principles learned from the study of known protein structures (Anishchenko et al., 2021).
Now, in eLife, Fyodor A Kondrashov (from the Institute of Science and Technology Austria and the Okinawa Institute of Science and Technology Graduate University) and colleagues – including Louisa Gonzalez Somermeyer as first author – have combined elements of both approaches to engineer new variants of naturally occurring green fluorescent proteins (GFP; Gonzalez Somermeyer et al., 2022). First, the team (who are based at various institutes in Austria, Japan, the United States, the United Kingdom, Germany and Russia) generated tens of thousands of GFP variants that differed from each other by three to four mutations on average, and measured their fluorescence. This was used to create a ‘fitness landscape’ showing how the genetic sequence of each mutant relates to its performance (Figure 1). The data was then fed in to a neural network that can expand the landscape by predicting the performance of variants that were not observed experimentally.
Figure 1
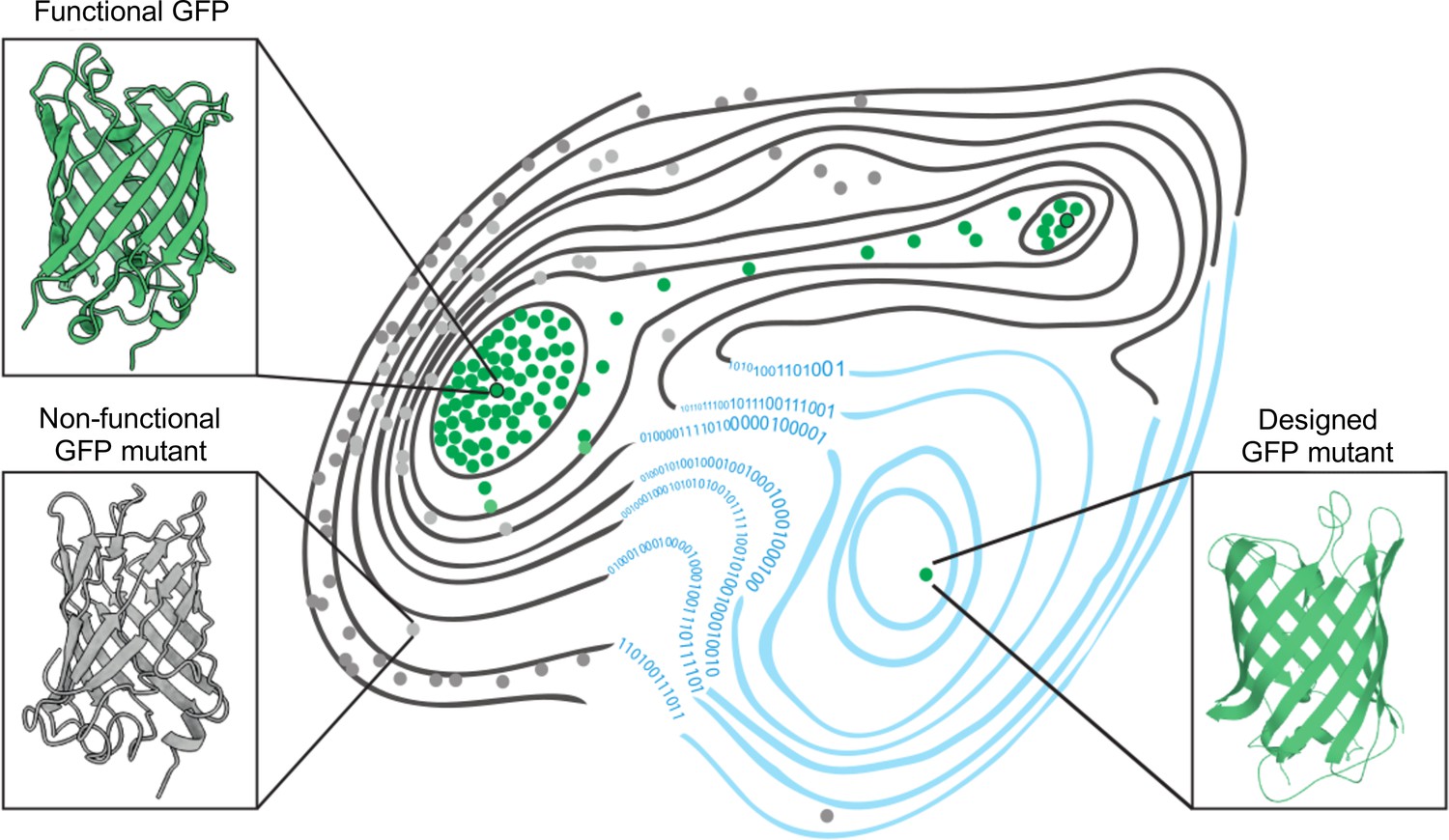
The fitness landscape of green fluorescent proteins.
Fitness landscapes provide a graphical representation of how a protein’s genetic sequence relates to its performance, leading to a multidimensional surface made up of peaks, ridges, and valleys. In the fitness landscape shown, horizontal distance represents the number of mutations that separate variants of a protein, while vertical elevation represented by contour lines indicates the fluorescence of each mutant. Two naturally occurring green fluorescent proteins (GFPs; dots outlined in black) reside on different peaks of the landscape (top left and top right) and are connected by a narrow ridge (area of high fitness). Mutant proteins at the peaks and ridges are all functional and able to fluoresce (green dots), whereas those in the valleys are non-functioning (grey dots). Application of a machine learning algorithm expanded the fitness landscape (right; blue contour lines) by including mutations that are not generated by evolution. This led to the creation of functional, synthetic variants (green dot, bottom right) that reside on different fitness peaks to variants that are naturally occurring.
Image credit: Marcin Plech & Grzegorz Kudla (CC BY 4.0).
Using this machine learning approach, Gonzalez Somermeyer et al. were able to design fluorescent proteins that differed from their closest natural relative by as many as 48 mutations. This is remarkable because in most protein mutagenesis experiments it only takes a few mutations before the function of the protein deteriorates. Evolution, on the other hand, can generate functional variants that differ by hundreds of mutations through a process of trial and error, which is akin to walking along a narrow ridge of high fitness one mutational step at a time. The neural network, however, appears to have jumped straight to a distant peak of high fitness (Figure 1). So, how did the network know where to take a leap?
To answer this, Gonzalez Somermeyer et al. experimented with three GFP proteins that originated from evolutionarily distant species. They found that machine learning was better at generating functional variants of cgreGFP than its two homologues, amacGFP and ppluGFP2 (a fourth homologue, avGFP, was also studied, but not in the machine learning experiment). This allowed the team to look for properties within each protein’s genetic sequence and fitness landscape which correlated with its machine learning performance.
Analysis of the fitness landscape revealed that the homologues differed in the number of mutations they could tolerate: it took on average three to four mutations until the fluorescence of cgreGFP and avGFP deteriorated, but seven to eight mutations were needed to compromise the function of amacGFP and ppluGFP2. The proteins also differed in their general sturdiness: ppluGFP2 was stable when exposed to high temperatures, whereas the structure of cgreGFP was more sensitive to changes in temperature.
Finally, Gonzalez Somermeyer et al. found that the increased mutational sensitivity of avGFP and cgreGFP (and to a lesser degree ppluGFP2) was due to negative epistasis – that is, when an individual mutation is well tolerated, but has a negative effect on the protein’s function when combined with other mutations (Bershtein et al., 2006; Domingo et al., 2019). The reduced fluorescence of amacGFP, however, could be ascribed almost entirely to additive effects, with each mutation incrementally making the protein less functional.
In order to generate functional variants, the network needs an opportunity to learn which properties of the fitness landscape are relevant from the data provided. The findings of Gonzalez Somermeyer et al. suggest that to predict a protein’s function, the algorithm only requires data on the effects of single-site mutations and low-order epistasis (interactions between small sets of mutations). This is good news for the protein engineering field as it suggests that prior knowledge of high-order interactions between large sets of mutations is not needed for protein design. Furthermore, it explains why the neural network is better at generating new variants of cgreGFP, which has a sharp fitness peak and high prevalence of epistasis.
In sum, these experiments provide a successful case study in protein engineering. An interesting extension would be to analyse the three-dimensional structures of the variants using AlphaFold, an algorithm which can predict a protein’s structure based on its amino acid sequence (Jumper et al., 2021). This would reveal if data from AlphaFold improves the prediction of functional variants, and help to identify structural features that rendered some of the variants non-fluorescent despite them being predicted to work. In the near future, assessing a new variant’s structure before it is synthesized could become a standard validation step in the design of new proteins. Furthermore, studying the fitness landscapes of multiple related variants, as done by Gonzalez Somermeyer et al., could reveal how a protein’s genetic sequence and structure changed over the course of evolution (Hochberg and Thornton, 2017; Mascotti, 2022). A better understanding of the evolution of proteins will help scientists to engineer synthetic molecules that carry out specific roles.
References
-
The causes and consequences of genetic interactions (epistasis)Annual Review of Genomics and Human Genetics 20:433–460.https://doi.org/10.1146/annurev-genom-083118-014857
-
Reconstructing ancient proteins to understand the causes of structure and functionAnnual Review of Biophysics 46:247–269.https://doi.org/10.1146/annurev-biophys-070816-033631
-
Resurrecting enzymes by ancestral sequence reconstructionMethods in Molecular Biology 2397:111–136.https://doi.org/10.1007/978-1-0716-1826-4_7
Article and author information
Author details
Publication history
Copyright
© 2022, Kudla and Plech
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,794
- views
-
- 327
- downloads
-
- 2
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Machine Learning: Lighting up protein design
eLife 11:e79310.
https://doi.org/10.7554/eLife.79310
Further reading
-
- Computational and Systems Biology
Plasmid construction is central to life science research, and sequence verification is arguably its costliest step. Long-read sequencing has emerged as a competitor to Sanger sequencing, with the principal benefit that whole plasmids can be sequenced in a single run. Nevertheless, the current cost of nanopore sequencing is still prohibitive for routine sequencing during plasmid construction. We develop a computational approach termed Simple Algorithm for Very Efficient Multiplexing of Oxford Nanopore Experiments for You (SAVEMONEY) that guides researchers to mix multiple plasmids and subsequently computationally de-mixes the resultant sequences. SAVEMONEY defines optimal mixtures in a pre-survey step, and following sequencing, executes a post-analysis workflow involving sequence classification, alignment, and consensus determination. By using Bayesian analysis with prior probability of expected plasmid construction error rate, high-confidence sequences can be obtained for each plasmid in the mixture. Plasmids differing by as little as two bases can be mixed as a single sample for nanopore sequencing, and routine multiplexing of even six plasmids per 180 reads can still maintain high accuracy of consensus sequencing. SAVEMONEY should further democratize whole-plasmid sequencing by nanopore and related technologies, driving down the effective cost of whole-plasmid sequencing to lower than that of a single Sanger sequencing run.
-
- Biochemistry and Chemical Biology
- Computational and Systems Biology
Protein–protein interactions are fundamental to understanding the molecular functions and regulation of proteins. Despite the availability of extensive databases, many interactions remain uncharacterized due to the labor-intensive nature of experimental validation. In this study, we utilized the AlphaFold2 program to predict interactions among proteins localized in the nuage, a germline-specific non-membrane organelle essential for piRNA biogenesis in Drosophila. We screened 20 nuage proteins for 1:1 interactions and predicted dimer structures. Among these, five represented novel interaction candidates. Three pairs, including Spn-E_Squ, were verified by co-immunoprecipitation. Disruption of the salt bridges at the Spn-E_Squ interface confirmed their functional importance, underscoring the predictive model’s accuracy. We extended our analysis to include interactions between three representative nuage components—Vas, Squ, and Tej—and approximately 430 oogenesis-related proteins. Co-immunoprecipitation verified interactions for three pairs: Mei-W68_Squ, CSN3_Squ, and Pka-C1_Tej. Furthermore, we screened the majority of Drosophila proteins (~12,000) for potential interaction with the Piwi protein, a central player in the piRNA pathway, identifying 164 pairs as potential binding partners. This in silico approach not only efficiently identifies potential interaction partners but also significantly bridges the gap by facilitating the integration of bioinformatics and experimental biology.




{kind=link}