An auto-inhibited state of protein kinase G and implications for selective activation
- Department of Pharmacology and Chemical Biology and Center for Drug Discovery, Baylor College of Medicine, United States
- Department of Biochemistry, University of Kassel, Germany
- Department of Chemistry and Chemical Biology, McMaster University, Canada
- Molecular Biophysics and Integrated Bioimaging, United States
- Department of Pathology and Immunology and Center for Drug Discovery, Baylor College of Medicine, United States
- Department of Medicine, University of California, San Diego, United States
- Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, United States
Figures
Figure 1 with 3 supplements
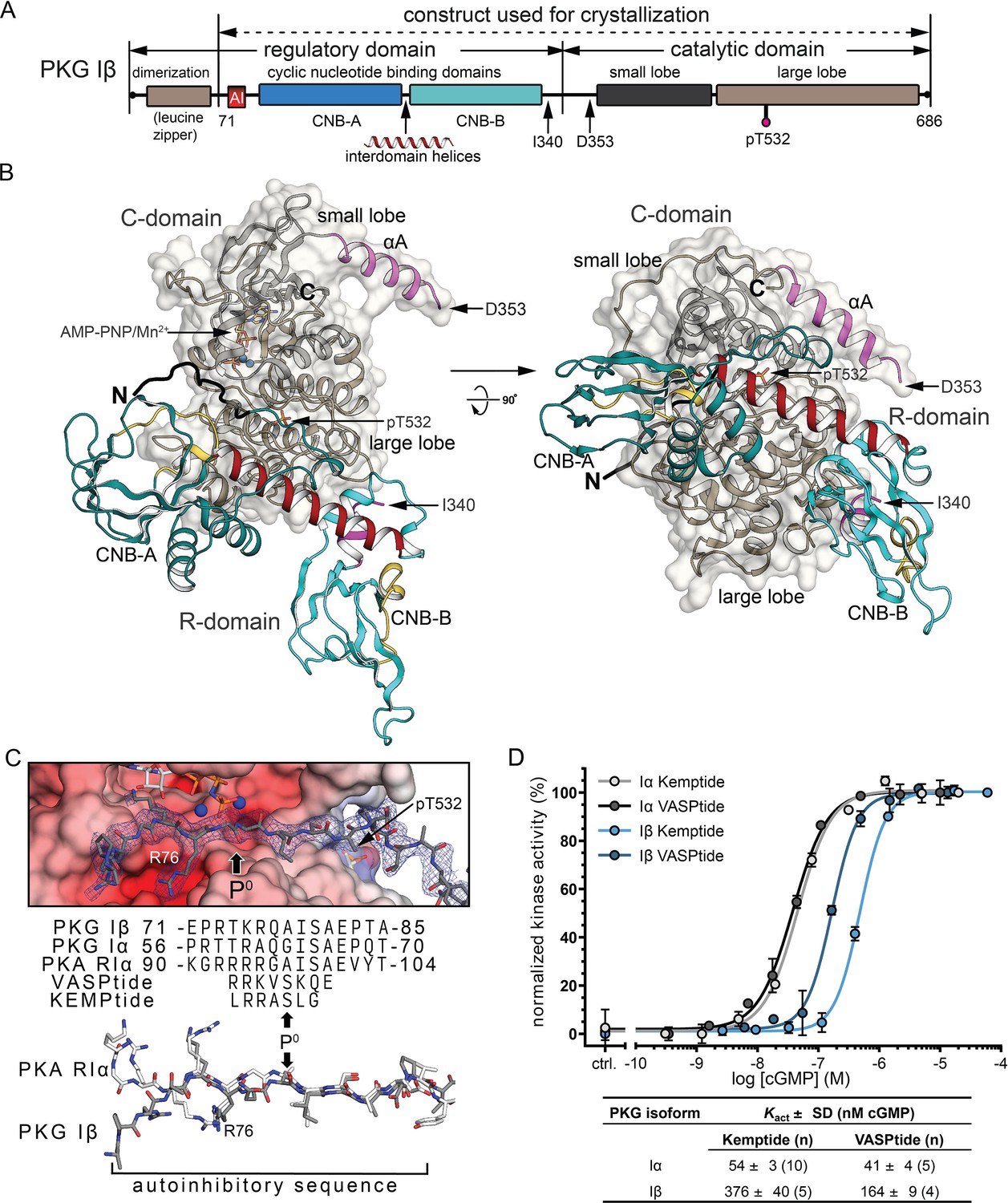
Overall structure of the R:C holoenzyme complex.
(A) Domain organization of PKG Iβ and the construct used for crystallization. AI, auto-inhibitory sequence; CNB, cyclic nucleotide-binding domain. (B) Overall structure of the PKG Iβ R:C complex (71–686). The R- and C-domains are shown in cartoons with a transparent surface on the C-domain. N and C termini are labeled. AI is colored in black. CNB-A and -B are colored in teal, phosphate-binding cassette (PBC) in yellow, αA-helix in magenta, and the interdomain helices in red. The αA helix of the C-domain is colored in magenta, the small lobe is in black and the large lobe is in dark tan. The phosphorylated T532 is shown in sticks. The last ordered residue in the R-domain and the first ordered residue in the C-domain are indicated with arrows. The entire C-domain shows clear density in both chains; besides the missing residues discussed in the text, the first two residues at the N-terminus (residues 71–72) of the R-domains and five residues that follow the AI sequence (residues 85–89) in one chain are missing. All structure images were generated using PyMOL (DeLano Scientific). (C) AI docking to the active site. Top: AI is shown with electron density (2Fo − Fc at σ = 1.0). The phosphorylation site (P0) in models and sequences is marked with arrows. Electrostatic potential surface is shown for the C-domain active site. Bottom: Alignment of AI sequences of PKG Iβ, Iα, and PKA RIα with substrates VASPtide and Kemptide. (D) Isozyme differences in cyclic guanosine monophosphate (cGMP)-dependent activation of PKG Iα (gray) and Iβ (blue) are revealed by activity measurements, and activation constants (Kact) vary with substrate. Both isoforms require less cGMP for half-maximal activation using VASPtide (dark gray and dark blue) compared to Kemptide (light gray and light blue). Data points show the mean of duplicates with error bars indication the standard deviation (SD). Kact values are given as mean of n measurements ± standard deviation (SD). Additional data related to these fits are presented in Figure 1—figure supplement 3.
Figure 1—figure supplement 1
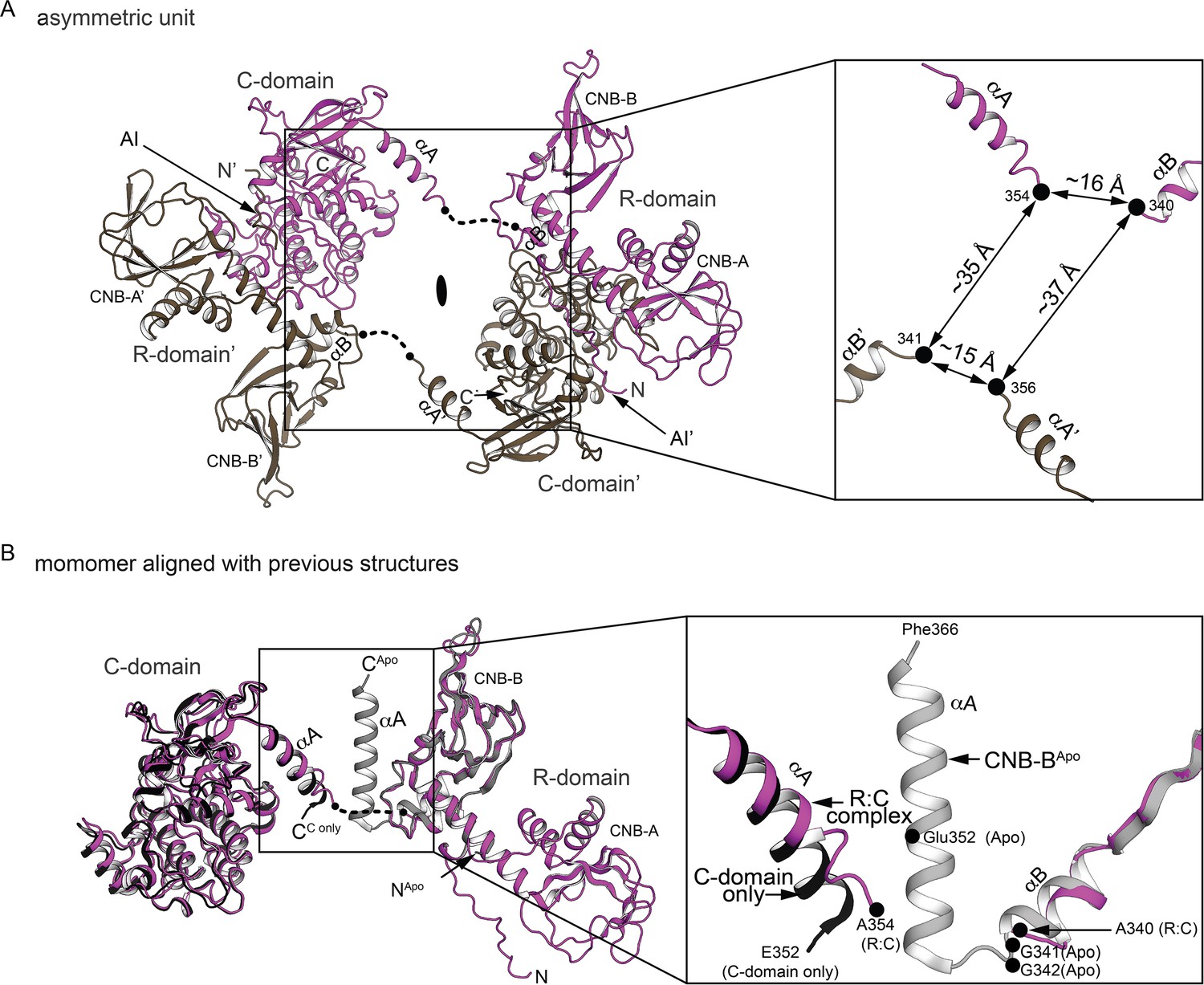
The R:C dimer captured in asymmetric unit and the R:C monomer alignment with previous structures.
(A) Asymmetric unit. Left: The dimer captured in asymmetric unit with each monomer colored in magenta or dark tan. Right: The zoom-in panel shows the R:C linker at the missing electron density with the distances between the last fitted atoms. (B) The R:C monomer alignment with previous structures. Left: Structural alignment of the R:C monomer with the C-domain bound to N46 (PDB ID: 6C0T) and the CNB-B apo structure (PDB ID: 4KU8). The missing linker region is shown as dotted line. Right: The zoom-in view shows the R:C linker region.
Figure 1—figure supplement 2
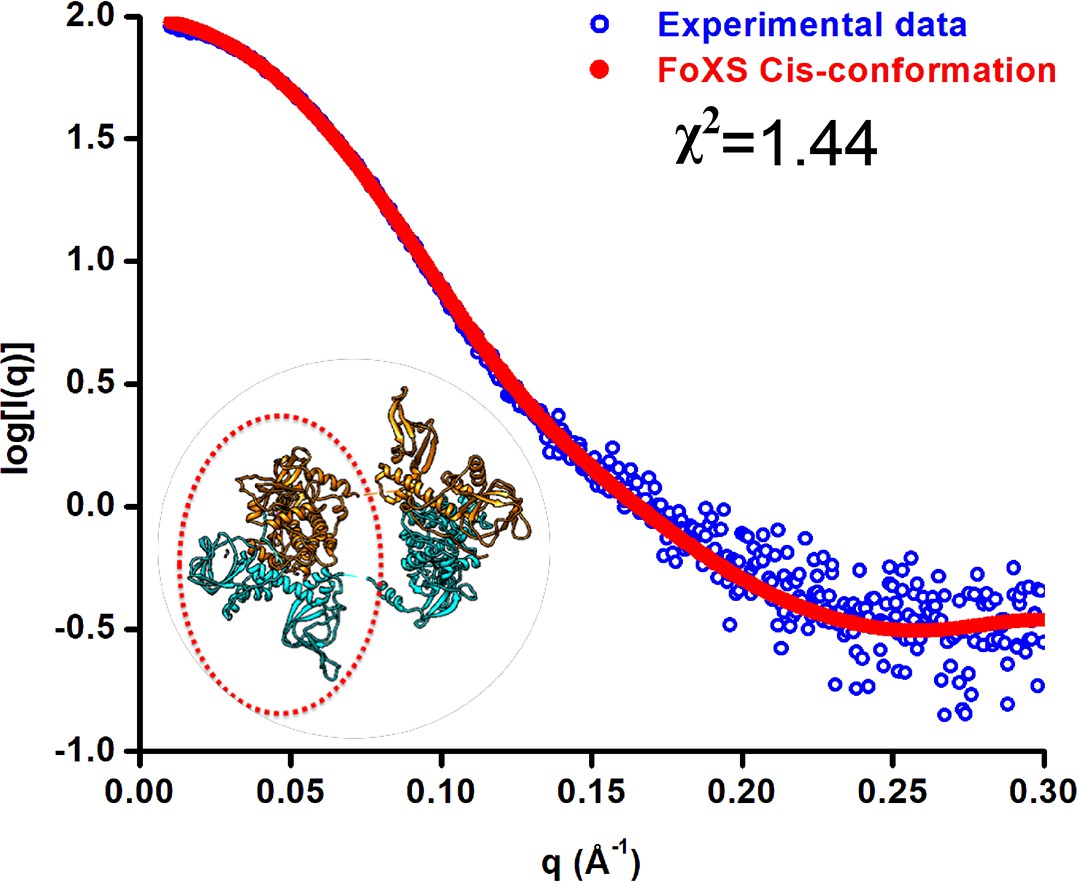
The FoXS webserver analysis shows that the theoretically calculated scattering profile from the cis-conformation (red) matches well with the experimentally observed scattering profile (blue) with a χ2 = 1.44.
Figure 1—figure supplement 3
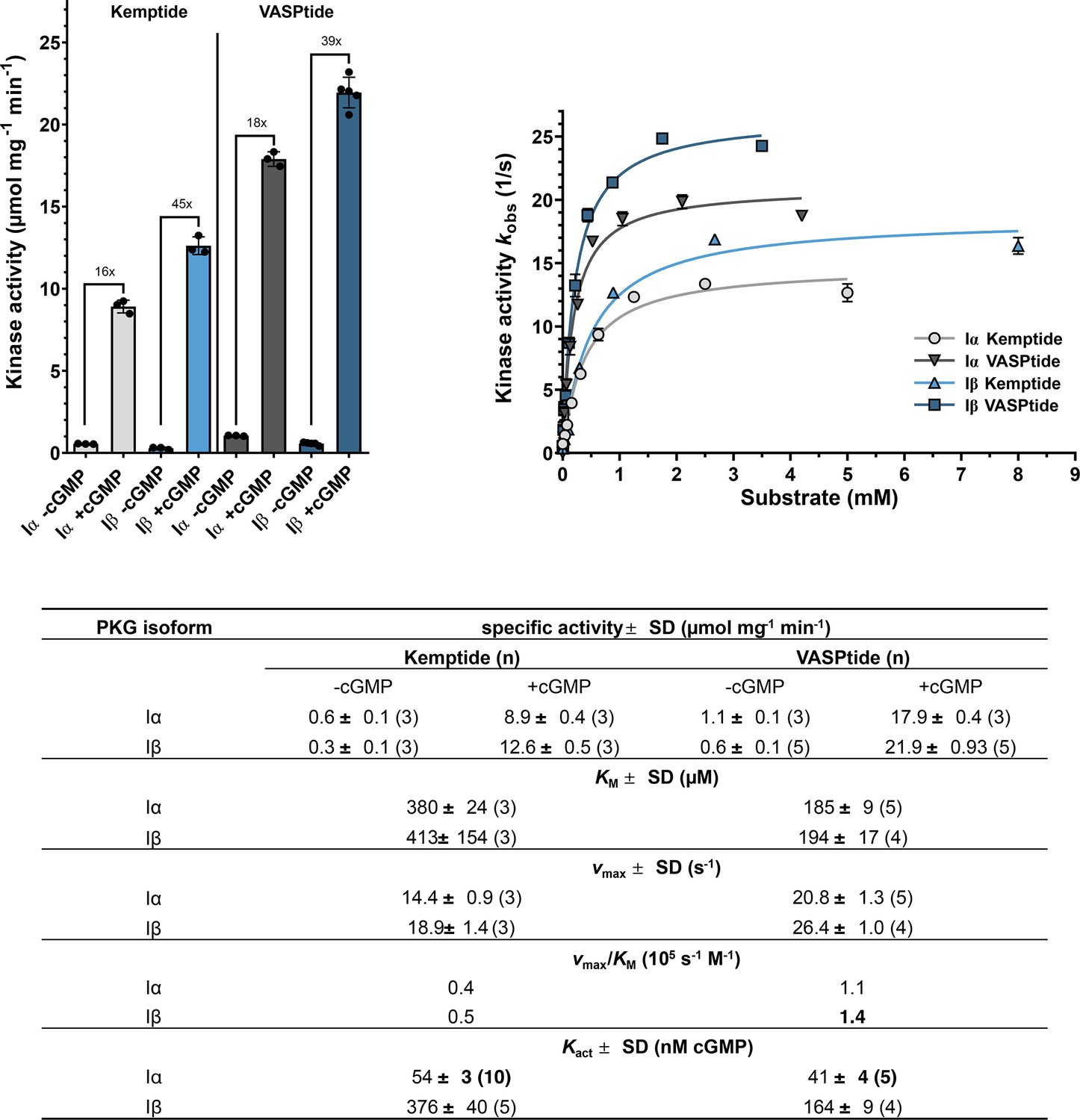
Isoform-specific differences between PKG Iα and Iβ kinetic parameters.
Top left: Specific kinase activities of PKG Iα (gray) and Iβ (blue) in unstimulated (−cGMP) and stimulated (+cGMP) condition using 1 mM of the peptide substrates VASPtide (RRKVSKQE, dark gray and dark blue) or Kemptide (LRRASLG, light gray and light blue). Comparison of both isozymes shows a higher basal activity for PKG Iα and PKG Iβ being more active when saturated with cyclic guanosine monophosphate (cGMP). Top right: The determination of the Michaelis–Menten constant KM and the maximum kinase activity vmax again show the higher kinase activity of PKG Iβ but most interestingly no difference in the KM for the respective substrate is detected. All measurements were performed as spectrophotometric kinase assays (Cook et al., 1982). Data points for Michaelis–Menten kinetics are depicted as mean of duplicates with error bars indicating the standard deviation (SD). All values are given as mean of n measurements ± SD.
Figure 2
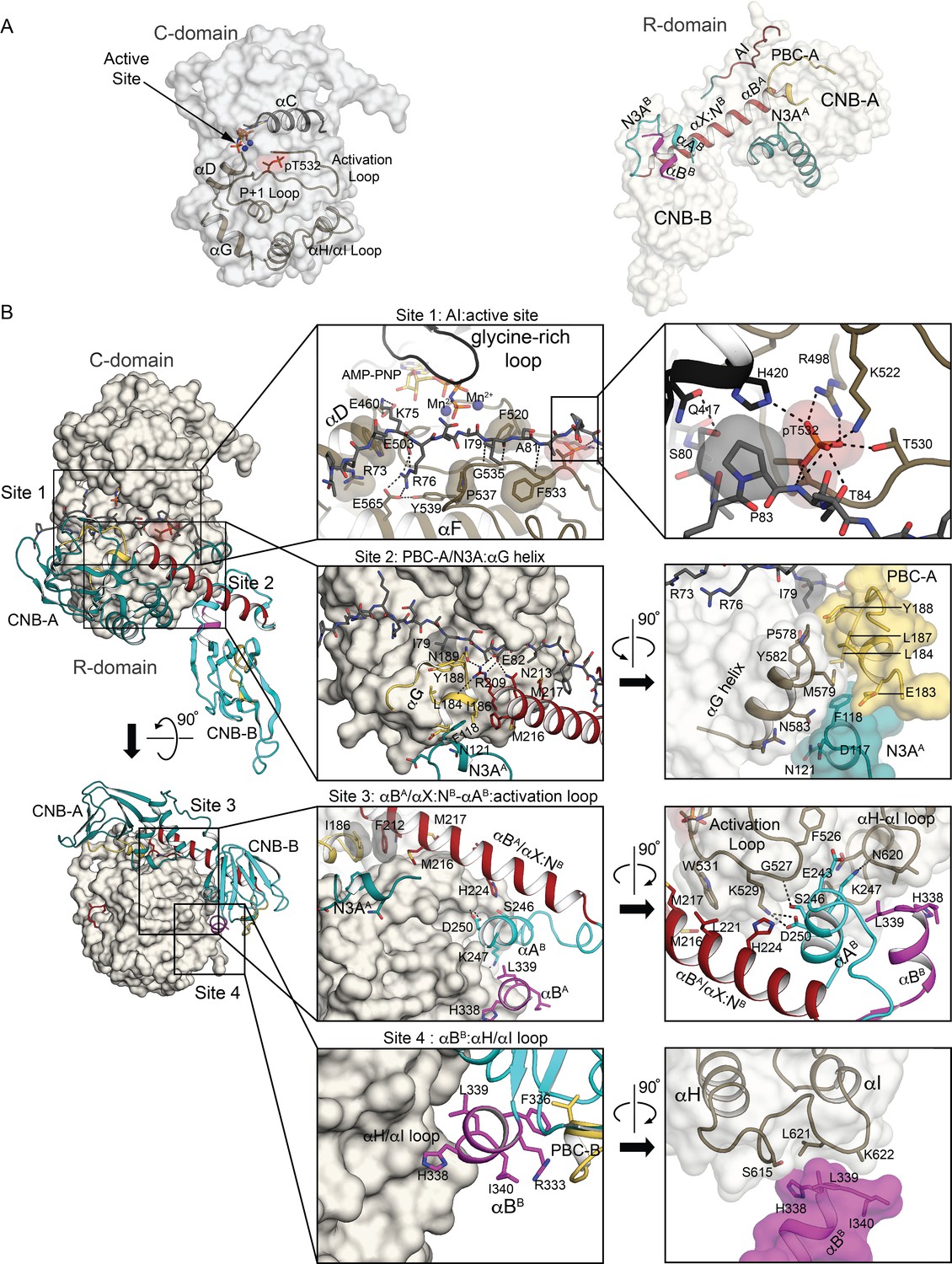
Interactions at the R:C interface.
(A) Key structural elements that form the R:C interface. The C- and R-domains (left and right) are displayed in isolation as transparent surface with key elements labeled and shown as cartoons using the same color scheme as in Figure 1B. (B) Detailed R:C interactions with the C-domain presented as a surface and the R-domain portrayed in cartoon using the same color scheme. Left two panels: Two orthogonal views of the AI complex with outlines identifying the regions discussed in the text as interaction sites 1 through 4. Middle four panels: Zoomed-in views of the four interaction sites with key residues labeled and shown as sticks, and hydrogen bonds shown as dotted lines. Right four panels: Further zoomed-in or rotated views of the four interaction sites. Detailed R:C contacts are summarized in Supplementary file 3.
Figure 3 with 1 supplement
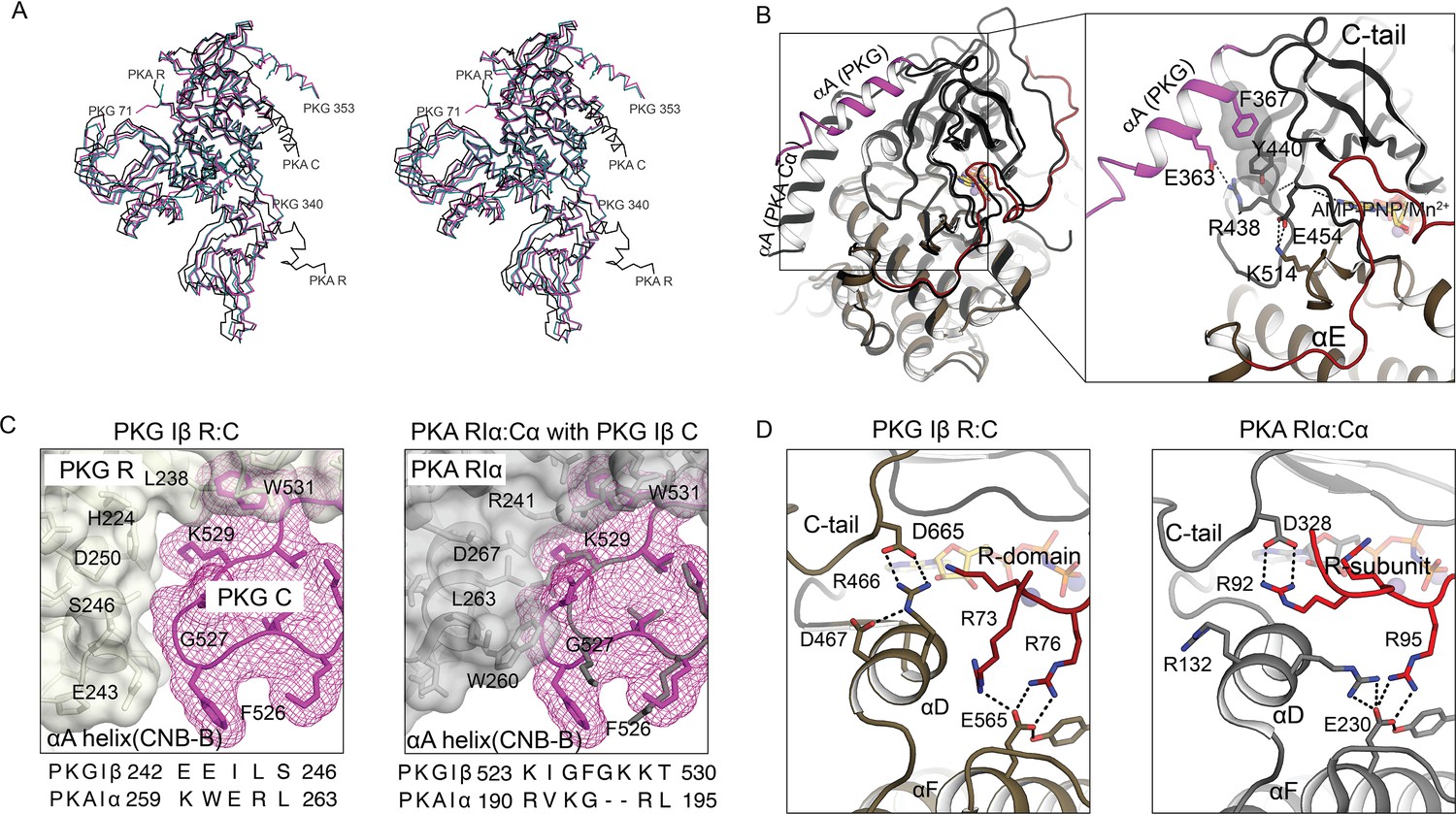
Comparison of AI PKG and PKA structures.
(A) Structural alignment of AI PKG Iβ (red, PDB ID: 7LV3) and PKA Iα (gray, PDB ID: 2QCS) in stereo-view. (B) C-domain αA helices are differently positioned in PKG Iβ and PKA. Left: AI PKG Iβ and PKA Iα structures are aligned using Cα atoms of the C-domains Right: Zoom-in view of interactions between the PKG Iβ αA helix and the catalytic core. The interacting residues and bound AMP-PNP:Mn2+ (ANP) are shown in sticks and labeled, and hydrogen bonds are shown as dotted lines. The short PKG Iβ αA-β1 loop allows αA to contact the small lobe of the C-domain via a salt bridge (E363:R438) and stacking interactions (F367:Y440) in a way that may be conveyed to the ribose pocket. (C) PKG Iβ contacts between the CNB-B αA helix and the C-domain near the activation loop imply that binding of PKA RIα would cause steric clashes. Left: PKG Iβ R:C interface at site 3 with sequences of PKG Iβ and PKA Iα at the CNB-B αA helix aligned. The activation loop is shown in mesh (magenta), the R-domain is shown in transparent surface (gray), and interacting residues are labeled. Right: Aligning AI PKA Iα on AI PKG Iβ using the C-domains (except the activation loop) shows that large PKA RIα side chains L263 and W260 that contact the shorter PKA Cα loop would clash with the larger PKG Iβ loop. Sequence alignment between PKG Iβ and PKA Iα at the activation loop is shown below. (D) In auto-inhibition, PKG Iβ and PKA C-terminal tails occupy similar positions but contact different partners. The C- and R-domains (or subunits) are colored in gray and red, respectively. Left: The C-tail of PKG interacts with the C-domain αD helix and the AI linker interacts with the C-domain αF helix. Right: The C-tail of PKA Iα interacts with the AI linker, and both the αD helix and the AI linker interact with the C-subunit αF helix.
Figure 3—figure supplement 1
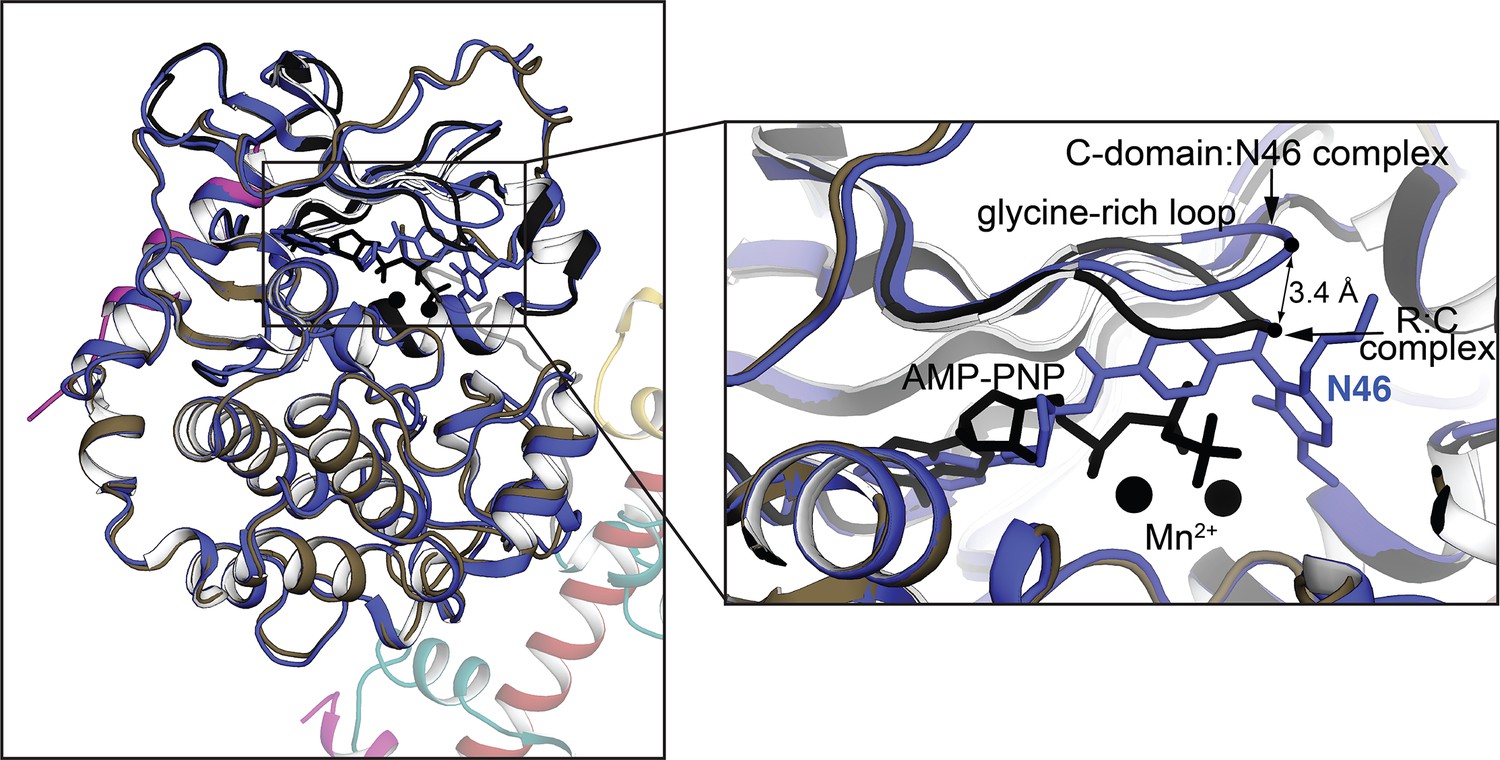
Structural alignment of AI PKG Iβ monomer with the isolated PKG I C-domain bound to N46 (PDB ID: 6C0T).
The isolated C-domain is in blue and the AI R–C complex is in the same color theme as Figure 1B. The zoom-in view on the right highlights the structural differences at the glycine-rich loop.
Figure 4
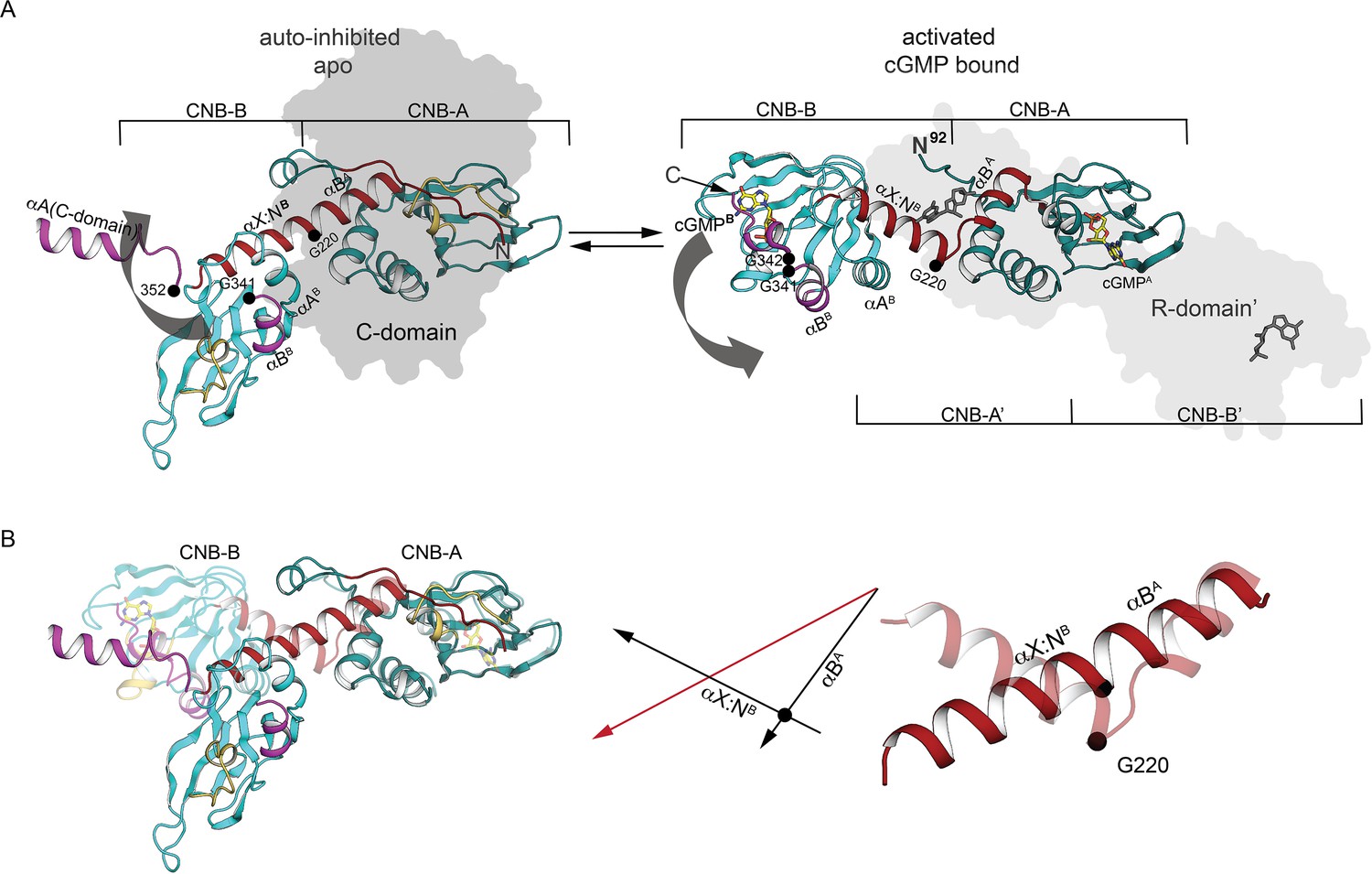
Comparison of PKG Iβ R-domain structures in activated and AI states.
(A) Cartoon of a single R-domain in either (left) the AI R:C state, bound to the C-domain depicted as a gray silhouette, or (right) the activated R:R state, bound to cyclic guanosine monophosphate (cGMP) and another R-domain shown as a light gray silhouette (PDB ID: 4Z07). The interdomain helices are labeled and shown in red, and G220, G341, and G342 are marked. (B) Structural alignment of interdomain helices in the AI and activated conformation. Left: The R-domain in the AI state (solid) and activated state (transparent) conformations are aligned using CNB-A (at right). Middle: orientations of the interdomain helical axes in the AI state (red) and the activated state (black). Right: Cartoon depiction of the interdomain helices only, with a black circle indicating the position of the G220 Cα atom in both the AI state (solid) and the activated state (transparent).
Figure 5 with 2 supplements
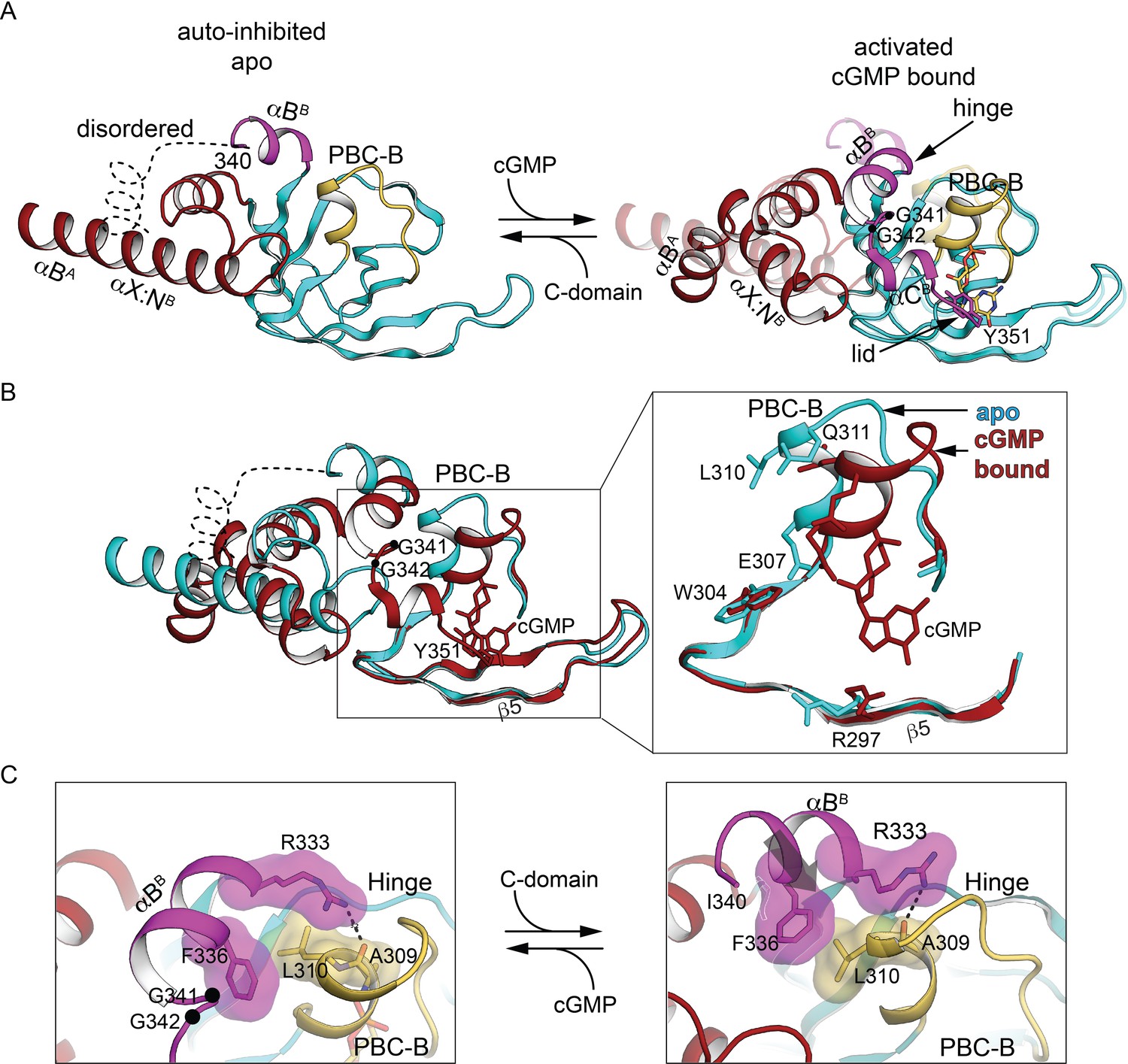
The CNB-B helical subdomains are structurally distinct in AI and activated conformation.
(A) Overall structure of the interdomain helices and CNB-B in the AI (left, PDB ID: 7LV3) and superposition of the cyclic guanosine monophosphate (cGMP)-bound, activated state of the tandem CNB dimer (solid) and AI (transparent) (right, PDB ID: 7LV3 and 4Z07). Hinge glycine Cα atoms are labeled and cGMPs are shown in stick. In the cGMP-bound ‘closed’ state, PBC-B moves along with αBB helix and the hydrogen bond interactions between αC helix (lid) and PBC-B provide cGMP capping interactions. (B) Superposition of CNB-B in the tandem CNB cGMP-bound dimer (red, PDB ID: 4Z07) and in the AI complex (cyan, PDB ID: 7LV3) using the invariant β domain elements. Left: β4, β5, and the β4–β5 loop superimpose well (bottom) but the interdomain helices adopt very different conformations. Right: Zoomed-in view of the differences between the phosphate-binding cassette (PBC) loops of the two states. In the AI ‘open’ state, the αB helix of CNB-B docks to the C-domain, the αC helix is disordered, and the more open PBC-B interacts directly with N3A. (C) Zoomed-in view of the hinge region. Key hinge residues are shown in stick with transparent surface.
Figure 5—figure supplement 1
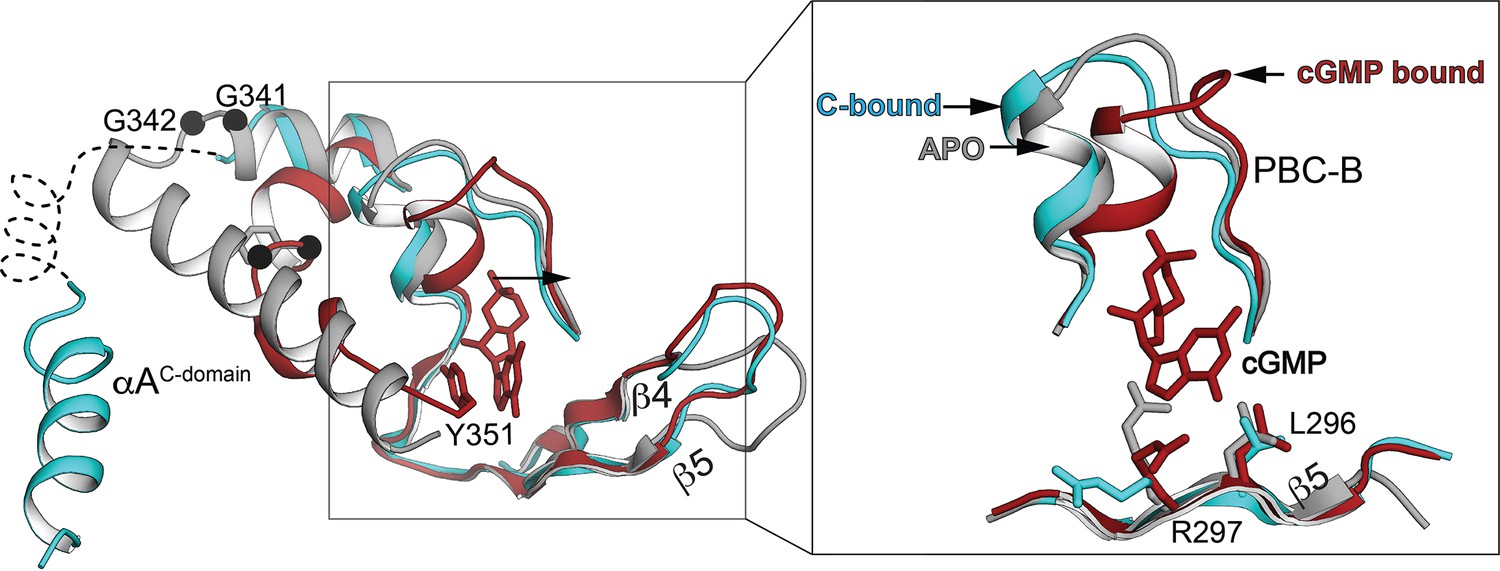
Superposition of the PKG Iβ CNB-B domain in the auto-inhibited state (teal, PDB ID: 7LV3) with the isolated CNB-B domain apo state (red, PDB ID: 4KU8) and cyclic guanosine monophosphate (cGMP)-bound state (gray, PDB ID: 4Z07).
Left: Only phosphate-binding cassette (PBC), β4, β5, and interdomain helices are shown. Right: Zoomed-in view showing PBC and B helix only. Key cGMP-interacting residues of β5 are shown in stick.
Figure 5—figure supplement 2

A conformation induced by cyclic guanosine monophosphate (cGMP) is prevented by RP-cGMPS.
Our structure of auto-inhibited PKG I suggests that the first steps in activation are phosphate-binding cassette reordering and closing of the CNB-B pocket on bound cGMP. Aligning the activated, auto-inhibited, and RP-cGMPS-bound CNB-B states (top left) shows that the protein backbone closes around cGMP, bringing T307 closer to A309 and G312 (bottom left) than in the auto-inhibited state (bottom right). The RP-cGMPS-bound state (top right) remains almost as open as the auto-inhibited state.
Figure 6 with 3 supplements
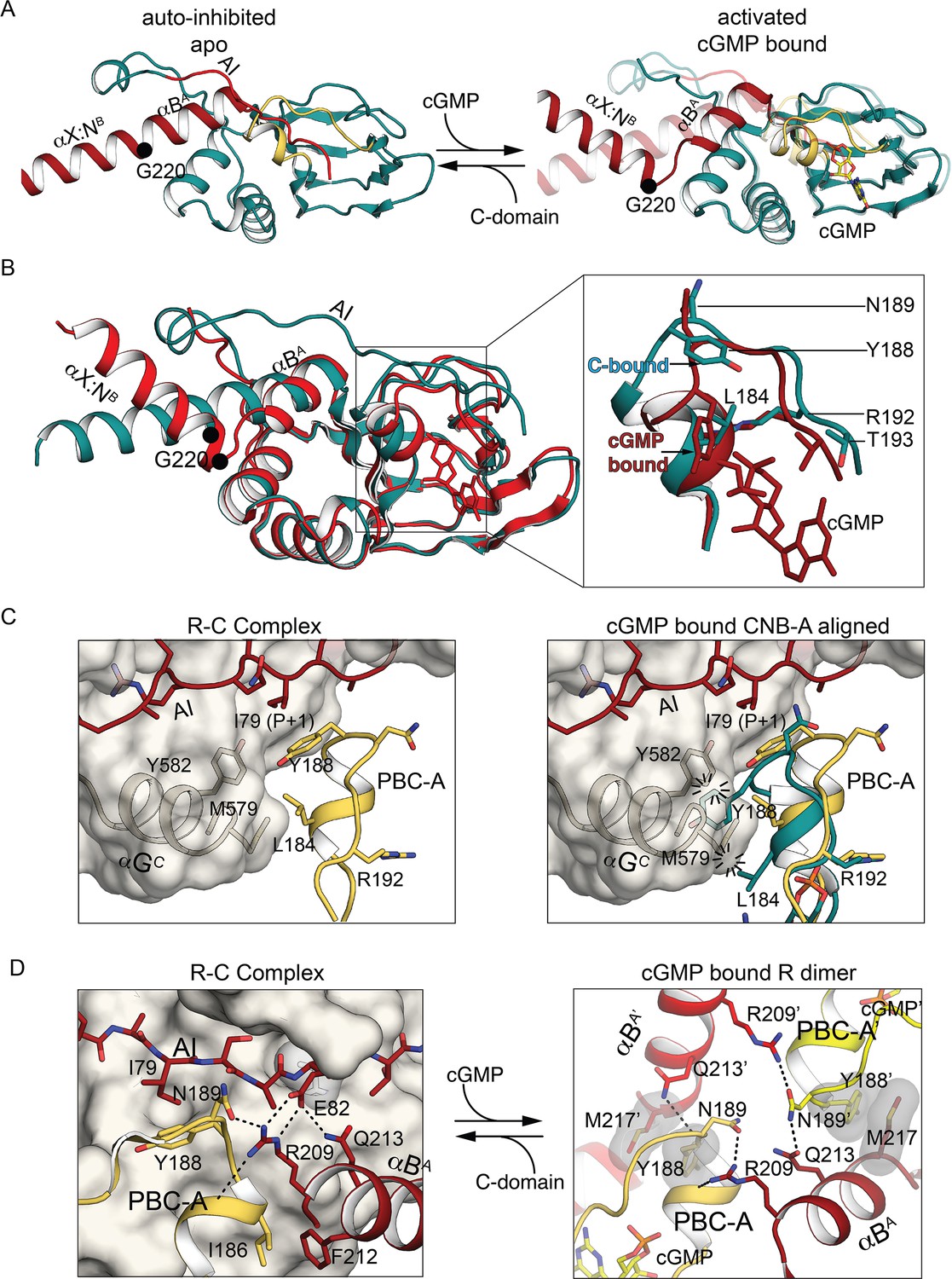
CNB-A phosphate-binding cassette (PBC) and interdomain linker differences in auto-inhibition compared to activation.
(A) Overall structure of the interdomain helices and CNB-A in the auto-inhibited (left, PDB ID: 7LV3) and superposition of the cyclic guanosine monophosphate (cGMP)-bound, activated state of the tandem CNB dimer (solid) and auto-inhibited (transparent) (right, PDB ID: 7LV3 and 4Z07). The same color scheme is used as in Figure 2. The Cα atom of G220 is marked with a sphere. (B) Structural alignment of cGMP- (PDB ID: 4Z07) and C-domain-bound conformation (PDB ID: 7LV3). CNB-A in cGMP- and C-bound conformations are colored in red and dark teal, respectively. Zoomed-in view at the right panel shows PBC and αB helix. Key hinge residues and R:C interface residues are shown in stick. (C) Alignment of CNB-A:cGMP with PKG holo complex. Left: R:C interface near the apo PBC-A is shown. PBC-A and the αG helix are shown in cartoon. The C-domain is shown as transparent surface. Key R:C interface residues are shown in sticks. Right: Structural alignment between CNB-A:cGMP with PKG Iβ holoenzyme. The residues L184 and Y188 of PBC-A of the cGMP-bound state show steric clashes with M579 and Y583 of the αG helix at the C-domain and suggest that CNB-A:cGMP is not compatible with the C-domain. (D) Dynamic AI region replaces the R:R contacts upon the R:C complex formation. Left: The R:R dimer interface near PBC-A is shown. Residues from the second chain are marked with ’. Right panel shows AI, PBC-B, and B helix of the R-domain in cartoon and the C-domain in surface. R209 helps assemble a docking surface consisting of AI, PBC-B, and B helix of the R-domain.
Figure 6—figure supplement 1
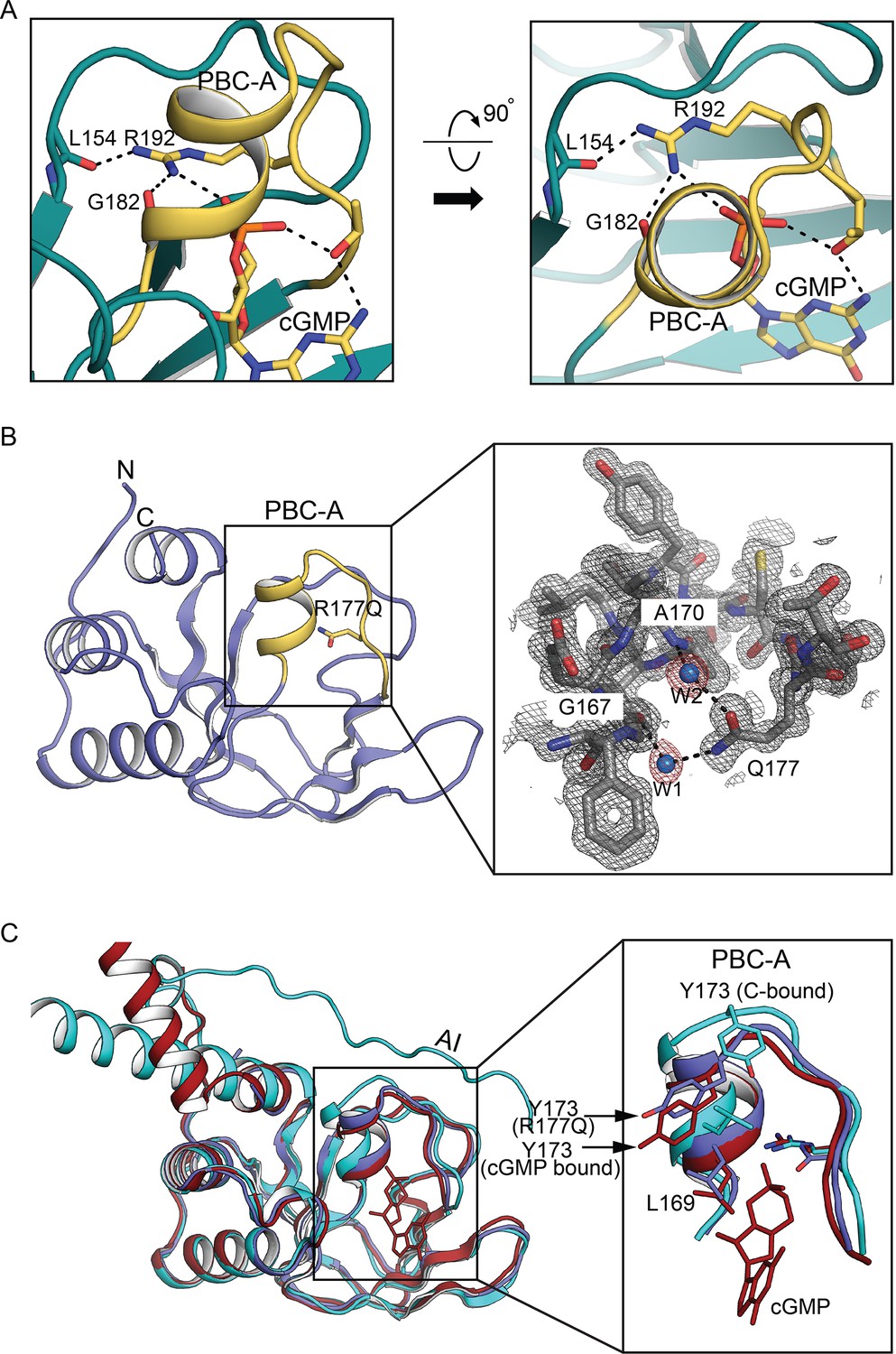
Structural basis of the constitutive activation in Thoracic Aortic Aneurysms and Dissections (TAAD) causing mutant.
(A) PBC-A R192 interactions in wildtype PKG Iβ (PDB ID: 3OD0). (B) Left: Overall structure of PKG Iα CNB-A R177Q (PDB ID: 7MBJ) determined at 1.26 Å. Structure is shown in cartoon with phosphate-binding cassette (PBC) in yellow and R177Q in stick. Right: Zoomed-in view of PBC-A with electron density (2Fo − Fc at σ = 1.0). PBC residues interacting with ordered water molecules and side chain of Q177 are shown in sticks. Ordered water molecules are shown as blue spheres. (C) Structural alignment of CNB-A R177Q with wildtype CNB-A bound to cyclic guanosine monophosphate (cGMP) and CNB-A contacting the C-domain (R:C complex). Left: R177Q mutant, cGMP- and C-bound CNB-A conformations. The cGMP-bound CNB-A is colored in red, C-domain bound in teal, and R177Q mutant in violet. Right: Zoomed-in view only showing PBC. The key R:C interface residues are shown in sticks.
Figure 6—figure supplement 2
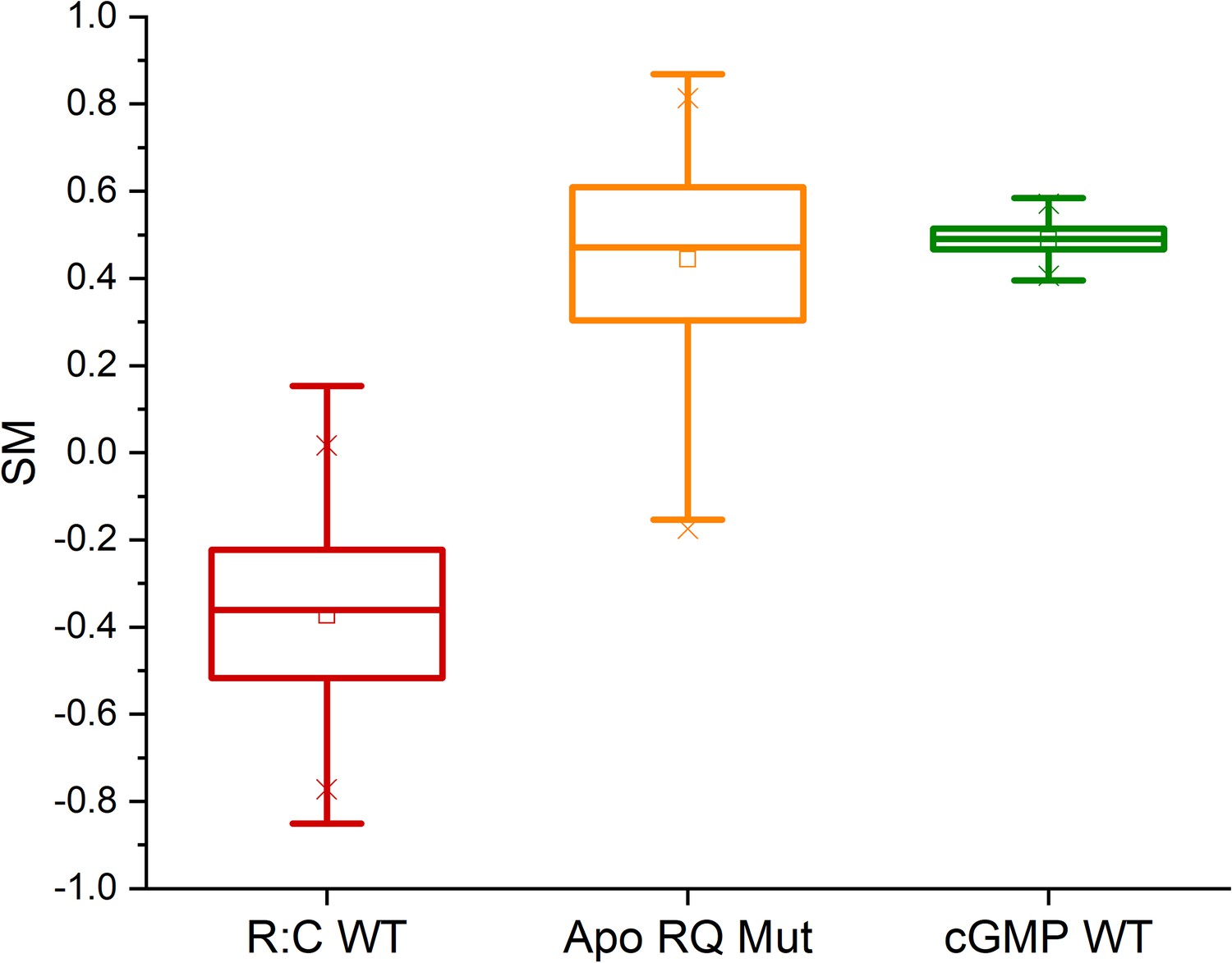
Molecular dynamics (MD) simulations.
CNB-A domain phosphate-binding cassette (PBC) similarity measures (SM) computed from the three replicate simulations of cyclic guanosine monophosphate (cGMP)-free dimeric PKG Iβ (‘R:C WT’), and from the simulations of the isolated apo RQ and cGMP-bound wildtype CNB-A domains (‘Apo RQ Mut’ and ‘cGMP WT’), using the CNB-A domain PBC from the ‘4Z07’ and cGMP-free dimer X-ray structures as active- and inactive-state reference structures, respectively. The statistics reported in each boxplot are as follows: the middle, bottom, and top lines of the central box represent the median, 25th and 75th percentiles of the dataset, respectively; the whiskers represent additional data falling within 1.5*IQR above the 75th percentile or below the 25th percentile (where IQR is the difference between the 75th and 25th percentiles); the ‘□’ symbol represents the mean of the dataset; and the two ‘×’ symbols represent the 1st and 99th percentiles of the dataset.
Figure 6—figure supplement 3
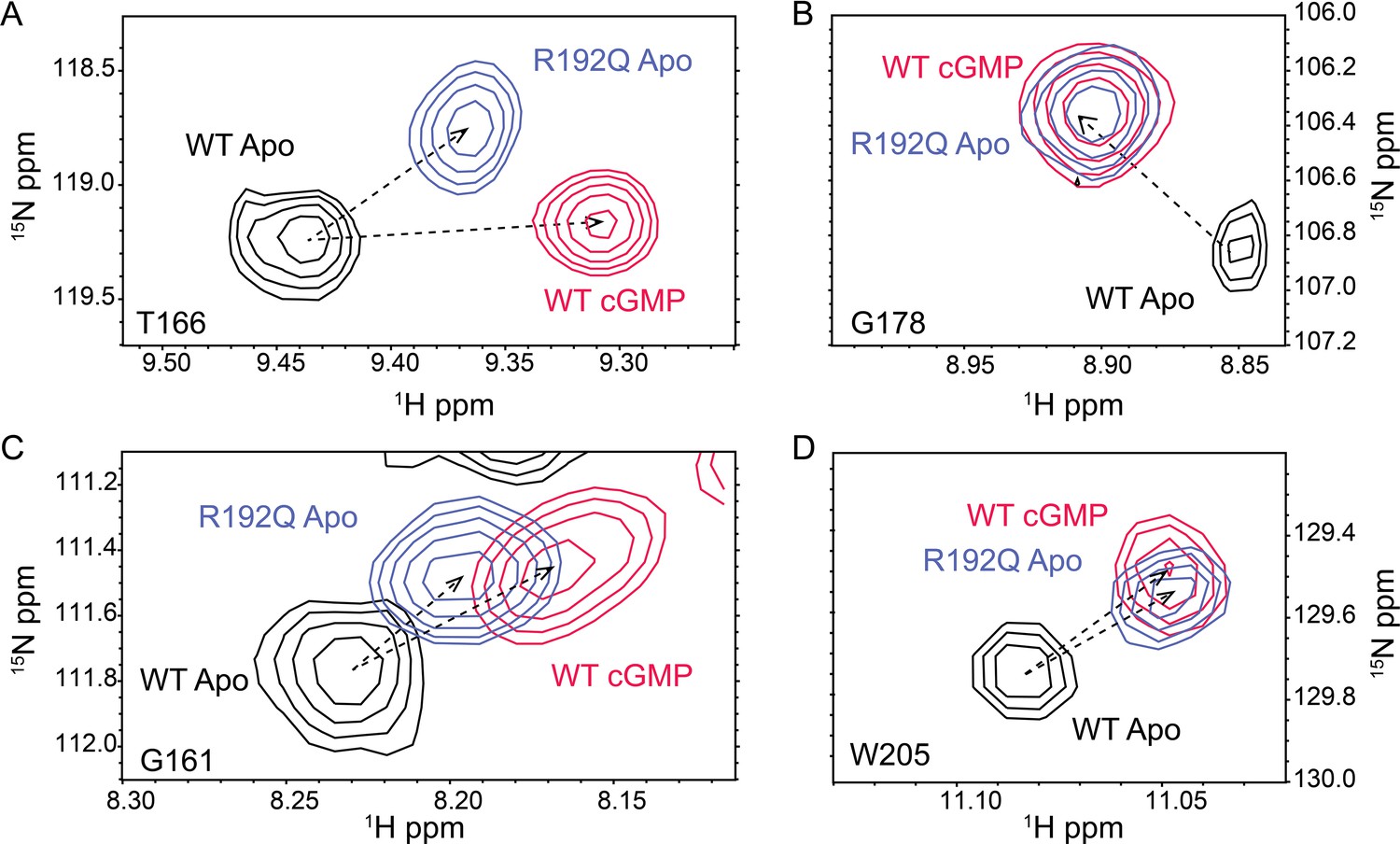
Representative 1H,15N HSQC cross-peaks of the apo wt (black), cyclic guanosine monophosphate (cGMP)-bound wt (red) and apo R192Q (purple) PKG1β (92–227) (A, B and C).
For W205 in panel (D), the side chain peak was selected.
Figure 7
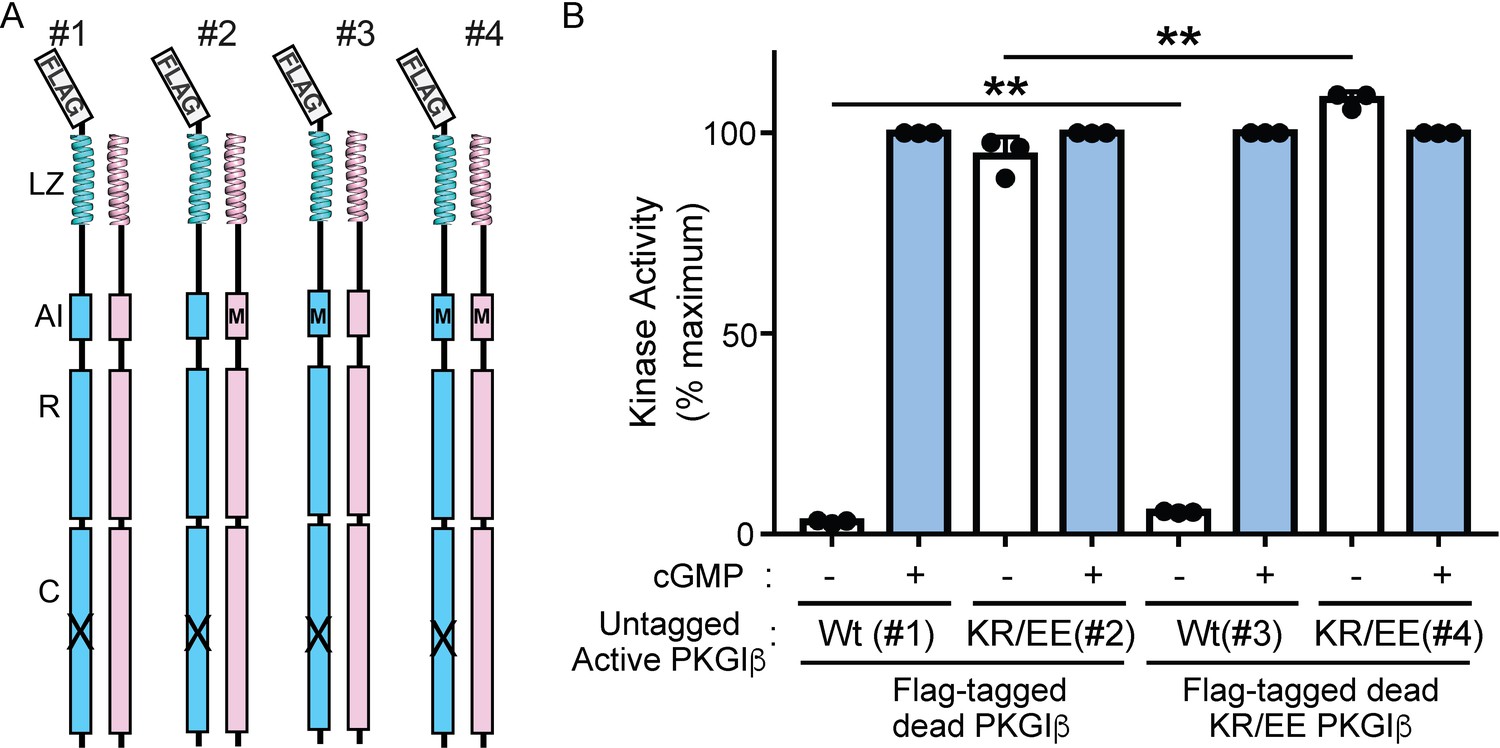
The PKG Iβ AI sequence does not contribute to activation through interchain contributions.
(A) Anti-Flag pulldown of a Flag-tagged (blue) catalytically ‘dead’ PKG Iβ (X) permits isolation of heterodimers containing an untagged ‘active’ PKG Iβ (pink) from cotransfected cells. Flag-tagged homodimers that are also present have no kinase activity. Introducing AI mutations K75E and R76E (KR/EE in the text, shown schematically as M) in the ‘dead’ and/or ‘active’ chain allows us to distinguish intrachain and interchain influences on auto-inhibition. (B) Kinase activity for four heterodimers at zero cGMP (−) or 10 μM cGMP (+) shows that AI mutations in the active monomer (heterodimers 2 and 4) confer constitutive activity and eliminate auto-inhibition. The presence of the AI mutation in the ‘dead’ monomer alone (heterodimer 3) does not eliminate auto-inhibition, though it may slightly enhance basal activity. Bars indicate mean ± standard deviation from three independent experiments. **p < 0.01, for comparisons between the indicated groups using unpaired two-tailed Student’s t-test.
Figure 8 with 1 supplement
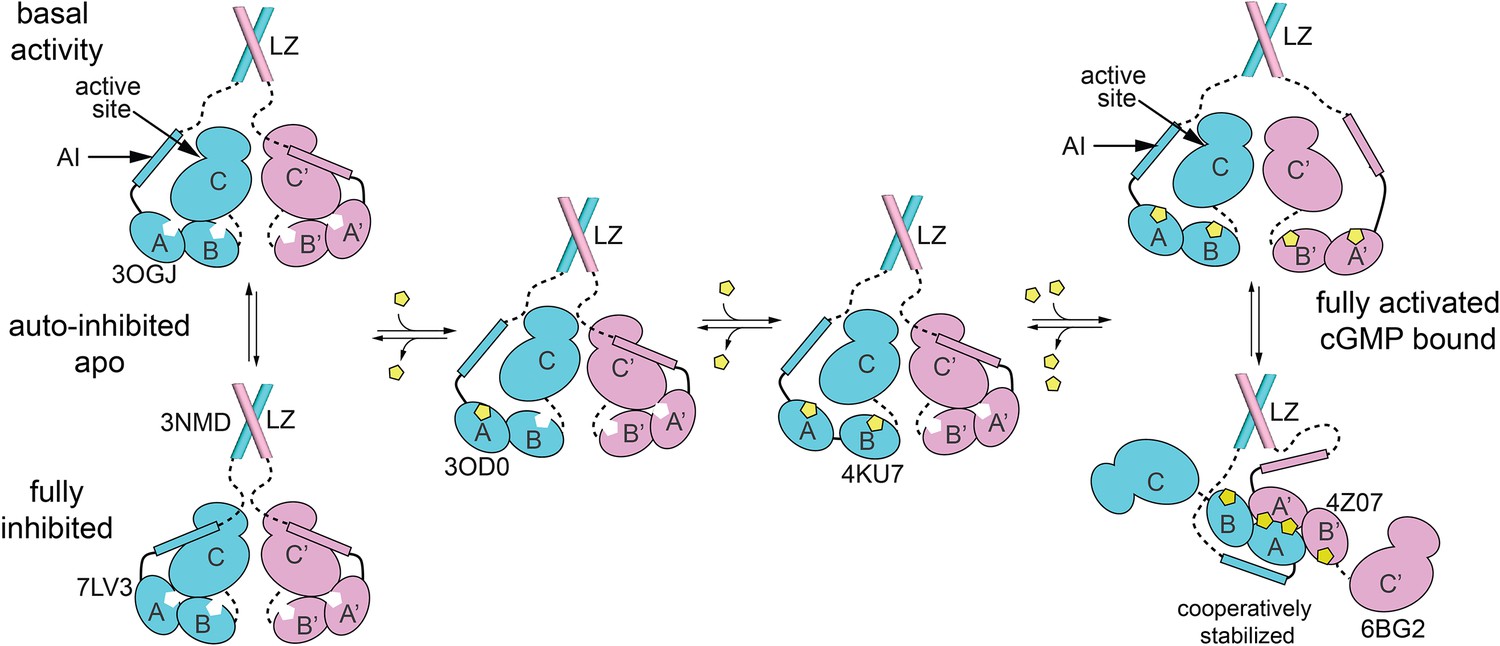
Schematic model for how equilibria between different states of PKG Iβ support auto-inhibition and activation.
Crystal structures that represent one or more domains in each state are indicated by PDB code. The auto-inhibited state (bottom left) dimerizes through leucine zipper domains (3NMD) that keep the cyclic guanosine monophosphate (cGMP)-free, auto-inhibited catalytic domains (7LV3) in close proximity. The AI sequence of cGMP-free PKG Iβ can transiently leave the active site and permit basal activity (top left). Binding of cGMP (yellow pentagon) to CNB-A (middle left; 3OD0) and/or CNB-B (middle right, 4KU7) lock the CNBs into conformations that are not compatible with the auto-inhibited state. Levels of cGMP that saturate CNBs of both monomers enable complete activation (top right) and support formation of a complex between the tandem CNB domains that provides cooperativity and may restrict the ability of the AI sequence to access the catalytic active site. The discussion further describes how peptide substrate and cGMP analogs, including cAMP, can influence the equilibria.
Figure 8—figure supplement 1
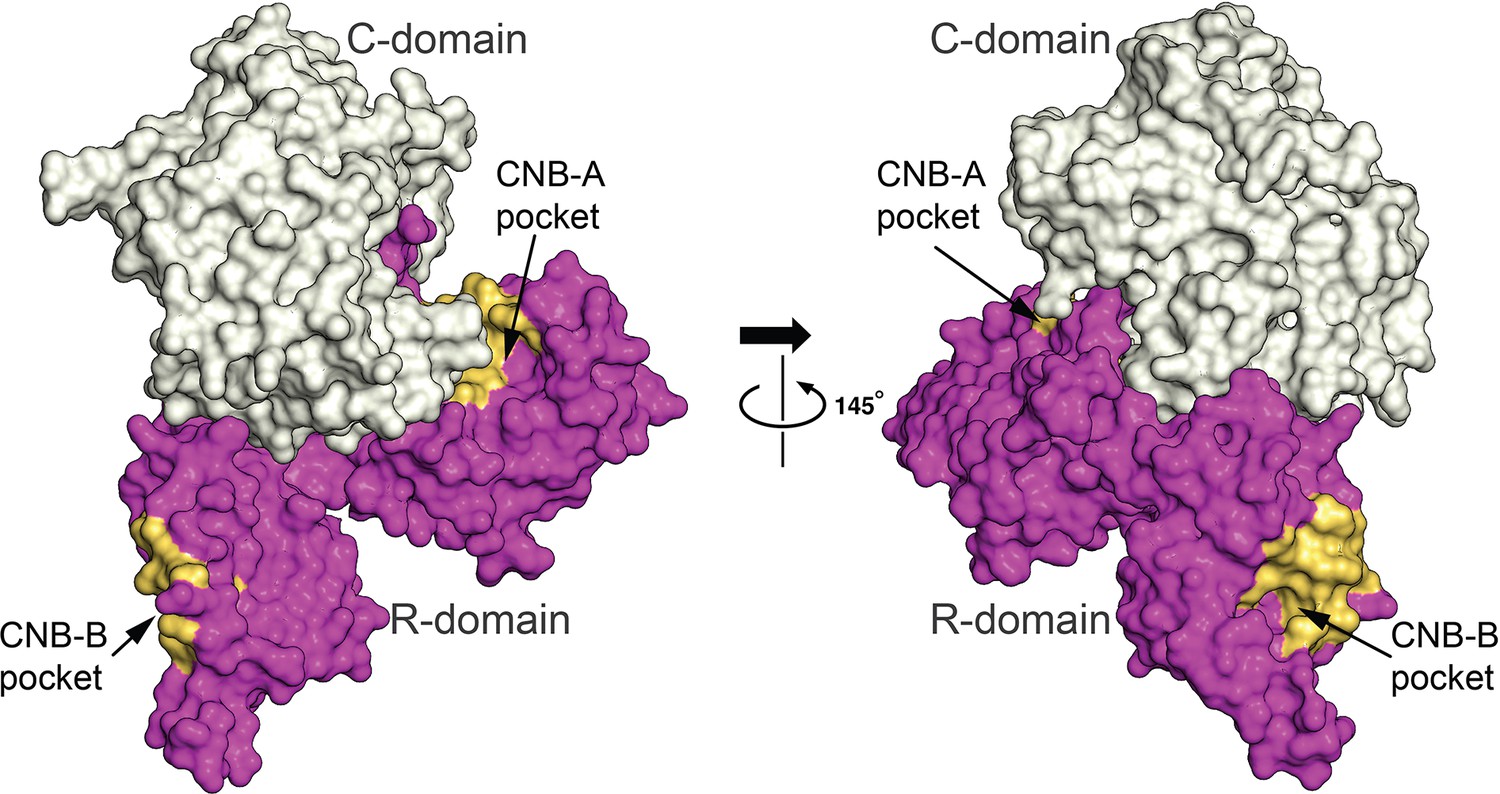
Solvent accessibility of cyclic guanosine monophosphate (cGMP)-binding pockets.
Surface view of the auto-inhibited monomer showing R-, C-domains and R-subdomain pockets (CNB-A and CNB-B). Both CNB-A and -B pockets are exposed to solvent. The C-domain is shown in gray, R-domain in magenta, CNB-A and CNB-B cGMP pockets in yellow.
Additional files
-
Supplementary file 1
Data collection and refinement statistics.
*Information for the highest resolution shell is shown in parenthesis.
†5.0% of the observed intensities were excluded from refinement for cross-validation purposes.
- https://cdn.elifesciences.org/articles/79530/elife-79530-supp1-v2.docx
-
Supplementary file 2
Small-angle X-ray scattering (SAXS) data collection and scattering-derived parameters for PKG Iβ 71–686.
- https://cdn.elifesciences.org/articles/79530/elife-79530-supp2-v2.docx
-
Supplementary file 3
Specific interactions between the R- and C-domains.
Specific interactions between the R- and C-domains. The location of each residue within the complex is listed alongside of each amino acid. Ion pair, hydrogen-bond, and van der Waals interactions are notated as ↔, →, and
 , respectively.
, respectively. - https://cdn.elifesciences.org/articles/79530/elife-79530-supp3-v2.docx
-
Supplementary file 4
Summary of the molecular dynamics (MD) simulations performed for PKG Iβ.
- https://cdn.elifesciences.org/articles/79530/elife-79530-supp4-v2.docx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/79530/elife-79530-mdarchecklist1-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
An auto-inhibited state of protein kinase G and implications for selective activation
eLife 11:e79530.
https://doi.org/10.7554/eLife.79530




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}