Gaucher Disease: Microglia orchestrate neuroinflammation
Experiments in genetically altered mice reveal that microglia play an important role in the neurological damage associated with neuro-nopathic Gaucher disease.
- Department of Microbiology and Immunology, University of Maryland School of Medicine, United States
Gaucher disease is a genetic disorder caused by mutations in the gene coding for the enzyme glucocerebrosidase. These mutations prevent cells from breaking down a lipid called glucosylceramide, which, together with its metabolite glucosylphingosine, promotes inflammation and other alterations that can harm the body’s tissues (Grabowski et al., 2021; Nagata et al., 2017; Nair et al., 2015).
There are three different forms of Gaucher disease: type 1 which is milder and affects the spleen, liver and bone; and types 2 and 3 which, in addition to these alterations, damage neurons and other cell types in the brain. However, the mechanisms responsible for the neurological effects associated with types 2 and 3 (which are collectively known as neuronopathic Gaucher disease) are poorly understood. This is largely because neurodegeneration is difficult to study as it involves complex interactions between neurons and multiple other cell types in the brain, including microglia and astrocytes, which support the function of neurons. Now, in eLife, Pramod Mistry and colleagues from Yale University – including Chandra Sekhar Boddupalli and Shiny Nair as joint first authors – report that microglia play a central role in the neurological damage associated with neuronopathic Gaucher disease (Boddupalli et al., 2022).
The team set out to find how glucocerebrosidase deficiency leads to neuroinflammation which is a major determinant of neurodegeneration in Gaucher disease. To do this, Boddupalli et al. studied a mouse model of neuronopathic Gaucher disease which lacked the gene for glucocerebrosidase (called Gba1) in all tissues except skin to avoid death soon after birth. They also generated three additional mouse models: neuronopathic Gaucher disease mice where Gba1 expression was restored in their microglia or neurons, and mice with Gba1 selectively deleted from their microglia, which mimics late onset neuronopathic Gaucher disease.
Boddupalli et al. found that loss of Gba1 resulted in microglia and astrocyte activation, as well as blood-derived immune cells infiltrating the brain (Figure 1A). Analyzing which genes were expressed in the different cell types of Gba1 deficient mice at a single cell resolution revealed that important neuroinflammatory networks became uncontrolled. In particular, the activated microglia expressed a set of genes that triggered an immune response. However, when the expression of Gba1 was restored in microglia, this reduced inflammation and astrocyte immune activation, stemmed the influx of immune cells, and improved mouse survival (Figure 1B). These results suggest that while the infiltration of immune cells into the brain is a key aspect of neuronopathic Gaucher disease, microglia activation plays a prominent role in the neuroinflammation associated with the disorder.
Figure 1
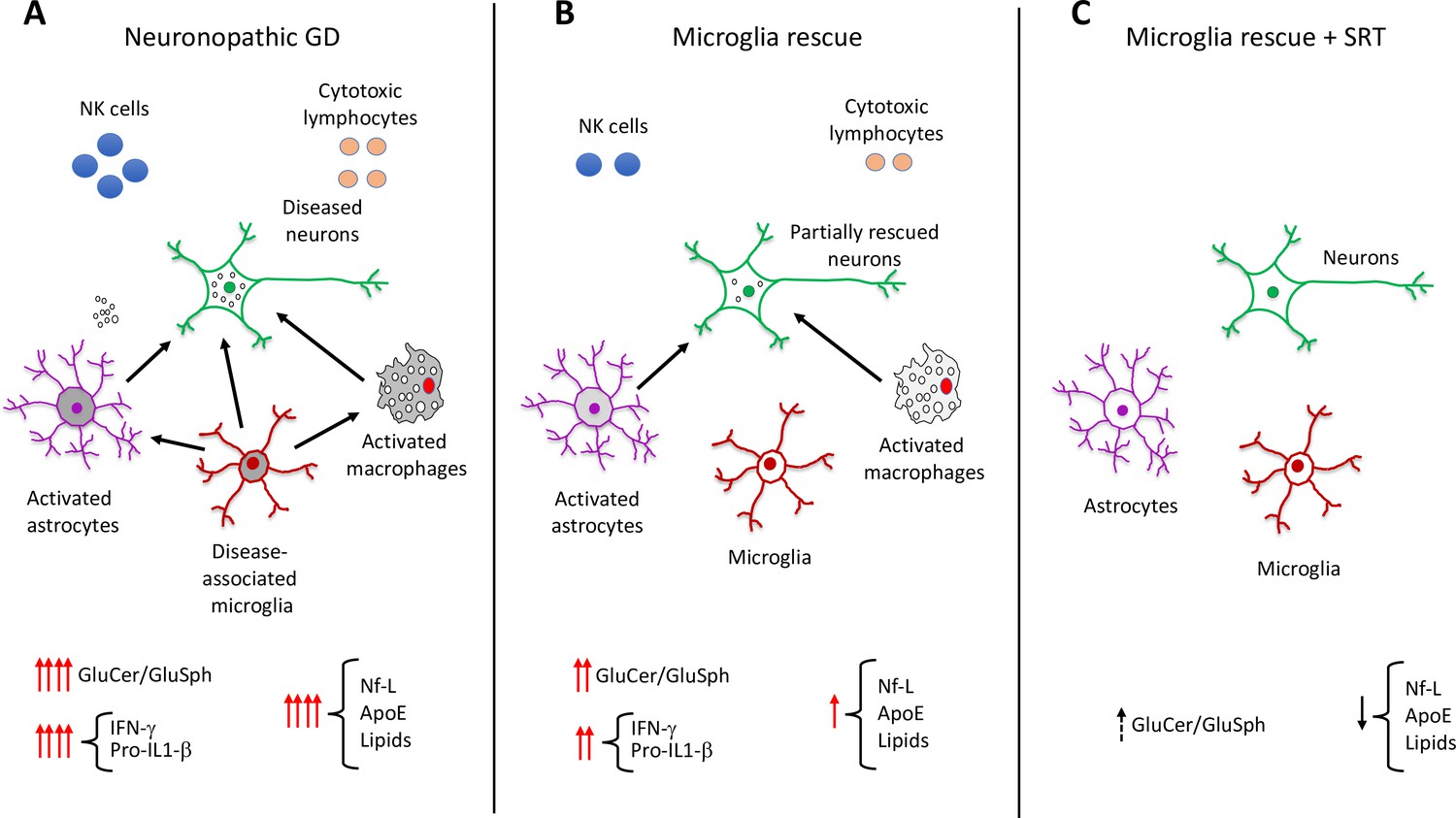
Role of elevated glucosphingolipids and activated microglia in neuronopathic Gaucher disease.
(A) In the brains of neuronopathic Gaucher disease mice are multiple interacting cell types, including neurons (green outline), astrocytes (purple outline), microglia (red outline), and immune cells from the blood, such as macrophages (grey), NK cells (blue circles), and cytotoxic lymphocytes (orange circles). Neuronopathic Gaucher disease mice, which lack the Gba1 gene, have higher levels of glucosylceramide and glucosylsphingosine. Elevation of these glucosphingolipids deregulates the interactions (black arrows) between neurons, microglia, astrocytes and macrophages, causing the immune cells to release inflammatory cytokines such as IFN-γ and Pro-IL-1β. The resulting inflammatory environment leads to neuronal injury and death. Injured neurons release the biomarker Nf-L, and there are also higher levels of the lipoprotein ApoE and certain lipids (such as hexosylceramides and lysophosphatidylcholine), which are released into the blood where they can be detected. (B) Selective rescue of Gba1 expression in the microglia of neuronopathic Gaucher disease mice reduced the influx of blood-derived immune cells, and the immune activity of macrophages and astrocytes. This decreased the levels of inflammatory cytokines, resulting in less neuronal injury and lower levels of the biomarkers Nf-L, ApoE, and lipids. (C) Substrate reduction therapy (SRT) using a drug that blocks the synthesis of glucosylceramide is approved to treat patients with type 1 Gaucher disease. When mice with Gba1 expression restored in their microglia were treated with a brain-permeable SRT drug, the level of glucosphingolipids and neuroinflammation was reduced even further, along with a decrease in the levels of Nf-L, ApoE and lipid biomarkers. GluCer, glucosylceramide; GluSph, glucosylsphingosine; Nf-L, neurofilament light chain; ApoE, apolipoprotein E; IFN-γ,Interferon gamma; Pro-IL-1-β, Pro-Interleukin 1 beta.
Image credit: Ricardo A. Feldman.
Next, Boddupalli et al. investigated the effects of substrate reduction therapy (SRT), a treatment strategy for certain metabolic disorders. SRT drugs block the synthesis of glucosylceramide, preventing this lipid and its metabolite glucosylsphingosine from accumulating inside cells (Blumenreich et al., 2021; Cabrera-Salazar et al., 2012). An SRT approach using a drug that cannot cross the blood-brain-barrier has been approved to treat the non-neurological symptoms of Gaucher disease. Boddupalli et al. found that administration of a brain-penetrant SRT drug to mutant mice that re-expressed Gba1 in their microglia, reduced neuroinflammation even further (Figure 1C). These findings suggest that the accumulation of glucosylceramide and glucosylsphingosine, and microglia activation, are the main drivers of neuroinflammation in Gaucher disease. A treatment that counteracts both these mechanisms could potentially ameliorate the neurological effects of the disorder.
Remarkably, the team also found a number of early indicators of neurological damage in mice lacking Gba1, including a marker of neuronal injury called Nf-L (short for neurofilament light chain; Gaetani et al., 2019; Weinhofer et al., 2021). Nf-L was 2,000 times higher in the blood of neuronopathic Gaucher mice, 100 times higher in mice with Gba1 selectively deleted from their microglia, and this correlated with the elevation of glucosylphingosine, a well-established marker of Gaucher disease. In addition, patients with Gaucher disease type 3 had greater amounts of Nf-L in their blood than patients with type 1, who do not exhibit overt neurological symptoms.
A protein that plays a critical role in lipid metabolism called ApoE (short for apolipoprotein E; Serrano-Pozo et al., 2021) was also highly elevated in the astrocytes and microglia of the mutant mice, as well as in the blood of untreated type 1 Gaucher patients. Furthermore, other lipids, namely hexosylceramides and lysophosphatidylcholine (Bodennec et al., 2002), were present at very high levels in the brains of mice which had Gba1 selectively deleted from their microglia. Boddupalli et al. found that SRT lowered the levels of Nf-L, ApoE, hexosylceramides and lysophosphatidylcholine in the mutant mice, providing further evidence that these molecules are relevant markers for neuronopathic Gaucher disease.
The lack of early biomarkers for neuronopathic Gaucher disease severely curtails clinicians’ ability to detect neurological damage before the onset of symptoms. Further validation of the markers identified in this study could help develop new therapies for preventing or delaying disease progression caused by glucocerebrosidase deficiency.
The potential impact of these findings extends well beyond Gaucher disease, as patients with this disorder have a five- to twenty-fold increased risk of developing Parkinson’s disease and Lewy Body Dementia. Furthermore, carriers of Gaucher disease, who have single allele mutations in the gene for glucocerebrosidase and no symptoms, are also equally susceptible to these neurodegenerative diseases (Aharon-Peretz et al., 2004; Sidransky and Lopez, 2012). Thus, early detection of the biomarkers identified could help clinicians detect which individuals are more likely to develop Parkinson’s disease and Lewy Body Dementia, and prevent or limit progression of these disorders.
References
-
Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi JewsThe New England Journal of Medicine 351:1972–1977.https://doi.org/10.1056/NEJMoa033277
-
Neurofilament light chain as a biomarker in neurological disordersJournal of Neurology, Neurosurgery, and Psychiatry 90:870–881.https://doi.org/10.1136/jnnp-2018-320106
-
Gaucher disease: basic and translational science needs for more complete therapy and managementMolecular Genetics and Metabolism 132:59–75.https://doi.org/10.1016/j.ymgme.2020.12.291
-
The link between the GBA gene and parkinsonismThe Lancet. Neurology 11:986–998.https://doi.org/10.1016/S1474-4422(12)70190-4
Article and author information
Author details
Publication history
Copyright
© 2022, Feldman
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,824
- views
-
- 318
- downloads
-
- 26
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Gaucher Disease: Microglia orchestrate neuroinflammation
eLife 11:e81890.
https://doi.org/10.7554/eLife.81890
Further reading
-
- Medicine
Background:
Approximately one-third of patients with HER2-positive breast cancer experienced recurrence within 10 years after receiving 1 year of adjuvant trastuzumab. The ExteNET study showed that 1 year of extended adjuvant neratinib after trastuzumab-based adjuvant therapy could reduce invasive disease-free survival (iDFS) events compared with placebo. This study investigated the efficacy and safety of pyrotinib, an irreversible pan-HER receptor tyrosine kinase inhibitor, after trastuzumab-based adjuvant therapy in patients with high-risk, HER2-positive early or locally advanced breast cancer.
Methods:
This multicenter phase II trial was conducted at 23 centers in China. After enrollment, patients received 1 year of extended adjuvant pyrotinib (400 mg/day), which should be initiated within 6 months after the completion of 1-year adjuvant therapy (trastuzumab alone or plus pertuzumab). The primary endpoint was 2-year iDFS rate.
Results:
Between January 2019 and February 2022, 141 eligible women were enrolled and treated. As of October 10, 2022, the median follow-up was 24 (interquartile range, 18.0–34.0) months. The 2-year iDFS rate was 94.59% (95% confidence interval [CI]: 88.97–97.38) in all patients, 94.90% (95% CI: 86.97–98.06) in patients who completed 1-year treatment, 90.32% (95% CI: 72.93–96.77) in patients who completed only 6-month treatment, 96.74% (95% CI: 87.57–99.18) in the hormone receptor (HR)-positive subgroup, 92.77% (95% CI: 83.48–96.93) in the HR-negative subgroup, 96.88% (95% CI: 79.82–99.55) in the lymph node-negative subgroup, 93.85% (95% CI: 86.81–97.20) in the lymph node-positive subgroup, 97.30% (95% CI: 82.32–99.61) in patients with adjuvant trastuzumab plus pertuzumab, and 93.48% (95% CI: 86.06–97.02) in patients with adjuvant trastuzumab. The most common adverse events were diarrhea (79.4%), fatigue (36.9%), lymphocyte count decreased (36.9%), nausea (33.3%), and hand-foot syndrome (33.3%).
Conclusions:
Extended adjuvant pyrotinib administrated after trastuzumab-based adjuvant therapy showed promising efficacy in patients with high-risk HER2-positive breast cancer. The follow-up is ongoing to determine the long-term benefit.
Funding:
No external funding was received for this work.
Clinical trial number:
ClinicalTrials.gov: NCT05880927
-
- Immunology and Inflammation
- Medicine
Preeclampsia (PE), a major cause of maternal and perinatal mortality with highly heterogeneous causes and symptoms, is usually complicated by gestational diabetes mellitus (GDM). However, a comprehensive understanding of the immune microenvironment in the placenta of PE and the differences between PE and GDM is still lacking. In this study, cytometry by time of flight indicated that the frequencies of memory-like Th17 cells (CD45RA−CCR7+IL-17A+CD4+), memory-like CD8+ T cells (CD38+CXCR3−CCR7+Helios−CD127−CD8+) and pro-inflam Macs (CD206−CD163−CD38midCD107alowCD86midHLA-DRmidCD14+) were increased, while the frequencies of anti-inflam Macs (CD206+CD163−CD86midCD33+HLA-DR+CD14+) and granulocyte myeloid-derived suppressor cells (gMDSCs, CD11b+CD15hiHLA-DRlow) were decreased in the placenta of PE compared with that of normal pregnancy (NP), but not in that of GDM or GDM&PE. The pro-inflam Macs were positively correlated with memory-like Th17 cells and memory-like CD8+ T cells but negatively correlated with gMDSCs. Single-cell RNA sequencing revealed that transferring the F4/80+CD206− pro-inflam Macs with a Folr2+Ccl7+Ccl8+C1qa+C1qb+C1qc+ phenotype from the uterus of PE mice to normal pregnant mice induced the production of memory-like IL-17a+Rora+Il1r1+TNF+Cxcr6+S100a4+CD44+ Th17 cells via IGF1–IGF1R, which contributed to the development and recurrence of PE. Pro-inflam Macs also induced the production of memory-like CD8+ T cells but inhibited the production of Ly6g+S100a8+S100a9+Retnlg+Wfdc21+ gMDSCs at the maternal–fetal interface, leading to PE-like symptoms in mice. In conclusion, this study revealed the PE-specific immune cell network, which was regulated by pro-inflam Macs, providing new ideas about the pathogenesis of PE.




{kind=link}