Immunogenetics: Untangling the immune basis of disease susceptibility
Interactions between immune cell receptors and proteins that determine disease susceptibility shed light on how different arms of the immune system are involved in three viral infections and Crohn's disease.
- Laboratório de Biomatemática, Instituto de Saúde Ambiental, Faculdade de Medicina, Universidade de Lisboa, Portugal
- Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Portugal
Why do people respond differently to the same infectious agent? A possible answer is that differences in the immune responses of individuals, caused by genetic variability between them, are responsible. Indeed, many studies have looked for associations between genes involved in immunity and disease outcome (Buniello et al., 2019), and it has been found that a gene complex called the human leukocyte antigen (HLA) system has a central role (Matzaraki et al., 2017).
The HLA genes encode the major histocompatibility complex (MHC). MHC proteins are found on the surface of cells and each one presents peptides from proteins within the cell (either proteins native to the cell or from foreign entities like bacteria) to immune cells. Thus, MHC proteins allow immune cells to recognize if a given cell is dangerous or not. For example, immune cells called CD8+ T lymphocytes recognize cells that have been infected with viruses because receptors on these T cells bind specific MHC proteins loaded with viral peptides on the surface of the infected cells. Likewise, other types of immune cells – such as natural killer cells, macrophages and dendritic cells – have receptors that bind to other parts of the MHC protein displayed by infected cells (Augusto and Petzl-Erler, 2015; Hudson and Allen, 2016).
The HLA is extremely diverse among individuals. It has been assumed that the association between some diseases and certain HLA alleles is evidence for a central role of CD8+ T cells in that disease. However, given that the HLA can also interact with receptors on other immune cells, disentangling the contributions of the different arms of the immune system remains a challenge. Now, in eLife, Becca Asquith of Imperial College and colleagues – including Bisrat Debebe as first author, researchers from various institutes in the UK and US, and the IAVI Protocol C Investigators – report the results of a new approach to disentangling these contributions (Debebe et al., 2020).
Debebe et al. reasoned that it may be possible to predict which immune cells are responsible for fighting a specific disease since receptors from particular immune cells interact with different regions of the MHC protein. For example, susceptibility to a disease may be associated with individuals carrying MHC proteins that are similar in the region that presents protein fragments to the T-cell receptor (which can be thought of as being similar in ‘T-cell receptor space’). If this is the case, then CD8+ T cells are likely involved in fighting the disease. The same reasoning can be made for associations with the other receptors, such as the killer immunoglobulin-like receptor (KIR) receptors in natural killer cells and leukocyte immunoglobulin-like receptor (LILR) in myeloid cells (Figure 1).
Figure 1
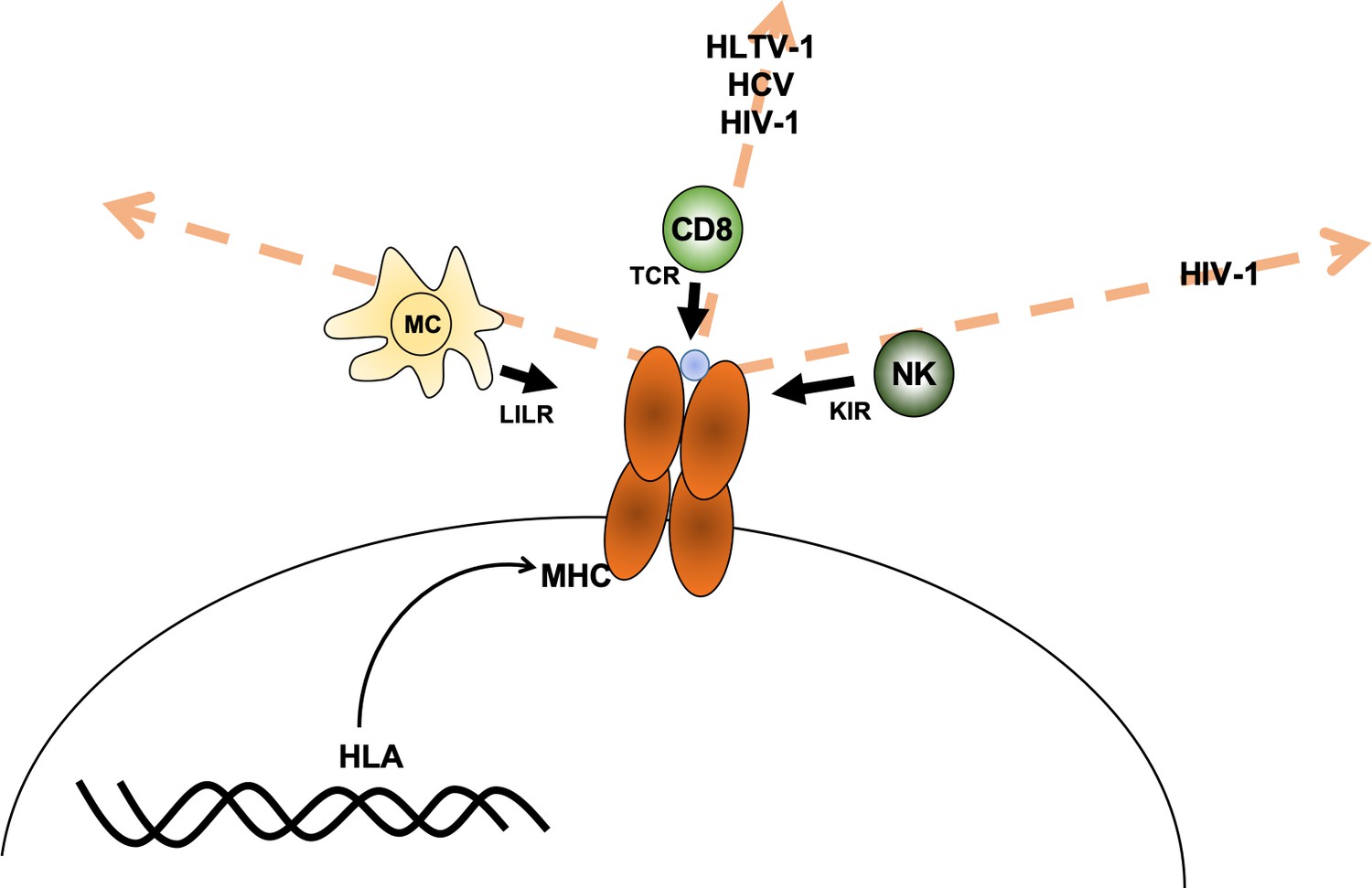
Axes of similarity in MHC proteins.
Debebe et al. showed that similarities in regions of the MHC molecule that interact with different immune cell receptors can be thought of in terms of different 'spaces' (which are defined by an axis for each type of receptor). They found that MHCs that are similar in the T-cell receptor (TCR) axis are associated with disease outcomes for HLTV-1, hepatitis C virus (HCV) and HIV-1. Disease outcomes for HIV-1 are also associated with MHCs that show similarity in the killer immunoglobulin-like receptor (KIR) axis. For the diseases tested, no MHCs were found to be similar in the region that interacts with the leukocyte immunoglobulin-like receptor (LILR) carried by myeloid cells (MC). CD8: CD8+ T cells; NK: natural killer cells; blue circle: peptide.
The challenge in this method is defining MHC similarity. Debebe et al. did so by taking an HLA known to associate with a specific disease outcome and predicting its ability to bind to different immune cell receptors. Next, they compared this predicted binding ability to that of all other HLAs, and found the HLAs with the highest binding similarity. Finally, they checked if these HLAs are also associated with disease susceptibility.
Debebe et al. applied this method to three infections (human T-cell leukemia virus type one or HTLV-1; hepatitis C virus; and HIV-1) and one autoimmune disease (Crohn’s disease). They used well-characterized cohorts where both the HLA and the outcome of the disease were known for each individual.
They studied three known HLA-outcome associations for HTLV-1 and found that other HLAs that were similar in T-cell receptor space were also associated with HTLV-1. However, other HLAs that were similar to these three HLAs in LILR space or KIR space (see Figure 1) were not associated with HTLV-1. Thus, they concluded that HTLV-1 is most likely controlled by CD8+ T cells. They also studied three HLA-outcome associations for hepatitis C, reaching the same conclusion: disease outcome is mostly controlled by CD8+ T cells. For HIV-1, Debebe et al. found that natural killer cells and CD8+ T cells both have a role, which is consistent with previous results (Bashirova et al., 2011; McBrien et al., 2018). They also studied two known HLA-outcome associations for Crohn’s disease in a large sample (2650 individuals), but other HLAs that were similar in T-cell, LILR or KIR space did not have significant effect on Crohn's disease outcome.
The approach suggested by Debebe et al. generates insights into which immune cells may be involved in fighting a disease. There are two issues that need to be explored further. First, while we know a great deal about T-cell receptors, we know less about KIR, and even less about LILR binding (Hirayasu and Arase, 2015). This knowledge (or lack thereof) is part of the definition of similarity used by Debebe et al., which can and should be updated as more data becomes available (Gwozdowicz et al., 2019). The second issue pertains to the fact that it may be possible to define similarity in other ways, leading to different results that provide further insights into how genetic variability affects the immune response.
References
-
HLA/KIR restraint of HIV: surviving the fittestAnnual Review of Immunology 29:295–317.https://doi.org/10.1146/annurev-immunol-031210-101332
-
KIR specificity and avidity of standard and unusual C1, C2, Bw4, Bw6 and A3/11 amino acid motifs at entire HLA:KIR interface between NK and target cells, the functional and evolutionary classification of HLA class I moleculesInternational Journal of Immunogenetics 46:217–231.https://doi.org/10.1111/iji.12433
-
Leukocyte Ig-like receptors - A model for MHC class I disease associationsFrontiers in Immunology 7:281.https://doi.org/10.3389/fimmu.2016.00281
-
Mechanisms of CD8+ T cell-mediated suppression of HIV/SIV replicationEuropean Journal of Immunology 48:898–914.https://doi.org/10.1002/eji.201747172
Article and author information
Author details
Publication history
Copyright
© 2020, Ribeiro and Graca
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,857
- views
-
- 297
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Immunogenetics: Untangling the immune basis of disease susceptibility
eLife 9:e56886.
https://doi.org/10.7554/eLife.56886
Further reading
-
- Computational and Systems Biology
In eukaryotes, protein kinase signaling is regulated by a diverse array of post-translational modifications, including phosphorylation of Ser/Thr residues and oxidation of cysteine (Cys) residues. While regulation by activation segment phosphorylation of Ser/Thr residues is well understood, relatively little is known about how oxidation of cysteine residues modulate catalysis. In this study, we investigate redox regulation of the AMPK-related brain-selective kinases (BRSK) 1 and 2, and detail how broad catalytic activity is directly regulated through reversible oxidation and reduction of evolutionarily conserved Cys residues within the catalytic domain. We show that redox-dependent control of BRSKs is a dynamic and multilayered process involving oxidative modifications of several Cys residues, including the formation of intramolecular disulfide bonds involving a pair of Cys residues near the catalytic HRD motif and a highly conserved T-loop Cys with a BRSK-specific Cys within an unusual CPE motif at the end of the activation segment. Consistently, mutation of the CPE-Cys increases catalytic activity in vitro and drives phosphorylation of the BRSK substrate Tau in cells. Molecular modeling and molecular dynamics simulations indicate that oxidation of the CPE-Cys destabilizes a conserved salt bridge network critical for allosteric activation. The occurrence of spatially proximal Cys amino acids in diverse Ser/Thr protein kinase families suggests that disulfide-mediated control of catalytic activity may be a prevalent mechanism for regulation within the broader AMPK family.
-
- Computational and Systems Biology
- Genetics and Genomics
Obesity is a major risk factor for type 2 diabetes, dyslipidemia, cardiovascular disease, and hypertension. Intriguingly, there is a subset of metabolically healthy obese (MHO) individuals who are seemingly able to maintain a healthy metabolic profile free of metabolic syndrome. The molecular underpinnings of MHO, however, are not well understood. Here, we report that CTRP10/C1QL2-deficient mice represent a unique female model of MHO. CTRP10 modulates weight gain in a striking and sexually dimorphic manner. Female, but not male, mice lacking CTRP10 develop obesity with age on a low-fat diet while maintaining an otherwise healthy metabolic profile. When fed an obesogenic diet, female Ctrp10 knockout (KO) mice show rapid weight gain. Despite pronounced obesity, Ctrp10 KO female mice do not develop steatosis, dyslipidemia, glucose intolerance, insulin resistance, oxidative stress, or low-grade inflammation. Obesity is largely uncoupled from metabolic dysregulation in female KO mice. Multi-tissue transcriptomic analyses highlighted gene expression changes and pathways associated with insulin-sensitive obesity. Transcriptional correlation of the differentially expressed gene (DEG) orthologs in humans also shows sex differences in gene connectivity within and across metabolic tissues, underscoring the conserved sex-dependent function of CTRP10. Collectively, our findings suggest that CTRP10 negatively regulates body weight in females, and that loss of CTRP10 results in benign obesity with largely preserved insulin sensitivity and metabolic health. This female MHO mouse model is valuable for understanding sex-biased mechanisms that uncouple obesity from metabolic dysfunction.




{kind=link}