PRC1 sustains the integrity of neural fate in the absence of PRC2 function
- Neuroscience Institute, Department of Neuroscience and Physiology, NYU School of Medicine, United States
- Department of Biology, New York University, United States
- Applied Bioinformatics Laboratories, Office of Science and Research, NYU School of Medcine, United States
Abstract
Polycomb repressive complexes (PRCs) 1 and 2 maintain stable cellular memories of early fate decisions by establishing heritable patterns of gene repression. PRCs repress transcription through histone modifications and chromatin compaction, but their roles in neuronal subtype diversification are poorly defined. We found that PRC1 is essential for the specification of segmentally restricted spinal motor neuron (MN) subtypes, while PRC2 activity is dispensable to maintain MN positional identities during terminal differentiation. Mutation of the core PRC1 component Ring1 in mice leads to increased chromatin accessibility and ectopic expression of a broad variety of fates determinants, including Hox transcription factors, while neuronal class-specific features are maintained. Loss of MN subtype identities in Ring1 mutants is due to the suppression of Hox-dependent specification programs by derepressed Hox13 paralogs (Hoxa13, Hoxb13, Hoxc13, Hoxd13). These results indicate that PRC1 can function in the absence of de novo PRC2-dependent histone methylation to maintain chromatin topology and postmitotic neuronal fate.
Editor's evaluation
This is an exciting and very well executed study, which will be of broad interest to the field of neuronal development. The demonstration of a similar logic in mouse as to what was reported earlier for Drosophila PRCs supports the idea of a deeply conserved mechanism of rostrocaudal patterning where PRC complexes control Hox gene expression.
https://doi.org/10.7554/eLife.72769.sa0Introduction
Accurate control of gene expression is essential for the specification and maintenance of neural fates during development. Studies of cell-type-restricted transcription factors have illuminated the mechanisms by which spatial and temporal regulation of gene expression gives rise to identifiable neuronal subtypes (Doe, 2017; Fishell and Kepecs, 2020; Hobert and Kratsios, 2019; Sagner and Briscoe, 2019; Venkatasubramanian and Mann, 2019). A parallel and critical mechanism of gene regulation is through the post-translational modification of histones, which enables and restricts transcription by modifying chromatin structure (Kishi and Gotoh, 2018; Schuettengruber et al., 2017). The Polycomb group (PcG) is key family of histone-associated proteins that play evolutionarily conserved roles in restricting gene expression during development (Blackledge et al., 2015; Gentile and Kmita, 2020; Simon and Kingston, 2009; Soshnikova and Duboule, 2009). In embryonic stem cells, cell fate determinants are repressed through PRC activities, and PRC-associated histone marks are subsequently removed from loci as cells differentiate (Boyer et al., 2006; Farcas et al., 2012; Tavares et al., 2012). PRC repression is maintained through cell division and after differentiation and is thought to contribute to stable cellular memories of early patterning events (Ciabrelli et al., 2017; Coleman and Struhl, 2017). In vertebrates, much of our knowledge of how PRCs regulate gene expression has emerged from biochemical studies of PcG proteins or from the activity of these factors in proliferating cells. Despite an in depth understanding of the mechanisms of PRC action, how PcG proteins interact with gene regulatory networks in the CNS remains poorly understood.
The specification of neuronal fates in the vertebrate spinal cord provides a tractable system to elucidate the function of PcG proteins, as the pathways that determine identities are well-defined, and the molecular signatures of many subtypes are known (Butler and Bronner, 2015; Sagner and Briscoe, 2019). One neuronal class where fate specification has been closely examined is the spinal MN. A core set of transcription factors, including Mnx1, Isl1/2, and Lhx3, determines class-specific features of MNs, including neurotransmitter identity (Shirasaki and Pfaff, 2002). The subsequent diversification of MNs into hundreds of muscle-specific subtypes is achieved through a conserved network of Hox transcription factors differentially expressed along the rostrocaudal axis (Philippidou and Dasen, 2013). During neural tube patterning, opposing gradients of retinoic acid (RA) and fibroblast growth factors (FGFs) provide spinal progenitors with a positional identity (Bel-Vialar et al., 2002; Dasen et al., 2003; Liu et al., 2001). These morphogens act, in part, by temporally and spatially depleting Polycomb-associated histone marks from Hox clusters (Mazzoni et al., 2013). As progenitors exit the cell cycle, MNs continue to express Hox genes, where they regulate repertoires of subtype-specific genes (Catela et al., 2016; Dasen et al., 2005; Mendelsohn et al., 2017). Although the role of Hox proteins in the CNS are well-characterized (Parker and Krumlauf, 2020), and are known targets of PRC activities (Gentile and Kmita, 2020), the specific contributions of PRC1 and PRC2 to CNS maturation remain unclear, as few studies have directly compared their functions during embryonic development.
Polycomb repression is initiated by PRC2, which methylates histone H3 at lysine-27 (H3K27me3), permitting recruitment of PRC1 through subunits that recognize this mark, leading to chromatin compaction at genes targeted for repression (Margueron and Reinberg, 2011; Schuettengruber et al., 2017). In embryonic stem (ES) cells, Hox gene clusters are initially covered by H3K27me3 and loss of PRC2 function leads to reduced PRC1 binding and ectopic Hox expression (Boyer et al., 2006). During mouse development, H3K27me3 marks are progressively removed from Hox clusters, allowing for the temporal and spatial activation of more caudal Hox genes during axis extension (Soshnikova and Duboule, 2009). The progressive removal of PRC2-associated histone marks is also recapitulated in ES cell-derived MNs (ESC-MNs), where RA functions to deplete H3K27me3 from rostral Hox1-Hox5 paralogs, while FGF removes H3K27me3 from more caudal Hox genes through Cdx proteins (Mazzoni et al., 2013). Although loss of PRC2 affects the viability of ESC-MNs, a hypomorphic mutation in the PRC2 component Suz12 leads to ectopic Hox expression (Mazzoni et al., 2013). Thus, during early phases of embryonic development, PRC2 appears to have a critical role in establishing the early profiles of Hox expression in MNs along the rostrocaudal axis.
PRC1 and PRC2 can also exist in a variety of configurations, which may contribute to neuronal subtype-specific activities. The core subunit of PRC1, Ring1, binds to one of six Polycomb group Ring finger (Pcgf) proteins (Gao et al., 2012). PRC1 containing Pcgf4 interacts with Cbx proteins (canonical PRC1) which recognize H3K27me3 (Bernstein et al., 2006; Morey et al., 2012). Variant forms of PRC1 containing Rybp can inhibit incorporation of Cbx proteins into PRC1, and bind target loci independent of H3K27me3 (Tavares et al., 2012; Wang et al., 2010). We previously found that a PRC1 component, Pcgf4 (Bmi1), is required to establish rostral boundaries of Hox expression in differentiating MNs (Golden and Dasen, 2012). By contrast, deletion of a core PRC2 component, Ezh2, from MN progenitors had no apparent impact on fate specification, possibly due to compensation by Ezh1 (Shen et al., 2008), or through use of variant PRC1 containing Rybp. These observations raise the question of what are the specific roles of canonical PRC1, variant PRC1, and PRC2 during MN subtype diversification.
To determine the function of PRCs during neural differentiation, we removed core components of these complexes from MN progenitors. We found that depletion of Ring1 proteins, essential constituents of all PRC1 complexes, causes pronounced changes in transcription factor expression and a loss of Hox-dependent MN subtypes. By contrast, neither PRC2 nor variant PRC1 activities are required to maintain rostrocaudal positional identities at the time of MN differentiation. Deletion of Ring1 leads to increased chromatin accessibility and derepression of a broad variety of cell fate determinants, while class-specific features of MNs are preserved. The derepression of caudal Hox genes in Ring1 mutants leads to the suppression of MN subtype diversification programs. These findings indicate that PRC1 function is essential during terminal differentiation to specify the transcriptional identities of motor neurons.
Results
PRC1 is essential for rostrocaudal patterning during neuronal differentiation
To determine the relative contributions of PRC1 and PRC2 to neuronal specification, we analyzed mice in which core subunit-encoding genes were selectively removed from MN progenitors. Ezh genes encode the methyltransferase activity of PRC2, while Eed is required to enhance this function. We first generated mice in which both Ezh genes are conditionally deleted, by breeding Ezh1 and Ezh2 floxed lines to Olig2Cre mice, which targets Cre to MN progenitors (EzhMNΔ mice) (Hidalgo et al., 2012; Su et al., 2003). We confirmed MN-restricted loss of PRC2 activity by examining the pattern of H3K27me3, which was selectively depleted from progenitors and post-mitotic MNs by E11.5 (Figure 1A and B, Figure 1—figure supplement 1A,B). Expression of Mnx1 and Slc18a3 (Vacht), two general markers of MN identity, were grossly unchanged in EzhMNΔ mice (Figure 1C, D and G). We next analyzed expression of Hox proteins in EzhMNΔ mice and found that the MN columnar subtype determinants Hoxc6, Hoxc9, and Hoxc10, were all expressed in their normal domains (Figure 1E and F, Figure 1—figure supplement 1C-E).
Figure 1 with 2 supplements see all
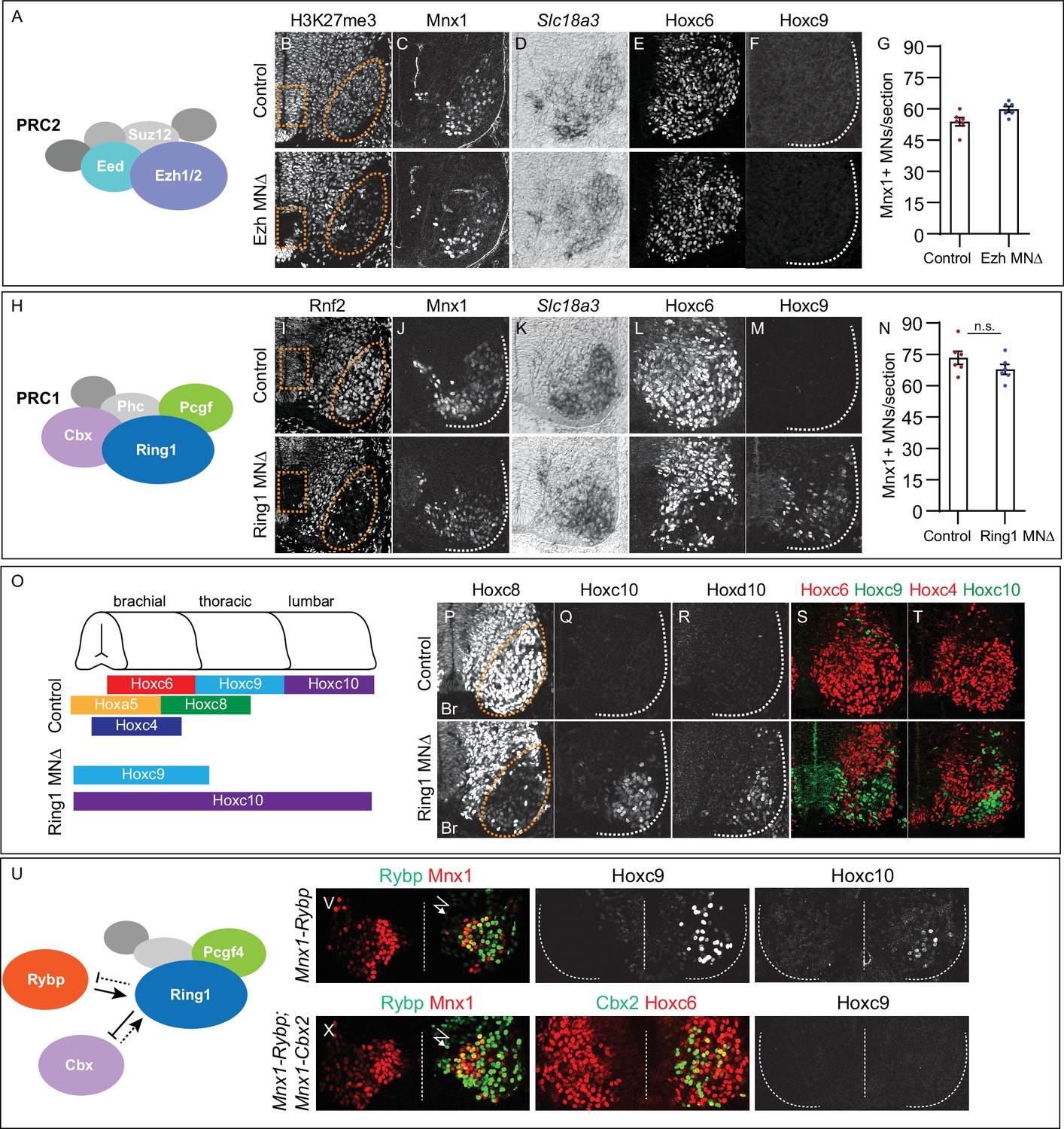
Roles of PRC1 and PRC2 in determining of Hox expression in spinal MNs.
(A) Core components of PRC2. (B) Brachial spinal sections showing H3K27me3 is depleted from progenitors (boxed region) and post-mitotic MNs (oval) in E11.5 EzhMNΔ (Ezh1flox/flox::Ezh2flox/flox, Olig2Cre) embryos. (C–D) MNs express Mnx1 and Slc18a3 (Vacht) in EzhMNΔ mice. (E–F) Brachial Hoxc6 expression is normal in EzhMNΔ mice, and no ectopic Hoxc9 is detected. (G) Quantification of MNs: 54±2 Mnx1+ MNs per section in brachial controls, versus 60±1 in EzhMNΔ mice, n = 6 sections, p = 0.0342, unpaired t-test. (H) Core components of PRC1. (I) Rnf2 (Ring1B) is selectively removed from progenitors (boxed region) and post-mitotic MNs (oval) in E12.5 Ring1MNΔ (Ring1-/-::Rnf2flox/flox, Olig2Cre) mice. (J–K) Ring1MNΔ mice express Mnx1 and Slc18a3. (L–M) Hoxc6 is lost from brachial MNs and Hoxc9 is ectopically expressed in Ring1MNΔ mice. (N) Quantification of MNs: 73±3 Mnx1+ MNs in controls, versus 68±2 in Ring1MNΔ mice, n = 6 sections, p = 0.1879, unpaired t-test. (O) Summary of changes in MN Hox expression of Ring1MNΔ mice. (P–R) Loss of Hoxc8 and ectopic Hoxc10 and Hoxd10 expression in brachial MNs of Ring1MNΔ mice. (S–T) Co-labeling of Hoxc6/Hoxc9 and Hoxc4/Hoxc10 in Ring1MNΔ mice, showing ectopically expressed caudal Hox proteins and loss of rostral Hox expression in brachial segments. (U) Schematic of Cbx and Rybp interactions in PRC1. (V) Misexpression of Rybp in postmitotic MNs under Mnx1 in chick leads to ectopic Hoxc9 and Hoxc10 expression in brachial MNs. Bolt symbol indicates electroporated side of spinal cord. (X) Co-expression of Rybp and Cbx under Mnx1 fails to induce Hoxc9 in brachial MNs. Panels B-F show brachial sections from E11.5 embryos; I-M, P-T brachial sections from E12.5 embryos; V,X brachial sections of HH st25 chick.
-
Figure 1—source data 1
Counts of Mnx1-positive MNs in Ezh and Ring1 mutants.
- https://cdn.elifesciences.org/articles/72769/elife-72769-fig1-data1-v2.xlsx
Removal of Ezh genes via Olig2Cre depletes PRC2 function in MN progenitors, raising the possibility that PRC2 regulates Hox expression at earlier stages. We therefore analyzed mice in which the core PRC2 component Eed was deleted using Sox1Cre (EedNEΔ mice), which targets Cre to neuroectoderm (Takashima et al., 2007; Yaghmaeian Salmani et al., 2018). In EedNEΔ mice, H3K27me3 was depleted from spinal progenitors and postmitotic neurons by E11.5, with some H3K27me3 present in the floor plate (Figure 1—figure supplement 1G). In EedNEΔ mice, the number of Mnx1+ cells, and pattern of Hoxc6, Hoxc9, and Hoxc10 in MNs were unaffected, similar to EzhMNΔ mice (Figure 1—figure supplement 1F, H-J). These observations indicate that depletion of PRC2 function at the time of differentiation does not affect MN class specification or rostrocaudal positional identities.
We next investigated the function of PRC1, which is thought to repress gene expression in a PRC2-dependent manner. We generated mice in which Rnf2 (also known as Ring1B) is conditionally deleted from MN progenitors using Olig2Cre in a global Ring1 (also known as Ring1A) mutant background (Ring1MNΔ mice) (Calés et al., 2008; del Mar Lorente et al., 2000). In Ring1MNΔ mice, Rnf2 (Ring1B) protein was selectively removed from progenitors and post-mitotic MNs (Figure 1H,I). The number of Mnx1+ MNs and pattern of Slc18a3 expression were similar to controls in Ring1MNΔ mice (Figure 1J, K and N). By contrast, Hox proteins normally expressed by forelimb-innervating brachial MNs were not detected, while thoracic and lumbar Hox proteins were ectopically expressed in more rostral MNs (Figure 1L, M and O–T, Figure 1—figure supplement 2C-E). Brachial-level Hox proteins (Hoxc4, Hoxa5, Hoxc6 and Hoxc8), were selectively depleted from MNs, while thoracic and lumbar Hox determinants, (Hoxc9, Hoxc10, and Hoxd10) were derepressed in brachial segments (Figure 1Q–T). Brachial MNs also co-expressed Hoxc9 and Hoxc10, which are normally restricted to thoracic and lumbar segments, respectively (Figure 1—figure supplement 2B). At thoracic levels, Hoxc9 expression was attenuated and Hoxd10 was ectopically expressed (Figure 1—figure supplement 2D). These results show that loss of Ring1 causes a derepression of caudal Hox genes, leading to co-expression of caudal Hox proteins in brachial MNs, without affecting pan-MN molecular features (Figure 1O).
Canonical PRC1 regulates Hox expression in MNs
Ring1 can interact with multiple Pcgf proteins, raising the question of which PRC1 configuration contributes to MN patterning. PRC1 containing Pcgf4 is required to establish Hox boundaries in MNs (Golden and Dasen, 2012), but can exist in two alternative configurations, depending on mutually exclusive incorporation of Cbx (canonical PRC1) or Rybp (variant PRC1) (Gao et al., 2012; Tavares et al., 2012). We examined the function of PRC1 isoforms first by manipulating Rybp and Cbx expression in MNs. We hypothesized that if canonical PRC1 regulates Hox expression then overexpression of Rybp would inhibit binding of Cbx to Ring1, leading to MN phenotypes similar to Ring1 mutants (Figure 1U). We used chick in ovo neural tube electroporation to express mouse Rybp in postmitotic MNs using the Mnx1 promoter (Mnx1-Rybp). Expression of Rybp under Mnx1 led to ectopic Hoxc9 and Hoxc10 expression at brachial levels (Figure 1V). If Rybp acts by displacing Cbx, then elevating Cbx levels should restore normal Hox expression. To test this, we co-electroporated Mnx1-Rybp and Mnx1-Cbx2 at equivalent plasmid concentration, and no longer observed ectopic Hoxc9 in brachial segments (Figure 1X).
These observations are consistent with canonical PRC1 containing Pcgf4, Cbx, and Ring1 restricting Hox expression in MNs. Rybp is expressed by MNs at the time of their differentiation (Figure 1—figure supplement 2F), raising the possibility that Rybp (or its paralog Yaf2) also plays regulatory roles during development. We therefore selectively deleted Rybp from MNs using Olig2Cre mice in a Yaf2-/- background (Rybp/Yaf2MNΔ mice). Combined deletion of Rybp and Yaf2 did not affect MN generation, Hoxc6, Hoxc9, or Hoxc10 expression (Figure 1—figure supplement 2F). These findings indicate that canonical PRC1 maintains appropriate Hox expression, while variant PRC1 and PRC2 do not contribute to rostrocaudal patterning at the time of MN differentiation.
PRC1 is required in MNs for subtype diversification
Hox transcription factors play central roles in establishing neuronal subtype identities through regulating expression of subtype-specific genes. To determine the consequences of altered Hox expression in Ring1MNΔ mice, we analyzed the molecular profiles and peripheral innervation pattern of MNs. A key Hox target in MNs is the transcription factor Foxp1, which is essential for the differentiation of limb-innervating lateral motor column (LMC) and thoracic preganglionic column (PGC) neurons (Dasen et al., 2008; Rousso et al., 2008). In Ring1MNΔ mice, Foxp1 expression is lost from brachial and thoracic MNs, and markedly reduced in lumbar MNs (Figure 2A, B and G, Figure 2—figure supplement 1C). By contrast, in EzhMNΔ, EedNEΔ, and Rybp/Yaf2MNΔ mice, Foxp1 expression was maintained, consistent with the preservation of normal Hox profiles in these mutants (Figure 1—figure supplement 2F; Figure 2—figure supplement 1A, B). In Ring1MNΔ mice, expression of the LMC marker Raldh2 was lost at brachial levels, and the thoracic PGC marker nNos was not detected (Figure 2C and D). Moreover, expression of determinants of respiratory phrenic MNs (Scip+ Isl1/2+), was markedly depleted, and likely contributes to the perinatal lethality of Ring1MNΔ mice (Figure 2—figure supplement 1D). These results indicate that Ring1 deletion leads to a loss of genes acting downstream of Hox function in MNs.
Figure 2 with 1 supplement see all
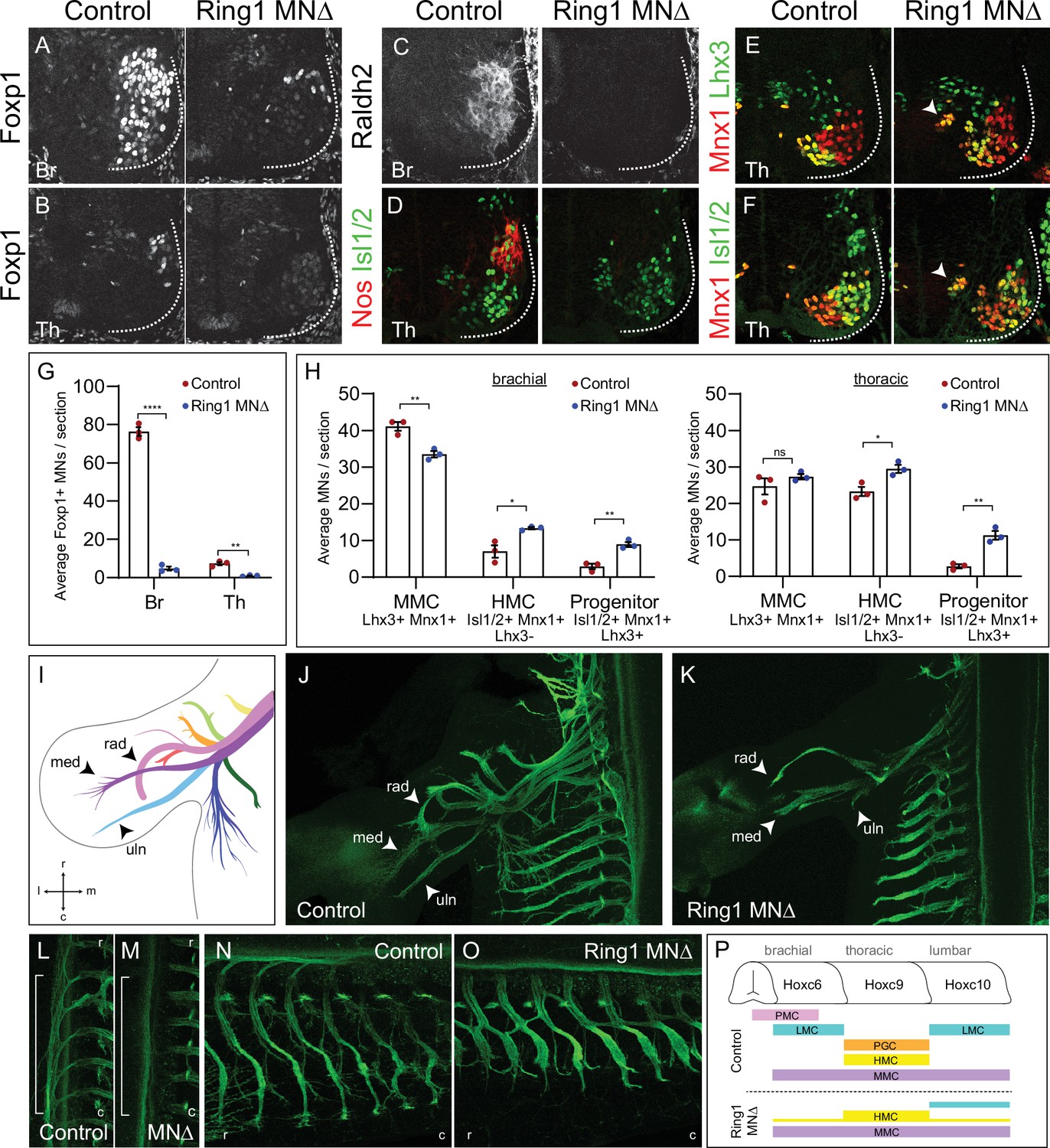
Ring1 is essential for the specification of Hox-dependent MN subtypes.
(A–B) Foxp1 expression is reduced in brachial (Br) and thoracic (Th) segments of Ring1MNΔ mice at E12.5. (C–D) Expression of the brachial LMC marker Raldh2 and thoracic PGC marker Nos were lost in Ring1MNΔ mice. (E–F) Staining of Mnx1+, Lhx3+ (MMC) and Mnx1, Isl1/2+ (HMC) neurons. In Ring1MNΔ mice, we also observed a population of medial neurons that coexpressed Isl1/2, Mnx1, and Lhx3 (indicated by arrow heads). (G) Quantification of Foxp1 reduction in Br and Th segments. (H) Quantification of MMC (Mnx1+, Lhx3+), HMC (Isl1/2+, Mnx1+, Lhx3-) and medial ‘progenitors’ (Isl1/2+, Mnx1+, Lhx3+) MNs arrow heads in E,F. Panels G-H show average from n = 3 mice, four sections each animal. Data shown in graphs shown as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001, unpaired t-test. (I) Schematic of nine primary nerves in E12.5 mouse forelimb (Adapted from Figure 1A Catela et al., 2016) med = median, rad = radial, uln = ulnar nerves. Rostral (r), caudal (c), medial (m), and lateral (l) orientation shown. (J–K) Forelimb motor axons of an E12.5 control and Ring1MNΔ mouse labeled by Mnx1-GFP. (L–M) Innervation of sympathetic chain ganglia (from PGC neurons) in control and Ring1MNΔ mice. Bracket shows region of PGC projections along rostrocaudal axis. (N–O) Innervation of dorsal and ventral axial muscles by MMC and HMC respectively. In Ring1MNΔ mice, HMC motor projections are shorter and thicker. (P) Summary of MN columnar organization of control and Ring1MNΔ mice.
-
Figure 2—source data 1
Quantification of MN subtypes in Ring1 mutants.
- https://cdn.elifesciences.org/articles/72769/elife-72769-fig2-data1-v2.xlsx
We next assessed the impact of Ring1 deletion on two MN columnar subtypes that are specified independent of Hox function, the hypaxial and median motor columns (HMC and MMC) (Dasen et al., 2008; Jung et al., 2014). In Ring1MNΔ mice, MMC neurons (Lhx3+ Mnx1+) were generated at normal numbers in thoracic segments, while the number of HMC neurons (Isl1/2+ Mnx1+ Lhx3-) was increased in thoracic and brachial segments (Figure 2E, F and H, Figure 2—figure supplement 1E,F). The increase in HMC neurons in Ring1 mutants is likely due to a reversion of presumptive Hox-dependent subtypes to an HMC fate, similar to mice in which Hox function is disrupted (Dasen et al., 2008; Hanley et al., 2016). We also observed a small population of medial neurons that coexpressed Isl1/2, Mnx1, and Lhx3 (Figure 2E, F and H). These cells likely represent undifferentiated MN precursors, but did not appear to express progenitor markers such as Olig2 and Nkx6.1 (Figure 1—figure supplement 2A). Mutation in Ring1 therefore depletes Hox-dependent subtypes, with the remaining MNs having a more ancestral axial or ambiguous subtype identity (Figure 2P).
As we observed a dramatic loss of segmentally restricted MN subtypes in Ring1MNΔ mice, we next assessed the impact on peripheral innervation pattern. To trace motor axon projections, we crossed Ring1MNΔ mice to a Mnx1-GFP transgenic reporter, in which all MN axons are labelled with GFP. In control mice, there are nine primary trajectories of forelimb-innervating motor axon at E12.5 (Figure 2I and J; Catela et al., 2016). In Ring1MNΔ mice, only three nerve branches, radial, median, and ulnar were visible but appeared prematurely truncated and unbranched (Figure 2K). In the trunk, innervation of sympathetic chain ganglia was lost, consistent with a loss of PGC fates (Figure 2L and M). Projections to dorsal and ventral axial muscles by MMC and HMC subtypes were maintained in Ring1MNΔ mice, and HMC axons were thicker and shorter than in controls (Figure 2N and O, Figure 2—figure supplement 1G). Loss of Ring1 therefore causes severe defects in innervation pattern, while the trajectories of axial MNs are relatively spared.
Loss of Ring1 causes derepression of developmental fate determinants
Polycomb proteins regulate diverse aspects of differentiation by restricting gene expression during development. To investigate changes in gene expression after deletion of Ring1 genes in an unbiased manner, we performed RNAseq on MNs isolated from Ring1MNΔ mice. We purified MNs from control and Ring1MNΔ::Mnx1-GFP embryos at E12.5 by flow cytometry (n = 4 Ring1 mutants, and n = 4 Cre- controls), and performed RNAseq. Because Ring1 mutants display segment-specific phenotypes, we collected MNs from brachial, thoracic, and lumbar levels and profiled each population independently.
We identified a total of 1001 upregulated and 641 downregulated genes in Ring1MNΔ mice (log2-FC > 2, FDR < 0.1), with 391 genes upregulated in all three segmental regions, compared to 46 genes that were commonly downregulated (Figure 3A–C, Figure 3—figure supplement 1A,C, Supplementary file 1). Thus, many upregulated genes are shared between each segment, while downregulated genes tended to be segment-specific. Strikingly, 76% of the top 100 derepressed genes (by log2-FC) at brachial levels encode transcription factors, and include genes normally involved in the specification of spinal interneurons (e.g. Gata3, Shox2, Lhx2), brain regions (Foxg1, Six6, Lhx8), and non-neuronal lineages (Figure 3B).
Figure 3 with 1 supplement see all
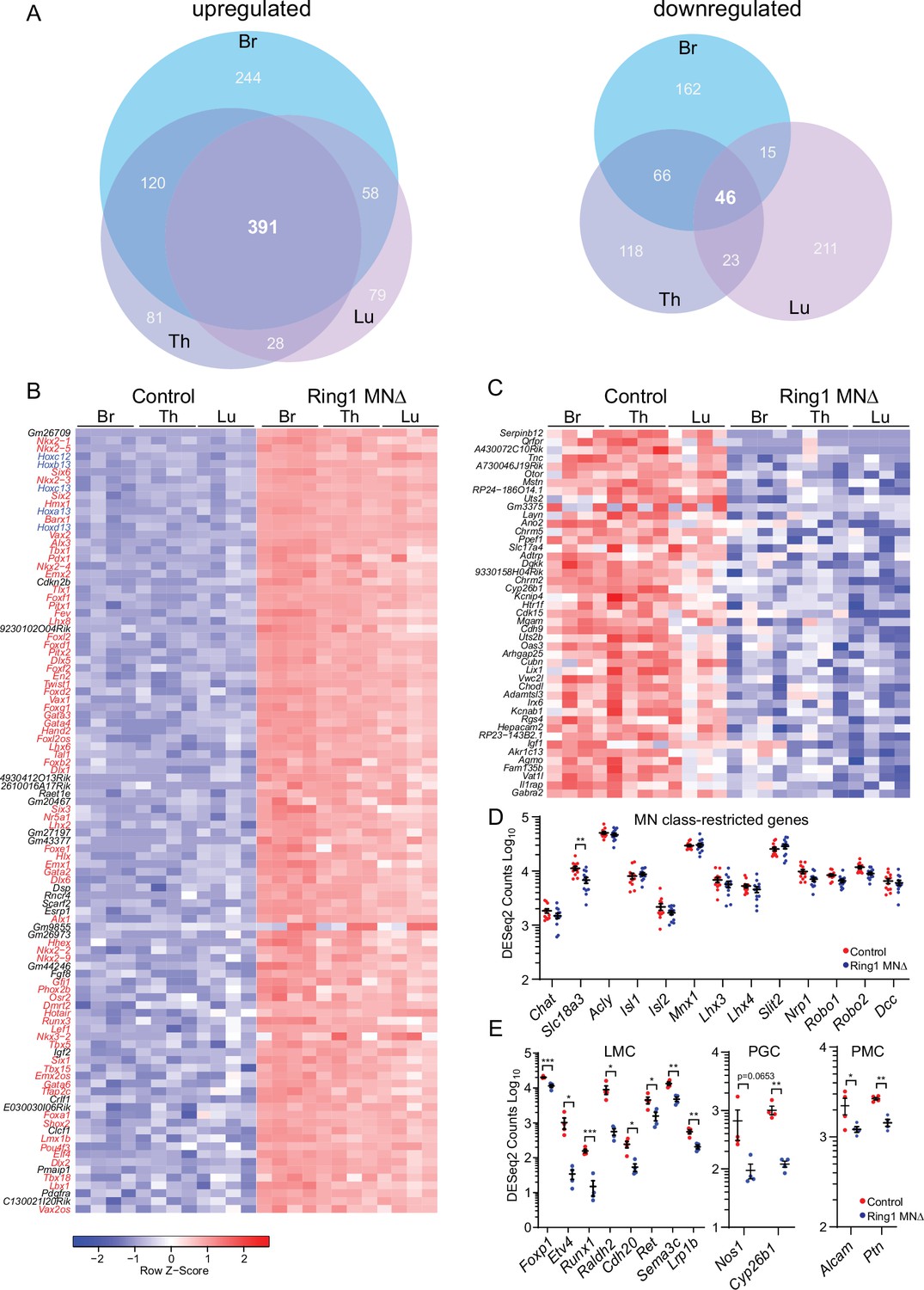
Ring1 is required to restrict transcription factor expression in spinal MNs.
(A) Venn diagrams showing the number of upregulated (left) and downregulated (right) genes in bracial (Br), thoracic (Th), and lumbar (Lu) MNs upon loss of Ring1 from MNs by RNAseq (log2-FC > 2, FDR < 0.1). (B) Heat map of top 100 upregulated genes (by log2-FC) in Ring1MNΔ mice and control MNs. Genes shown in blue are Hox genes and other transcription factor are shown in red. (C) Heat map of 45 genes downregulated in Ring1MNΔ mice in comparison to control MNs. (D) Plots of DESeq2 counts of genes associated with MN class identity. Each data point shows DESeq2 counts for each sample, and segment-specific counts are plotted together. Expression of Slc18a3 (Vacht) is reduced (padj. = 0.026477) in Ring1MNΔ mice. (E) DESeq2 counts of genes associated with specific MN subtype identities were reduced in Ring1MNΔ mice. Counts for LMC and PMC markers are from Br segments, PGC from Th segments. Black bars shown in graphs indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, unpaired t-test.
Although a variety of cell fate determinants were derepressed in Ring1MNΔ mice, our RNAseq analyses provide further evidence that core molecular determinants of MN class identity are preserved in Ring1 mutants. We found no significant changes in transcription factors (Isl1, Isl2, Mnx1, Lhx3, Lhx4), guidance molecules (Slit2, Robo1, Robo2, Dcc) and neurotransmitter genes (Chat, Acly) associated with pan-MN features (Figure 3D). There was a modest decrease in expression of Slc18a3 (Figure 3D). Expression of genes that mark other excitatory (Slc17a7, Slc17a6) or inhibitory (Slc32a1, Gad2, Gad1,) neuronal classes were not markedly derepressed in Ring1 mutants (Figure 3—figure supplement 1B). By contrast, expression of genes associated with MN subtype identities was decreased in Ring1MNΔ mice, including genes that mark limb-innervating (e.g. Foxp1, Etv4, Raldh2, Runx1), thoracic-specific (Nos1, Cyp26b), and respiratory (Alcam, Ptn) subtypes (Figure 3E).
To validate these changes in gene expression, we performed mRNA in situ hybridization on a subset of upregulated or downregulated genes. We found that Gata3, Lhx8, and Lhx2 were markedly upregulated in MNs of Ring1MNΔ mice (Figure 4A–F), while other genes (Foxg1, Six6, Pitx2, Shox2, Nkx2.1) showed less prominent, but detectable, ectopic expression (Figure 4—figure supplement 1A-J). We also examined expression of previously uncharacterized genes that were downregulated in all three segmental regions. We found Rgs4, Uts2b, Vat1, and Gabra2 were expressed by MNs of control embryos, and were downregulated in Ring1 mutants (Figure 4G–L, Figure 4—figure supplement 1K,L). These results indicate that despite the derepression of multiple fate determinants in Ring1MNΔ mice, core features of MN identity are preserved, but that subtype diversification programs are selectively disrupted.
Figure 4 with 1 supplement see all
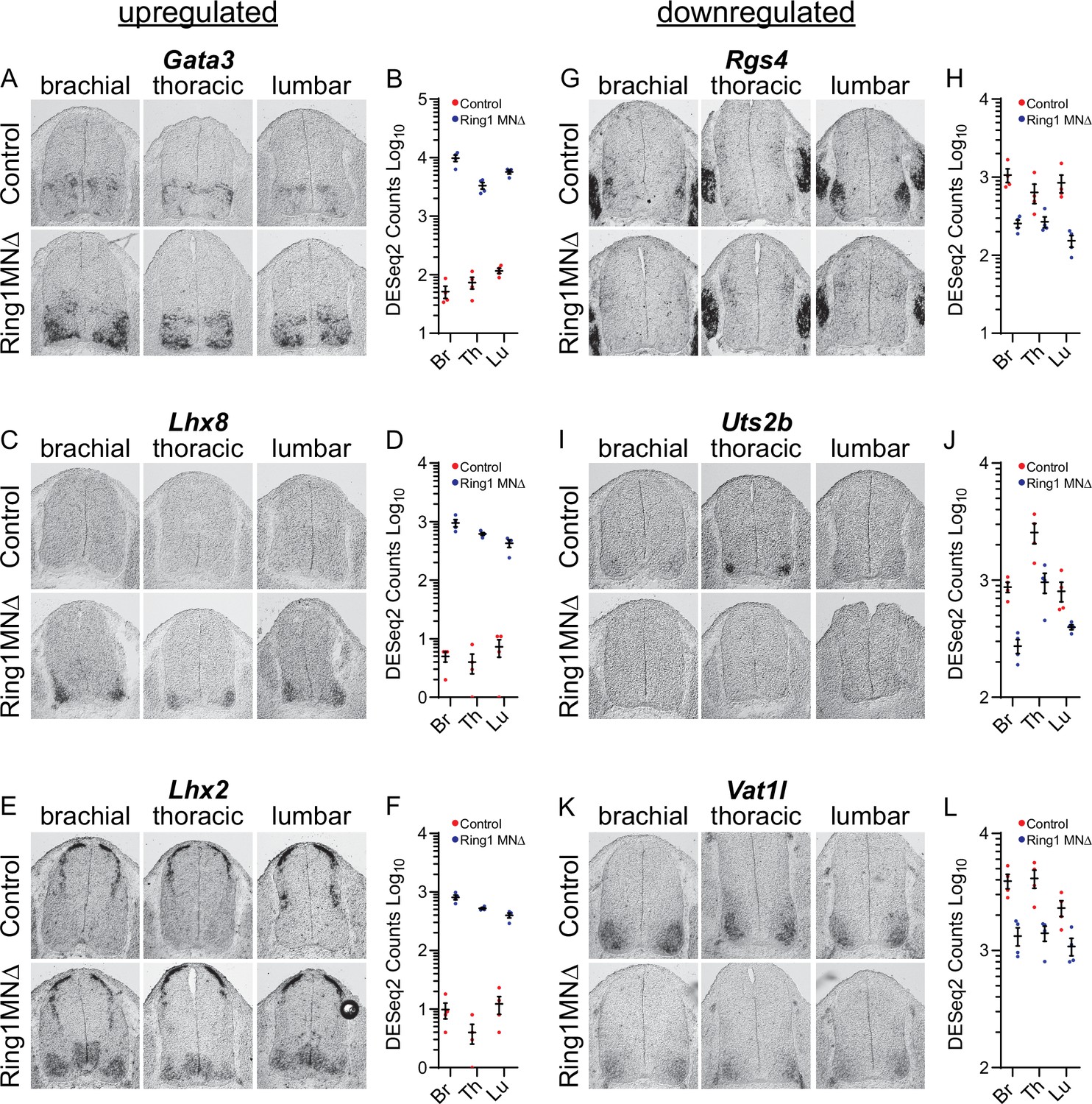
Analyses of misregulated genes in Ring1 mutants.
(A,C,E) In situ mRNA hybridization of selected upregulated genes from Ring1MNΔ RNAseq. Images show sections of brachial, thoracic, and lumbar segments from E12.5 control and Ring1MNΔ mice. (B,D,F) Graphs of DESeq2 counts for upregulated gene in each segment. Data points show DESeq2 counts from MNs of individual animals from indicated segments. (G, I, K) Analyses of downregulated genes by in situ hybridization. Rgs4 and Uts2b displayed elevated expression in specific segmental levels in controls, suggesting that a subset of the commonly downregulated genes are also Hox-dependent. Rgs4 expression is normally elevated in LMC neurons (panel G), while Uts2b is elevated in thoracic segments of controls (panel I) (H,J,L) Graphs of DESeq2 counts for each downregulated gene in each segment.
Selective derepression of caudal Hox genes in Ring1MNΔ mice
As our preliminary analyses revealed altered expression in a subset of Hox genes in Ring1MNΔ mice, we further evaluated expression of all 39 Hox genes in our RNAseq dataset. Consistent with the analysis of Hox protein expression, rostral Hox genes (Hox4-Hox8 paralogs) were reduced in brachial MNs of Ring1MNΔ mice, while caudal Hox genes (Hox10-Hox13 paralogs) were derepressed (Figure 5A, Figure 5—figure supplement 1A-C). We observed derepression of caudal Hox genes from each of the four vertebrate Hox clusters, including genes not normally detectable in the ventral spinal cord (e.g. HoxB genes) (Dasen et al., 2005; Figure 5A, Figure 5—figure supplement 1A-C). The extent of caudal Hox derepression correlated with the relative position of genes within a cluster, with Hox13 paralogs (Hoxa13, Hoxb13, Hoxc13, Hoxd13) displaying the most pronounced derepression in Ring1MNΔ mice (by FC), relative to other caudal Hox genes (Figure 5B, Figure 3—figure supplement 1C). The marked derepression of Hox13 paralogs was observed in each of the three segmental levels we analyzed, while Hox10 paralogs (Hoxa10, Hoxc10, and Hoxd10) were selectively derepressed in brachial and thoracic segments (Figure 5A, Figure 5—figure supplement 1A-C).
Figure 5 with 2 supplements see all
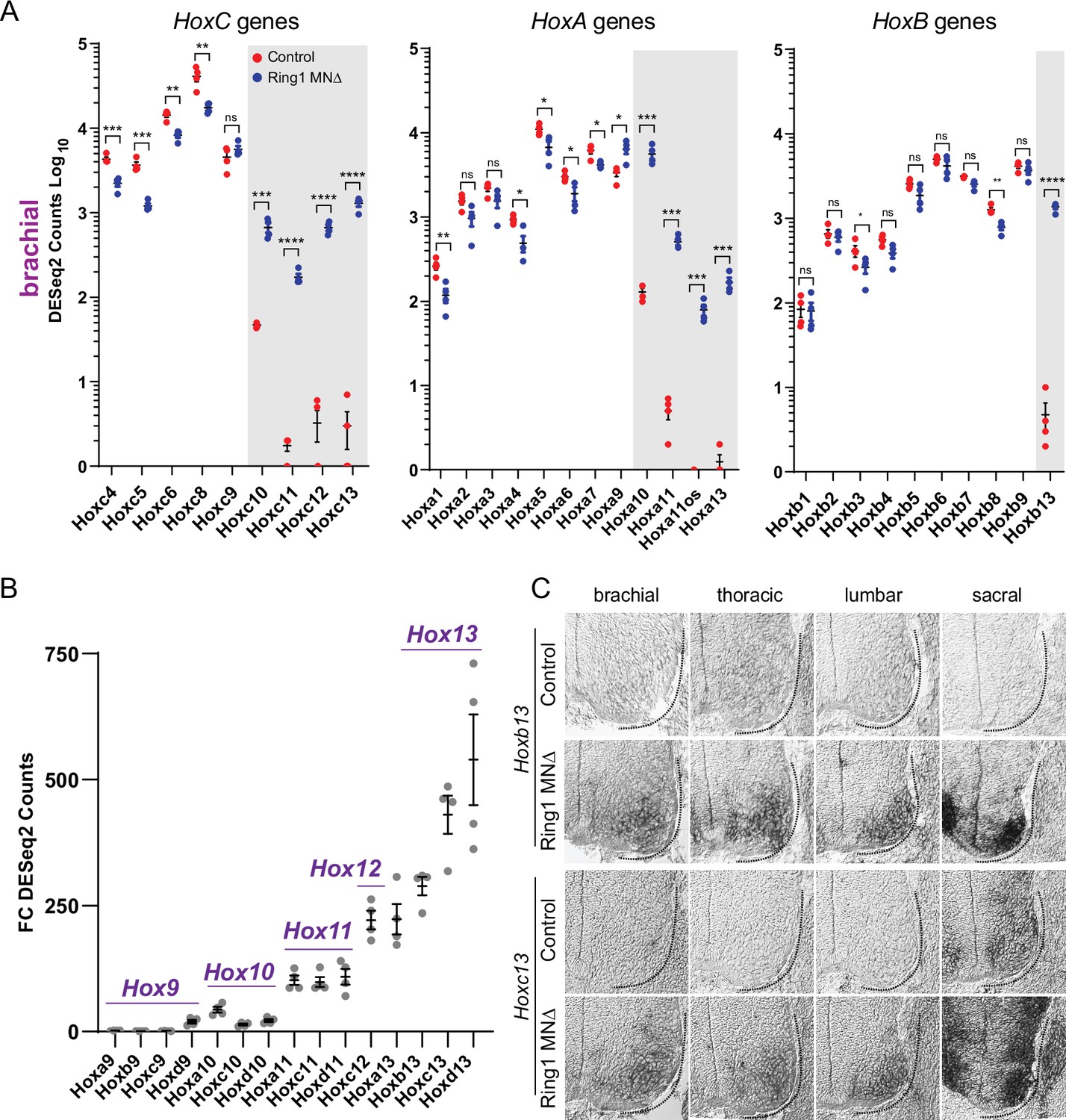
Derepression of caudal Hox genes in Ring1 mutants.
(A) DESeq2 counts of HoxC, HoxA, and HoxB cluster genes in brachial segments in control and Ring1MNΔ mice showing derepression of caudal Hox genes. Gray shaded regions highlight Hox genes that are derepressed in Ring1MNΔ mice. Hoxc9 does not show significant derepression, likely because it is normally expressed by caudal brachial MNs. Black bars shown in graphs indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, unpaired t-test. (B) Comparison of Hox9-Hox13 paralog gene derepression in brachial segments. Graph shows absolute fold changes of DESeq2 counts, showing a marked increase for caudal Hox13 paralogs in Ring1MNΔ mice. Each data point shows individual counts for Ring1 mutants/average of controls. (C) In situ of Hoxb13 and Hoxc13 mRNA transcripts in E12.5 embryos. Hoxb13 is normally not detectable in spinal cord, but is derepressed in MNs in Ring1MNΔ mice. Hoxc13 transcripts are normally restricted to sacral segments but derepressed in rostral segments in Ring1MNΔ mice.
To further validate these findings, we analyzed Hoxc13 and Hoxb13 expression by in situ hybridization. In controls Hoxc13 is restricted to sacral segments, while Hoxb13 is not detected in MNs (Figure 5C). In Ring1MNΔ mice, both Hoxb13 and Hoxc13, were de-repressed in MNs throughout the rostrocaudal axis (Figure 5C). In addition, in situ hybridization of Hoxc6 and Hoxc9 expression revealed reduced expression at brachial and thoracic levels, respectively (Figure 5—figure supplement 1D). By contrast, ectopic expression of Hoxb13 and Hoxc13 was not observed in EzhMNΔ mice (Figure 5—figure supplement 2A,B). Thus, ectopic expression of Hox13 paralogs in Ring1 mutants is associated with a loss of rostral Hox gene expression.
Ring1 is essential to maintain MN chromatin topology
Our findings indicate that in the absence of Ring1 genes, a broad variety of cell fate determinants are ectopically expressed in MNs, while only a subset of caudal Hox genes are derepressed. As PRC1 restricts gene expression through chromatin compaction, we investigated whether removal of Ring1 leads to changes in DNA accessibility at derepressed loci. We used Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATACseq) to identify genomic regions which have gained or lost accessibility in Ring1MNΔ mice. We purified Mnx1-GFP MNs at E12.5 from control and Ring1MNΔ embryos at brachial, thoracic, and lumbar levels, and performed ATACseq. In control samples, we observed a progressive opening of caudal Hox genes in more rostral segments. For example, the accessibility of Hoxc9 and Hoxa9 increases from brachial to thoracic segments, while Hoxc10, Hoxc11, and Hoxa10 are more accessible in lumbar segments (Figure 6—figure supplement 1A, B).
To determine which genomic regions gained accessibility in Ring1 mutants, we compared ATACseq profiles between MNs of control and Ring1MNΔ mice. We identified a total of 2305 loci that gained accessibility in Ring1MNΔ embryos, 324 (14%) of which were common to all three segmental levels (Figure 6A, Supplementary file 2). Common genes included caudal Hox genes (Hoxa13, Hoxc13) and other transcription factors (e.g. Foxg1, Lhx2, Pitx2) that were derepressed in our RNAseq analyses (Figure 6C). We also identified 1264 loci that lost accessibility in Ring1MNΔ mice, 39 (3%) of which were common to all three segments (Figure 6A). Thus, similar to our RNAseq results, loss of Ring1 leads to increased accessibility in many genes that are shared among each segment, while genes that lose accessibility tend to be segment-specific.
Figure 6 with 1 supplement see all
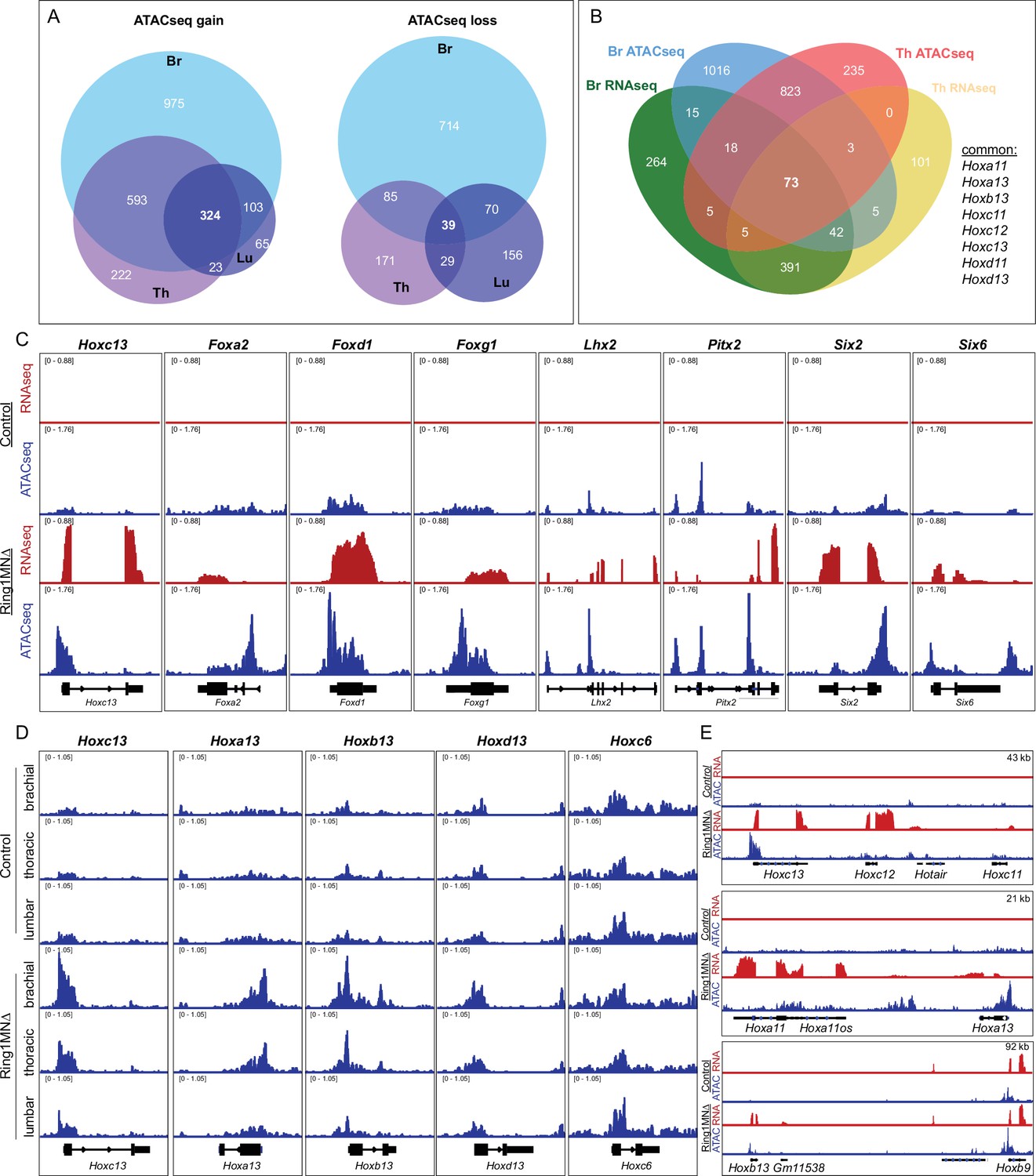
Ring1 is essential for maintaining chromatin topology at cell fate determining genes.
(A) Proportional Venn diagram showing the number of genes that gain (left) or lose (right) chromatin accessibility in brachial, thoracic and lumbar segments in Ring1MNΔ mice. (B) Venn diagram of upregulated genes from RNAseq and genes that gained accessibility in ATACseq of brachial and thoracic segments. (C) IGV browser views of selected genes that are upregulated and gained chromatin accessibility in controls and Ring1MNΔ mice in brachial segments. (D) IGV browser views of ATACseq tracks in controls and Ring1MNΔ mice in brachial, thoracic, and lumbar segments. Hox13 paralogs gain chromatin accessibility in each segment, while reduced brachial Hoxc6 expression is not associated with a loss of chromatin accessibility. (E) Comparison of IGV browser views of RNAseq and ATACseq tracks of caudal HoxC, HoxA, and HoxB genes in brachial segments.
We next examined the overlap between transcripts that were upregulated and loci that gained accessibility in Ring1MNΔ mice. We found that 18% (148/813) of genes that were ectopically expressed at brachial segments also gained accessibility in Ring1MNΔ mice (Figure 6B). We found 19 genes, including Hoxa13 and Hoxc13, were derepressed and gained accessibly in all three segments (Figure 6—figure supplement 1C). Since lumbar segments still retain features of Hox-dependent subtypes, we also compared the overlap between genes that were upregulated and gained accessibility in brachial and thoracic MNs. We identified 73 genes, 56 (77%) of which encode transcription factors, including each of the caudal Hox genes we found by RNAseq (Figure 6B, Figure 6—figure supplement 1C). The gain of accessibility at transcription factor-encoding genes was prominent near transcription start sites (Figure 6C), and regions that gained accessibility in Ring1MNΔ neurons correspond to regions shown to be bound by Rnf2 (Ring1B) in ES cells (Figure 6—figure supplement 1D; Bonev et al., 2017).
In agreement with our RNAseq analysis, Hox13 paralogs showed pronounced increases in accessibility, with each of the four Hox13 paralogs gaining accessibility in each of the three segmental levels (Figure 6D and E). By contrast, rostral Hox4-Hox8 paralogs, which are transcriptionally downregulated in Ring1 mutants, were not among that targets that lost accessibility (Figure 6D). These results suggest that reduced expression of rostral Hox4-Hox8 paralogs in Ring1 mutants is not due to a loss in chromatin accessibility.
Hox13 paralogs repress rostral Hox genes by engaging accessible chromatin domains
As PRC1-mediated chromatin compaction is essential for Hox repression, it is surprising that Hox4-Hox8 paralogs are diminished in Ring1 mutants, without a noticeable reduction in chromatin accessibility at these loci. Because cross-repressive interactions between Hox genes themselves are an important regulatory mechanism determining Hox boundaries (Philippidou and Dasen, 2013), it is possible that ectopically expressed Hox13 paralogs directly repress multiple Hox genes in Ring1MNΔ mice. To test this, we first analyzed MNs derived from ES cells (ESC-MNs) in which Hoxc13 expression can be induced upon doxycycline (Dox) treatment (Bulajić et al., 2020). ESC-MNs differentiated via RA and Sonic Hedgehog agonist are characterized by expression of anterior Hox paralogs. RNAseq analyses revealed that Hoxa4, Hoxa5, Hoxc4, and Hoxc5 expression were markedly reduced after Dox-induced Hoxc13 expression in MN progenitors, compared to non-induced ESC-MNs (Figure 7A, Figure 7—figure supplement 1A-C). This repressive effect also appears to be direct, as ChIPseq analysis indicates that Hoxc13 can bind at multiple Hox genes (Figure 7A).
Figure 7 with 1 supplement see all
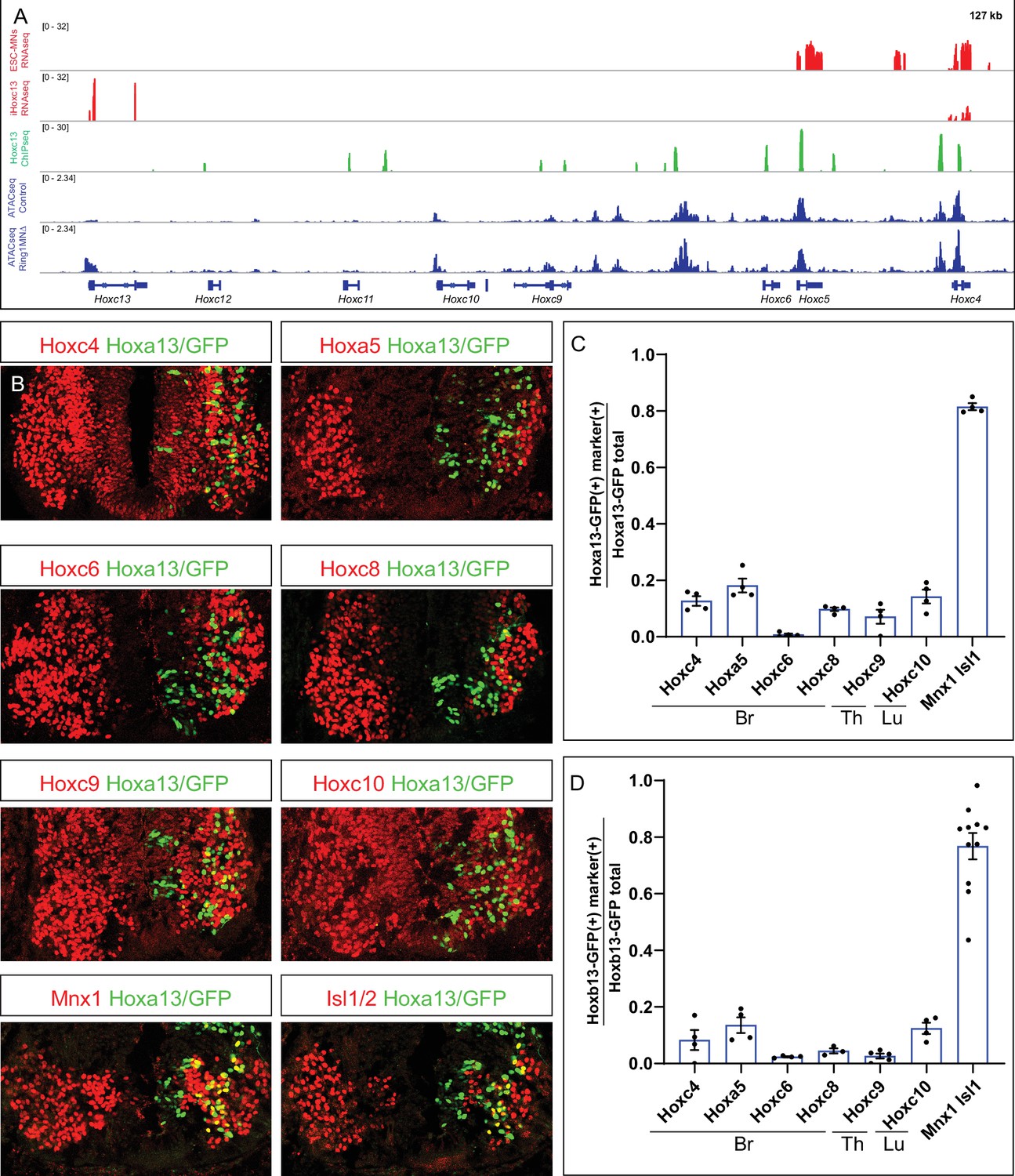
Hoxa13 and Hoxb13 repress multiple Hox4-Hox10 paralogs.
(A) Effects of Hoxc13 induction in ESC-MNs. Top panels show IGV browser views (log scale) of RNAseq (red) and ChIPseq tracks (green) at the HoxC cluster. Induced Hoxc13 binds near multiple HoxC genes and represses expression of Hoxc4 and Hoxc5. Bottom panels show ATACseq in brachial control and Ring1 mutant MNs at the HoxC cluster (blue). (B) Misexpression of Hoxa13-ires-nucGFP represses more rostral Hox genes. Hoxc4, Hoxa5, Hoxc6, and Hoxc8 were analyzed in brachial segments, Hoxc9 in thoracic segments, and Hoxc10 in lumbar segments. Mnx1 and Isl1 images are from brachial segments. (C,D) Quantification of percentage of electroporated cells which retained the expression of indicated Hox protein and MN markers upon Hoxa13 or Hoxb13 misexpression. Data in graphs show percentages of GFP+ neurons that express the markers indicated on x-axis. Data are from at least four embryos, four sections each embryo, and show mean ± SEM. Quantified electroporated cells were selected from the ventrolateral spinal cord, where MNs reside. Mnx1 and Isl1/2 quantification shows percentage of GFP+ neurons that express either marker, and taken from Br segments in panel C, and Br and Th segments in D.
-
Figure 7—source data 1
Quantification of Hoxa13- and Hoxb13-electroporated embryos.
- https://cdn.elifesciences.org/articles/72769/elife-72769-fig7-data1-v2.xlsx
Hoxc13 and Hoxa13 can target inaccessible chromatin (Bulajić et al., 2020; Desanlis et al., 2020), while repression by the Drosophila Hox protein Ubx is associated with chromatin compaction (Loker et al., 2021). To examine the possible effects of caudal Hox proteins on local chromatin structure, we compared accessibility at Hox loci in both control and Ring1 mutants, relative to the location of Hoxc13 binding sites. At Hoxc4, Hoxc5, Hoxa4, and Hoxa5, Hoxc13 bound sites correspond to regions that are accessible in both control and Ring1 mutant MNs (Figure 7A, Figure 7—figure supplement 1A-C). Hox13 proteins therefore appear to repress rostral Hox genes, in part, through binding at pre-existing accessible regions.
To test whether Hox13 paralogs can repress multiple Hox paralogs in vivo, we used chick neural tube electroporation to express Hoxa13 and Hoxb13 within brachial, thoracic, and lumbar segments, and assessed Hox protein expression. We electroporated pCAGGs-Hoxa13-ires-nucGFP or pCAGGs-Hoxb13-ires-nucGFP and found that both cell-autonomously repressed expression of brachial, thoracic, and lumbar Hox proteins (Hoxc4, Hoxa5, Hoxc6, Hoxc8, Hoxc9, and Hoxc10) (Figure 7B–D). By contrast, misexpression of Hoxa13 and Hoxb13 did not affect expression of the general MN markers Isl1/2 and Mnx1 (Figure 7B–D). These results indicate that Hox13 paralogs can repress multiple Hox genes, and likely contribute to the MN fate specification defects of Ring1MNΔ mice.
Discussion
The Polycomb group encompass a large and diverse family of proteins essential for maintaining epigenetic memory of early patterning events. Classically, PcG-mediated repression is thought to depend on recruitment of PRC1 through recognition of histone methylation marks deposited by PRC2 activity. Although alternative, H3K27me3-independent, mechanisms of PRC1 repression have been described (Tavares et al., 2012), the relative contribution of PRC1 and PRC2 to neuronal fate specification have not been resolved. In this study, we found that genetic depletion of PRC2 components has no discernable impact on neural class or subtype diversification programs at the time of MN differentiation. By contrast, a core PRC1 subunit, Ring1, is required to restrict transcription factor expression in the CNS, maintain neuronal subtype-specific chromatin topology, and determine rostrocaudal boundaries of Hox expression. Our findings indicate PRC1 plays a key role in regulating expression of cell fate determinants during MN differentiation, safeguarding neurons from acquiring inappropriate gene regulatory programs.
PRC functions in Neural development
Polycomb proteins function in diverse aspects of CNS maturation, including the temporal transition from neurogenesis to gliogenesis, maintenance of adult neural stem cell fates, and restriction of gene expression in mature neurons (Di Meglio et al., 2013; Fasano et al., 2009; Hirabayashi et al., 2009; Molofsky et al., 2003; von Schimmelmann et al., 2016). The role of PRCs in neural subtype diversification has been challenging to study, in part, due the complexity and dynamic temporal regulation of its constituents. By selectively removing core PRC1 and PRC2 subunits from neural progenitors, we determined the relative contributions of these chromatin-associated complexes to neuronal diversification. We found that PRC1 represses a broad variety of cell fate determinants, with the majority of highly derepressed genes in Ring1 mutants encoding transcription factors. Ectopic expression of Ring1-regulated targets is associated with a disruption in chromatin topology, resulting in a gain in accessibility of derepressed genes.
Despite the derepression of multiple fate determinants in the absence of PRC1 function, markers of MN class identity (e.g. Mnx1, Isl1/2, and Lhx3) are largely unchanged, and MNs do not appear to acquire features of alternate neuronal class fates, beyond expression of fate-defining transcription factors. This observation suggests that once a class identity has been established, ectopic expression of fate determinants is insufficient to respecify basic neuronal features, such as neurotransmitter identity. In C. elegans the ability of transcription factors to reprogram neuronal class identity requires removal of histone deacetylase proteins (Tursun et al., 2011), suggesting that additional regulatory constraints, such as histone acetylation and/or DNA-methylation, restricts the ability of ectopic transcription factors to interfere with class-specific programs in Ring1 mutants.
While basic elements of MN fate are maintained in the absence of PRC1, their differentiation into molecularly distinct subtypes is severely compromised. The loss of subtype-specific programs in brachial and thoracic segments can be attributed to the attenuation of expression of rostral HoxA and HoxC cluster paralogs, as mice lacking these Hox genes show a similar loss in MN subtype features (Jung et al., 2010; Jung et al., 2014; Philippidou et al., 2012). Selective loss of Hox function in MNs also appears to account for the observation that the majority of downregulated genes in Ring1 mutants are segment-specific.
Our results indicate that in Ring1 mutants brachial MNs ectopically express Hox9-Hox13 paralogs, while expression of Hox genes normally expressed by brachial MNs is diminished. As a consequence, MNs fail to acquire a limb-innervating LMC molecular fate, and express markers indicative of MMC and HMC subtypes. These observations are consistent with a model in which the Hox code of MNs is scrambled in Ring1 mutants, reverting to the fate of ancestral axial subtypes. However, we cannot formally rule out the possibility that some MNs acquire a sacral Hox13+ fate, although we did not observe ectopic expression of markers of sacral PGC identity (e.g. nNos, Foxp1).
We previously found that acute depletion of the canonical PRC1 component Pcgf4 (Bmi1) in chick leads to ectopic Hoxc9 expression in brachial segments and a switch of LMC neurons to a thoracic PGC fate (Golden and Dasen, 2012). By contrast in Ring1 mutants, multiple caudal Hox genes (Hox9-Hox13 paralogs) are derepressed, and the remaining MNs have an axial subtype fate. While the exact mechanisms of these difference are unclear, they could reflect differences in the timing and spatial extent of the manipulations. In Ring1 mutants PRC1 function is depleted from MN progenitors throughout the rostrocaudal axis, whereas Bmi1 knockdown was performed in brachial segments just prior to differentiation.
PRC-dependent and independent mechanisms of rostrocaudal patterning in the CNS
Our results show that mutation in Ring1 genes leads to ectopic expression and increased accessibility of many transcription factor-encoding genes that are common to each segment, while genes that are downregulated and lose accessibility tend to be segment-specific. These observations indicate that PRC1 is required to restrict expression of multiple cell-fate determinants throughout the rostrocaudal axis. By contrast, the downregulation of segment-specific genes can be attributed to the loss of Hox-dependent MN columnar subtypes, due to altered Hox gene expression in PRC1 mutants.
We found that removal of PRC1 leads to the derepression and increased accessibility of caudal Hox genes. Loss of PRC1 does not result in a sustained derepression of all Hox genes, as Hox genes normally expressed by brachial- and thoracic-level MNs are attenuated in the absence of Ring1. These findings are reminiscent of the function of PRCs in Drosophila, where loss of PRC function leads to ectopic expression of the caudal Hox gene Abd-b and repression of Ubx expression (Struhl and Akam, 1985; Struhl and White, 1985), likely reflecting cross-repressive interactions between caudal Hox proteins and rostral Hox genes. Notably, reduced rostral Hox gene expression in Ring1 mutants is not a reflection of reduced chromatin accessibility at these targets, consistent with a model in which this form of repression is not associated with global changes in chromatin structure.
In Ring1 mutants, Hox13 paralogs are derepressed in MNs, and appear to contribute to the loss of MN subtypes. Derepression of Hox13 paralogs alone does not appear to account for all of the specification defects in Ring1 mutants, as Hox10 paralogs (Hoxa10, Hoxc10, and Hoxd10) were expressed normally in lumbar segments. Hoxc9 can also repress Hox4-Hox8 paralogs in MNs through direct binding (Jung et al., 2010), and likely contributes to the Ring1 mutant phenotype. Derepression of multiple caudal Hox paralogs therefore appears to have a cumulative effect on repressing brachial and thoracic Hox genes. In conjunction with studies in Drosophila, these finding indicate a conserved mechanism of rostrocaudal patterning, in which PRCs establish rostral Hox boundaries, while Hox cross-repressive interactions define caudal boundaries.
PRC1 functions in maintaining neuronal subtype fate
Studies of neuronal and non-neuronal development provide compelling evidence that PRC2 activity is required to restrict gene expression in early development (Ezhkova et al., 2009; Gentile et al., 2019; Snitow et al., 2015; Yaghmaeian Salmani et al., 2018). Our studies show that Hox boundaries are unaffected after genetic removal of PRC2 function, which leads to diminished H3K27me3 by the time of MN differentiation. Mutation of Ezh genes or Eed depletes H3K27me3 from progenitors and postmitotic neurons by E11.5, without appreciably affecting MN generation, Hox expression, or downstream Hox effectors. As the axial identity of progenitor cells appears to be shaped prior to neurogenesis (Metzis et al., 2018), our PRC2 manipulations likely remove H3K27me3 after Polycomb repression has been initiated. Consistent with this idea, our analyses of Ezh mutants as well as previous analyses of EedNEΔ mice (Yaghmaeian Salmani et al., 2018), showed detectable levels of H3K27me3 in the MN domain up until E10.5, indicating that some H3K27me3 remains during the phase in which anterior Hox boundaries are established.
How does PRC1 restrict expression of fate determinants in the absence of PRC2 activity? One plausible scenario is that residual H3K27me3 is transmitted through cell division in PRC2 mutants and is sufficient to recruit canonical PRC1 and maintain target gene repression. As cells divide, newly synthesized histones are presumably devoid of H3K27me3 in PRC2 mutants, leading to replication-coupled dilution of H3K27me3. During neural differentiation, the rate of cell division decreases (Kicheva et al., 2014; Wilcock et al., 2007), potentially limiting H3K27me3 depletion, and enabling PRC1 to bind at target loci, even in the absence of de novo H3K27 methylation. Consistent with this idea, recent studies on the effects of PRC2 depletion in intestinal stem cells indicate that ~40% of residual H3K27me3 can maintain PRC repression (Jadhav et al., 2020), although the extent of derepression varies with gene target. Although we observed no MN phenotypes in Ezh or Eed mutants, later-born oligodendrocytes derived from the MN progenitor domain have been shown to depend on PRC2 function for their differentiation (Wang et al., 2020). This more pronounced effect on glial development likely reflects further dilution of H3K27me3 through cell division in PRC2 mutants.
Stabilization of PRC1 at repressed loci could also maintain target repression independently of de novo H3K27 methylation. Repression by canonical PRC1 has been shown to depend on the formation of phase-separated subnuclear structures (Polycomb bodies) assembled through polymerization of Polyhomeotic-like (Phc) and/or Cbx proteins (Isono et al., 2013; Plys et al., 2019; Tsuboi et al., 2018). Mutation of Phc2 in mice leads to ectopic expression of Hox genes, with Hoxb13 among the most robustly derepressed targets (Isono et al., 2013). One possibility is that PcG-mediated repression may not exclusively depend on anchoring of PRC1 through H3K27me3, but is also maintained through Phc-mediated chromatin compaction at specific loci. Alternatively, PRC2-independent repression by PRC1 could be facilitated through its H2A-ubiquitination activity, as has been suggested in other systems (Tsuboi et al., 2018).
We suggest that in the early phases of development PRC2 activity defines the sites of maintained H3K27 tri-methylation, initiates PRC1 recruitment, and restricts transcription factor expression in MNs. These activities are likely initiated prior to MN progenitor specification, as recent studies have demonstrated that the establishment of rostrocaudal positional identities can be specified before neural induction (Metzis et al., 2018). Patterning morphogens, such as RA and FGF, may act on stem cells to establish the pattern of H3K27me3 at Hox loci prior to neurogenesis. This early rostrocaudal patterning step may ultimately serve to coordinate Hox expression in the neural tube and surrounding mesodermally-derived tissues. The subsequent switch to reliance on PRC1 function could reflect a general mechanism of gene regulation in neuronal subtypes that become terminally differentiated during development.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (M. musculus) | Ezh1flox/flox | PMID:23122289 | MGI:1097695 | |
| Genetic reagent (M. musculus) | Ezh2flox/flox | PMID:12496962 | MGI:107,940 | |
| Genetic reagent (M. musculus) | Olig2Cre | PMID:18046410 | MGI: 3774124 | |
| Genetic reagent (M. musculus) | Ring1-/-::Rnf2flox/flox | PMID:18039844, 11060235 | MGI:1101770, MGI:1101759 | |
| Genetic reagent (M. musculus) | Yaf2-/-::Rybpflox/flox | PMID:27705745 | MGI:1914307MGI:1929059 | |
| Genetic reagent (M. musculus) | Mnx1-GFP | PMID:10482234 | ||
| Biological sample (chicken eggs) | SPF Eggs | Charles River | 10100332 | |
| Antibody | Anti-Hoxc4 (Rabbit polyclonal) | PMID:16269338 | (1:16,000) | |
| Antibody | Anti-Hoxa5 (Rabbit polyclonal) | PMID:16269338 | (1:16,000) | |
| Antibody | Anti-Hoxc6 (Guinea pig polyclonal) | PMID:11754833 | RRID: AB_2665443 | (1:16,000) |
| Antibody | Anti-Hoxc6 (Rabbit polyclonal) | Aviva Systems Biology | Cat# ARP38484; RRID: AB_10866814 | (1:32,000) |
| Antibody | Anti-Hoxc8 (Mouse monoclonal) | Covance | RRID: AB_2028778 | (1:4000) |
| Antibody | Anti-Foxp1 (Rabbit polyclonal) | PMID:18662545 | RRID: AB_2631297 | (1:32,000) |
| Antibody | Anti-Isl1/2 (Guinea pig polyclonal) | Jessell lab | (1:10,000) | |
| Antibody | Anti-Rnf2 (Goat polyclonal) | Abcam | Cat# ab3832, RRID: AB_304100 | (1:2000) |
| Antibody | Anti-Rnf2 (Rabbit polyclonal) | Abcam | Cat# ab101273, RRID: AB_10711495 | (1:5000) |
| Antibody | Anti-Rybp (Rabbit monoclonal) | Abcam | Cat# ab185971 | (1:2000) |
| Antibody | Anti-Cbx2 (Rabbit polyclonal) | Bethyl | Cat# A302-524A, RRID:AB_1998943 | (1:5000) |
| Antibody | Anti-H3K27me3 (Rabbit polyclonal) | Cell Signaling | Cat# 9733, RRID: AB_2616029 | (1:2000) |
| Antibody | Anti-Hoxc9 (Guinea pig polyclonal) | PMID:20826310 | RRID:AB_2636809 | (1:64,000) |
| Antibody | Anti-Hoxc10 (Rabbit polyclonal) | PMID:31141687 | (1:64,000) | |
| Antibody | Anti-Hoxd10 (Guinea pig polyclonal) | Abcam | Cat# ab172865 | (1:16,000) |
| Antibody | Anti-Nos (Rabbit polyclonal) | Immunostar | Cat# 24431, RRID:AB_572255 | (1:10,000) |
| Antibody | Anti-Raldh2 (Guinea pig polyclonal) | PMID:18662545 | RRID: AB_2631299 | (1:32,000) |
| Antibody | Anti-Scip (Rabbit polyclonal) | PMID:28190640 | RRID:AB_2631304 | (1:8000) |
| Antibody | Anti-Lhx3 (Rabbit polyclonal) | Jessell lab | (1:16,000) | |
| Antibody | Anti-Mnx1 (Mouse monoclonal) | DSHB | Cat# MNR2, RRID: AB_2314625 | (1:100) |
| Antibody | Anti-GFP (Rabbit polyclonal) | Invitrogen | Cat# A-6455, RRID: AB_221570 | (1:5000) |
| Antibody | Anti-DIG AP | Sigma-Aldrich | Cat# 11093274910, RRID: AB_2734716 | (1:5000) |
| Recombinant DNA reagent | pGEM-Mnx1-Rybp(plasmid) | This paper | See Materials and methods, and Figure 1U legend | |
| Recombinant DNA reagent | pGEM-Mnx1-Cbx2(plasmid) | This paper | See Materials and methods, and Figure 1U legend | |
| Recombinant DNA reagent | pCAGGS-mouseHoxa13-IRES2-nucEGFP(plasmid) | This paper | See Materials and methods, and Figure 7B legend | |
| Recombinant DNA reagent | pCAGGS-mouseHoxb13-IRES2-nucEGFP(plasmid) | This paper | See Materials and methods, and Figure 7B and D legend | |
| Commercial assay or kit | DIG RNA Labeling Kit | Sigma-Aldrich | Cat# 11175025910 | |
| Commercial assay or kit | One Taq One-Step RT-PCR | NEB | Cat# E5315S | |
| Commercial assay or kit | SMARTer Stranded RNA-Seq Kit | Takara | Cat# 634,837 | |
| Commercial assay or kit | Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-121–1,030 | |
| Commercial assay or kit | Papain Dissociation System | Worthington | PDS | |
| Commercial assay or kit | Arcturus Picopure RNA Isolation Kit | Applied Biosystems | KIT0204 | |
| Commercial assay or kit | NEBNext High-Fidelity 2 X PCR Master Mix | NEB | M0541S | |
| Commercial assay or kit | Qiagen MinElute | Qiagen | 28,204 | |
| Chemical compound, drug | Glycergel | Agilent | Cat# C0563 | |
| Chemical compound, drug | AMPure XP | Beckman Coulter Life Sciences | A63881 | |
| Chemical compound, drug | Turbo DNase | Invitrogen | AM2238 | |
| Chemical compound, drug | SYBR Green I Nucleic Acid Gel Stain | Invitrogen | S7563 | |
| Software, algorithm | Integrative Genomics Viewer | RRID: SCR_011793 | https://software.broadinstitute.org/software/igv/ | |
| Software, algorithm | Picard | RRID: SCR_006525 | ||
| Software, algorithm | BEDTools | RRID:SCR_006646 | https://github.com/arq5x/bedtools2, Quinlan, 2021 | |
| Software, algorithm | GraphPad Prism | GraphPad Software | RRID: SCR_002798 | https://www.graphpad.com/ |
| Software, algorithm | MACS | RRID: SCR_013291 | https://github.com/macs3-project/MACS, Liu, 2021 |
Mouse genetics
Request a detailed protocolEzh1flox/flox (Hidalgo et al., 2012), Ezh2flox/flox (Su et al., 2003), Olig2Cre (Dessaud et al., 2007), Mnx1GFP (Arber et al., 1999) mice have been previously described. Rybpflox/flox::Yaf2-/- (Hisada et al., 2012; Rose et al., 2016) mice were generated by microinjection of the mouse ESCs with these alleles into blastocysts followed by implantation into pseudopregnant female mice. Generation of EzhMNΔ mice was performed by crossing Ezh1flox/flox, Ezh2flox/flox and Olig2Cre mice. EzhMNΔ mice are viable at birth but do not survive beyond P20. Generation of Ring1MNΔ mice was performed by crossing Ring1-/-::Rnf2flox/flox and Olig2Cre. Ring1MNΔ mice perish at birth. Generation of Ring1MNΔ::Mnx1-GFP mice was performed by crossing Ring1-/-::Rnf2 flox/flox, Olig2Cre mice to Mnx1-GFP mice. Animal work was approved by the Institutional Animal Care and use Committee of the NYU School of Medicine in accordance to NIH guidelines (Protocol IA16-00045).
Slide immunohistochemistry
Request a detailed protocolEmbryos were fixed in 4% PFA for 1.5–2 hr at 4 °C, washed 5–6 times in cold PBS for 15–30 minutes each wash, and incubated overnight in 30% sucrose. Tissue was embedded in OCT, frozen in dry ice, and sectioned at 16 µm on a cryostat. For antibody staining of sections, slides of cryosections were placed in PBS for 5 min to remove OCT. Sections were then transferred to humidified trays and blocked for 20–30 min in 0.75 ml/slide of PBT (PBS, 0.1% Triton) containing 1% Bovine serum albumin (BSA). The blocking solution was replaced with primary staining solution containing antibodies diluted in PBT with 0.1% BSA. Primary antibody staining was performed overnight at 4 °C. Slides were then washed three times for 5 min each in PBT. Fluorophore-conjugated secondary antibodies were diluted 1:500-1:1000 in PBT and filtered through a 0.2 µm syringe filter. Secondary antibody solution was added to slides (0.75 ml/slide) and incubated at room temperature for 1 hr. Slides were washed three times in PBT, followed by a final wash in PBS. Coverslips were placed on slides using 110 µl of Vectashield (Vector Laboratories).
Antibodies against Hox proteins and MN subtypes were generated as described (Dasen et al., 2008; Dasen et al., 2005). Additional antibodies were used as follows: goat anti-Rnf2 (Abcam, 1:2000), rabbit anti-Rnf2 (Abcam, 1:5000), rabbit anti-Rybp (Abcam1:2000), rabbit anti-Yaf2 (Abcam, 1:2000), rabbit anti-Cbx2 (Bethyl, 1:5000), rabbit anti-H3K27me3 (Cell Signaling, 1:2000).
In situ mRNA hybridization
Request a detailed protocolProbe templates were generated by RT-PCR and incorporated a T7 promoter sequence in the antisense strand. Total RNA was first extracted from eviscerated E12.5 embryos using TRIzol (Invitrogen). Genes of interest were amplified with the One Taq One-Step RT-PCR kit (NEB) using 1 µg of RNA. After amplification by RT-PCR, a second PCR was performed to incorporate a T7 promoter sequence. Antisense riboprobes were generated using the Digoxigenin-dUTP (SP6/T7) labeling kit (Sigma-Aldrich). For in situ hybridization, sections were first dried for 10–15 min at room temperature, placed in 4% PFA, and fixed for 10 min at room temperature. Slides were then washed three times for 3 min each in PBS, and then placed in Proteinase K solution (1 µg/ml) for 5 min at room temperature. After an additional PFA fixation and washing step, slides were treated in triethanolamine for 10 min, to block positive charges in tissue. Slides were then washed three times in PBS and blocked for 2–3 hr in hybridization solution (50% formamide, 5 X SSC, 5 X Denhardt’s solution, 0.2 mg/ml yeast RNA, 0.1 mg/ml salmon sperm DNA). Prehybridization solution was removed, and replaced with 100 µl of hybridization solution containing 100 ng of DIG-labeled antisense probe. Slides were then incubated overnight (12–16 hr) at 72 °C. After hybridization, slides were transferred to a container with 400 ml of 5 X SSC and incubated at 72 °C for 20 min. During this step, coverslips were removed using forceps. Slides were then washed in 400 ml of 0.2 X SSC for 1 hr at 72 °C. Slides were transferred to buffer B1 (0.1 M Tris pH 7.5, 150 mM NaCl) and incubated for 5 min at room temperature. Slides were then transferred to staining trays and blocked in 0.75 ml/slide of B1 containing 10% heat inactivated goat serum. The blocking solution was removed and replaced with antibody solution containing 1% heat inactivated goat serum and a 1:5000 dilution of anti-DIG-AP antibody (Roche). Slides were then incubated overnight at 4 °C in a humidified chamber. The following day, slides were washed three times, 5 min each, with 0.75 ml/slide of buffer B1. Slides were then transferred to buffer B3 (0.1 M Tris pH 9.5, 100 mM NaCl, 50 mM MgCl2) and incubated for 5 min. Slides were then developed in 0.75 ml/slide of B3 solution containing 3.5 µl/ml BCIP and 3.5 µl/ml NBT for 12–48 hr. After color development, slides were washed in ddH20 and coverslipped in Glycergel (Agilent). A more detailed in situ hybridization protocol is available on our lab website (http://www.dasenlab.com).
Wholemount immunohistochemistry
Request a detailed protocolFor wholemount immunohistochemistry embryos were fixed in PFA for 2 hr, then bleached for 24 hr at 4 °C in a 10% H2O2, 10% DMSO solution prepared in methanol. Embryos were washed three times for 10 min each in methanol, followed by five washes for 10 min in PBS. Primary antibodies were diluted in staining solution (5% BSA, 20% DMSO in PBS) and specimens were incubated in staining solution on a rotator overnight at room temperature. Samples were then washed three times for 5 min each in PBS, followed by four 1 hr washes in PBS. Specimens were then incubated in secondary antibodies diluted in staining solution overnight at room temperature. Samples were then washed three times for 5 min each in PBS, followed by four 1 hr washes in PBS, a single 10 min wash in 50% methanol, and three 20 min washes in 100% methanol. Samples were transferred to glass depression slides and tissue was cleared by incubating samples in BABB solution (1-part benzyl alcohol: 2-parts benzyl benzoate). Confocal images of embryos were obtained from Z-stacks using Zen software (Zeiss). Further details of wholemount staining protocols are available on our lab website: (http://www.dasenlab.com).
In ovo chick electroporation
Request a detailed protocolIn ovo electroporation were performed on Hamburger Hamilton (HH) stage 13–14 chick embryos and analyzed at HH stage 24–25. Fertilized chicken eggs (Charles River) were incubated in a humidified incubator at 39 °C for 40–48 hr until they reached HH13-14. The top of the egg shell was removed and a 1 µg/µl DNA (150–500 ng/µL expression plasmid and pBKS carrier DNA) containing ~0.02% Fast green was injected into the central canal of the neural tube using a sharpened glass capillary tube. Electrodes (Platinum/Iridium (80%/20%), 250 μm diameter, UEPMGBVNXNND, FHC Inc) were placed on both sides of the neural tube (4 mm separation) and DNA was electroporated using an ECM 830 electroporator (ECM 830, BTX; 25 V, 4 pulses, 50ms duration, 1 s interval). Eggs were sealed with parafilm and incubated for 48 hr prior to fixation. Results shown in figures are representative of at least three electroporated embryos from two or more experiments in which electroporation efficiency in MNs was above 60%.
RNA preparation and library preparation
Request a detailed protocolRNA was extracted from FACS purified MNs dissected from E12.5 mouse embryo (10–20,000 cells/segment), using the Arcturus Picopure RNA isolation kit. For on-column DNase treatment, Turbo DNase was used (ambion, AM2238). Each samples used separated bar code for libraries. RNA quality and quantity were measured with an Agilent Picochip using a Bioanalyzer, all samples had quality scores between 9–10 RIN. For library preparation 10 ng of total RNA was used to generate cDNA, which was amplified with SMARTer Stranded RNA-Seq kit. 100 ng of cDNA were used as input to prepare the libraries (Takara, #634837), and amplified by 10 PCR cycles. Samples were run in four 50-nucleotide paired-end read rapid run flow cell lanes with the Illumina HiSeq 4000 sequencer.
RNAseq data analyses
Request a detailed protocolSequencing reads were mapped to the assembled reference genome (mm10) using the STAR aligner (v2.5.0c) (Dobin et al., 2013). Alignments were guided by a Gene Transfer Format (GTF) file. The mean read insert sizes and their standard deviations were calculated using Picard tools (v.1.126) (http://broadinstitute.github.io/picard; Broad Institute, 2019). The read count tables were generated using HTSeq (v0.6.0) (Anders et al., 2015), normalized based on their library size factors using DEseq2 (Love et al., 2014), and differential expression analysis was performed. The Read Per Million (RPM) normalized BigWig files were generated using BEDTools (v2.17.0) (Quinlan and Hall, 2010) and bedGraphToBigWig tool (v4). To compare the level of similarity among the samples and their replicates, we used two methods: principal-component analysis and Euclidean distance-based sample clustering. All the downstream statistical analyses and generating plots were performed in R environment (v3.1.1) (https://www.r-project.org/).
ATACseq
Request a detailed protocolATACseq was performed following previously described protocols (Buenrostro et al., 2015). DNA was extracted from purified dissected mouse embryonic MNs. Cells were aliquoted and washed twice in ice-cold 1× PBS. Cell pellets were resuspended in 10 mM Tris (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40 (v/v), 0.1% tween20, 0.01% Digitonin and 1% BSA, centrifuged at 500 g for 5 min at 4 °C. Pellets were resuspended in 12.5 μl of 2× tagmentation DNA buffer, 1.25 μl Tn5 (Nextera DNA Sample Preparation Kit, FC-121–1030) and 11.25 μl of water, and incubated at 37 °C for 30 min. The sample was purified using the MinElute PCR Purification Kit (Qiagen, 28004). PCR enrichment of the library was performed with custom-designed primers and 2× NEB Master Mix. A qPCR reaction with 1× SYBR Green (Invitrogen), custom-designed primers and 2× NEB Master Mix (New England Labs, M0541) was performed to determine the optimal number of PCR cycles (one third of the maximum measured fluorescence) (Buenrostro et al., 2013). The libraries were purified using the AMPure XP beads (Beckman Coulter, A63880). High Sensitivity DNA ScreenTape (Agilent, 5067–5584) was used to verify the fragment length distribution of the library. Library quantification was performed using the KAPA Library Amplification kit on a Roche LightCycler 480. The libraries were sequenced on an Illumina NovaSeq (100 cycles, paired-end).
ATACseq data analysis
Request a detailed protocolAll of the reads from the Sequencing experiment were mapped to the reference genome (mm10) using the Bowtie2 (v2.2.4)(Langmead and Salzberg, 2012) and duplicate reads were removed using Picard tools (v.1.126) (http://broadinstitute.github.io/picard/). Low-quality mapped reads (MQ < 20) were removed from the analysis. The read per million (RPM) normalized BigWig files were generated using BEDTools (v.2.17.0) (Quinlan and Hall, 2010) and the bedGraphToBigWig tool (v.4). Peak calling was performed using MACS (v1.4.2)(Zhang et al., 2008) and peak count tables were created using BEDTools. Differential peak analysis was performed using DESeq2 (Love et al., 2014). ChIPseeker (v1.8.0) (Yu et al., 2015) R package was used for peak annotations and motif discovery was performed using HOMER (v4.10)(Heinz et al., 2010). ngs.plot (v2.47) and ChIPseeker were used for TSS site visualizations and quality controls. To compare the level of similarity among the samples and their replicates, we used two methods: principal-component analysis and Euclidean distance-based sample clustering. The downstream statistical analyses and generating plots were performed in R environment (v3.1.1) (https://www.r-project.org/).
Statistics
Samples sizes were determined based on previous experience and the number of animals and definitions of N are indicated in the main text and figure legends. In figures where a single representative image is shown, results are representative of at least two independent experiments, unless otherwise noted. No power analysis was employed, but sample sizes are comparable to those typically used in the field. Data collection and analysis were not blind. Graphs of quantitative data are plotted as means with standard error of mean (SEM) as error bars, using Prism 8 (Graphpad) software. Unless noted otherwise, significance was determined using unpaired t-test in Prism eight software, or using adjusted p-values. Exact p-values are indicated, where appropriate, in the main text, figures, and figure legends.
Data availability
RNAseq and ATACseq data are available through GEO (GSE175503).
-
NCBI Gene Expression OmnibusID GSE175503. PRC1 sustains the integrity of neural fate in the absence of PRC2 function.
-
NCBI Gene Expression OmnibusID GSE96107. Multi-scale 3D genome rewiring during mouse neural development.
References
-
Initiating Hox gene expression: in the early chick neural tube differential sensitivity to FGF and RA signaling subdivides the HoxB genes in two distinct groupsDevelopment 129:5103–5115.
-
Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatinMolecular and Cellular Biology 26:2560–2569.https://doi.org/10.1128/MCB.26.7.2560-2569.2006
-
Targeting Polycomb systems to regulate gene expression: modifications to a complex storyNature Reviews. Molecular Cell Biology 16:643–649.https://doi.org/10.1038/nrm4067
-
ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-WideCurrent Protocols in Molecular Biology 109:21.https://doi.org/10.1002/0471142727.mb2129s109
-
Loss- and gain-of-function mutations show a polycomb group function for Ring1A in miceDevelopment 127:5093–5100.
-
STAR: ultrafast universal RNA-seq alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Temporal Patterning in the Drosophila CNSAnnual Review of Cell and Developmental Biology 33:219–240.https://doi.org/10.1146/annurev-cellbio-111315-125210
-
Interneuron Types as Attractors and ControllersAnnual Review of Neuroscience 43:1–30.https://doi.org/10.1146/annurev-neuro-070918-050421
-
RYBP represses endogenous retroviruses and preimplantation- and germ line-specific genes in mouse embryonic stem cellsMolecular and Cellular Biology 32:1139–1149.https://doi.org/10.1128/MCB.06441-11
-
Neuronal identity control by terminal selectors in worms, flies, and chordatesCurrent Opinion in Neurobiology 56:97–105.https://doi.org/10.1016/j.conb.2018.12.006
-
Evolving Hox activity profiles govern diversity in locomotor systemsDevelopmental Cell 29:171–187.https://doi.org/10.1016/j.devcel.2014.03.008
-
Regulation of Chromatin Structure During Neural DevelopmentFrontiers in Neuroscience 12:874.https://doi.org/10.3389/fnins.2018.00874
-
Fast gapped-read alignment with Bowtie 2Nature Methods 9:357–359.https://doi.org/10.1038/nmeth.1923
-
Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signalsNature Neuroscience 16:1191–1198.https://doi.org/10.1038/nn.3490
-
A Hox gene regulatory network for hindbrain segmentationCurrent Topics in Developmental Biology 139:169–203.https://doi.org/10.1016/bs.ctdb.2020.03.001
-
Sustained Hox5 gene activity is required for respiratory motor neuron developmentNature Neuroscience 15:1636–1644.https://doi.org/10.1038/nn.3242
-
Transcriptional codes and the control of neuronal identityAnnual Review of Neuroscience 25:251–281.https://doi.org/10.1146/annurev.neuro.25.112701.142916
-
Mechanisms of polycomb gene silencing: knowns and unknownsNature Reviews. Molecular Cell Biology 10:697–708.https://doi.org/10.1038/nrm2763
-
The development and assembly of the Drosophila adult ventral nerve cordCurrent Opinion in Neurobiology 56:135–143.https://doi.org/10.1016/j.conb.2019.01.013
-
Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegenerationNature Neuroscience 19:1321–1330.https://doi.org/10.1038/nn.4360
-
Model-based analysis of ChIP-Seq (MACS)Genome Biology 9:R137.https://doi.org/10.1186/gb-2008-9-9-r137
Article and author information
Author details
Funding
National Institutes of Health (R35 NS116858)
- Jeremy S Dasen
National Institutes of Health (R01 NS062822)
- Jeremy S Dasen
National Institutes of Health (R01 NS097550)
- Jeremy S Dasen
National Institutes of Health (NS 100897)
- Esteban Orlando Mazzoni
National Institutes of Health (T32 GM007238)
- Ayana Sawai
National Institutes of Health (F31 NS087772)
- Ayana Sawai
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Kristen D’Elia, Sara Fenstermacher, Jessica Treisman, and Ed Ziff for discussion and comments on the manuscript, and Rachel Kim and Orly Wapinski for assistance. We thank Isabel Hidalgo and Susana Gonzalez for providing Ezh1flox/flox mice, Alexander Tarakhovsky for Ezh2flox/flox mice, Stefan Thor for Eed mutant embryos, Haruhiko Koseki for Ring1-/-::Rnf2flox/flox ES cells, and Robert Klose for Rybyflox/flox::Yaf2-/- ES cells. We also thank Genome Technology Center and Cytometry and Cell Sorting Laboratory at NYU Langone. This work was supported by NIH NINDS grants T32 GM007238, F31 NS087772 to AS, R01 NS 100897 to EOM, R35 NS116858, R01 NS062822 and R01 NS097550 to JD.
Ethics
Animals work was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal work was approved by the Institutional Animal Care and use Committee of the NYU School of Medicine in accordance to NIH guidelines.
Copyright
© 2022, Sawai et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,233
- views
-
- 330
- downloads
-
- 16
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
PRC1 sustains the integrity of neural fate in the absence of PRC2 function
eLife 11:e72769.
https://doi.org/10.7554/eLife.72769
Further reading
-
- Cancer Biology
- Developmental Biology
Missense ‘hotspot’ mutations localized in six p53 codons account for 20% of TP53 mutations in human cancers. Hotspot p53 mutants have lost the tumor suppressive functions of the wildtype protein, but whether and how they may gain additional functions promoting tumorigenesis remain controversial. Here, we generated Trp53Y217C, a mouse model of the human hotspot mutant TP53Y220C. DNA damage responses were lost in Trp53Y217C/Y217C (Trp53YC/YC) cells, and Trp53YC/YC fibroblasts exhibited increased chromosome instability compared to Trp53-/- cells. Furthermore, Trp53YC/YC male mice died earlier than Trp53-/- males, with more aggressive thymic lymphomas. This correlated with an increased expression of inflammation-related genes in Trp53YC/YC thymic cells compared to Trp53-/- cells. Surprisingly, we recovered only one Trp53YC/YC female for 22 Trp53YC/YC males at weaning, a skewed distribution explained by a high frequency of Trp53YC/YC female embryos with exencephaly and the death of most Trp53YC/YC female neonates. Strikingly, however, when we treated pregnant females with the anti-inflammatory drug supformin (LCC-12), we observed a fivefold increase in the proportion of viable Trp53YC/YC weaned females in their progeny. Together, these data suggest that the p53Y217C mutation not only abrogates wildtype p53 functions but also promotes inflammation, with oncogenic effects in males and teratogenic effects in females.
-
- Developmental Biology
Stem cell self-renewal often relies on asymmetric fate determination governed by niche signals and/or cell-intrinsic factors but how these regulatory mechanisms cooperate to promote asymmetric fate decision remains poorly understood. In adult Drosophila midgut, asymmetric Notch (N) signaling inhibits intestinal stem cell (ISC) self-renewal by promoting ISC differentiation into enteroblast (EB). We have previously shown that epithelium-derived Bone Morphogenetic Protein (BMP) promotes ISC self-renewal by antagonizing N pathway activity (Tian and Jiang, 2014). Here, we show that loss of BMP signaling results in ectopic N pathway activity even when the N ligand Delta (Dl) is depleted, and that the N inhibitor Numb acts in parallel with BMP signaling to ensure a robust ISC self-renewal program. Although Numb is asymmetrically segregated in about 80% of dividing ISCs, its activity is largely dispensable for ISC fate determination under normal homeostasis. However, Numb becomes crucial for ISC self-renewal when BMP signaling is compromised. Whereas neither Mad RNA interference nor its hypomorphic mutation led to ISC loss, inactivation of Numb in these backgrounds resulted in stem cell loss due to precocious ISC-to-EB differentiation. Furthermore, we find that numb mutations resulted in stem cell loss during midgut regeneration in response to epithelial damage that causes fluctuation in BMP pathway activity, suggesting that the asymmetrical segregation of Numb into the future ISC may provide a fail-save mechanism for ISC self-renewal by offsetting BMP pathway fluctuation, which is important for ISC maintenance in regenerative guts.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}