Drug targeting Nsp1-ribosomal complex shows antiviral activity against SARS-CoV-2
- Department of Molecular Reproduction, Development and Genetics, Indian Institute of Science, India
- Microbiology & Cell Biology Department, Centre for Infectious Disease Research, Indian Institute of Science, India
- Department of Biochemistry, Indian Institute of Science, India
Abstract
The SARS-CoV-2 non-structural protein 1 (Nsp1) contains an N-terminal domain and C-terminal helices connected by a short linker region. The C-terminal helices of Nsp1 (Nsp1-C-ter) from SARS-CoV-2 bind in the mRNA entry channel of the 40S ribosomal subunit and blocks mRNA entry, thereby shutting down host protein synthesis. Nsp1 suppresses host immune function and is vital for viral replication. Hence, Nsp1 appears to be an attractive target for therapeutics. In this study, we have in silico screened Food and Drug Administration (FDA)-approved drugs against Nsp1-C-ter. Among the top hits obtained, montelukast sodium hydrate binds to Nsp1 with a binding affinity (KD) of 10.8 ± 0.2 µM in vitro. It forms a stable complex with Nsp1-C-ter in simulation runs with –95.8 ± 13.3 kJ/mol binding energy. Montelukast sodium hydrate also rescues the inhibitory effect of Nsp1 in host protein synthesis, as demonstrated by the expression of firefly luciferase reporter gene in cells. Importantly, it shows antiviral activity against SARS-CoV-2 with reduced viral replication in HEK cells expressing ACE2 and Vero-E6 cells. We, therefore, propose montelukast sodium hydrate can be used as a lead molecule to design potent inhibitors to help combat SARS-CoV-2 infection.
Editor's evaluation
This study reports on the repurposing of Montelukast, an FDA-approved drug, with Nsp-1. The Non-structural protein (Nsp)-1 from SARS-CoV2 mimics the binding mode of eukaryotic initiation factor 3 (eIF3j) to the mRNA entry tunnel of the 40S ribosomal subunits and blocks the entry of mRNA, which shuts down host protein synthesis. As a result, the host immune function is suppressed. This makes Nsp-1 an attractive target for therapeutic intervention. In the revised manuscript the authors' claims are supported using Biophysical and cellular assays. They also rationalize their findings using molecular dynamics simulations.
https://doi.org/10.7554/eLife.74877.sa0Introduction
SARS-CoV-2, the causative agent of severe coronavirus disease-19 (COVID-19) pandemic, is an enveloped positive-strand RNA-containing virus and belongs to beta coronavirus family (V’kovski et al., 2021). The virus contains nearly 30 kb RNA genome with 5'-cap and 3' poly-A tail (Finkel et al., 2021; V’kovski et al., 2021). The SARS-CoV-2 genome encodes for 14 open reading frames (ORFs). Upon entry into host cells, ORF1a and ORF1b encode for two polyproteins, which are later auto-proteolytically cleaved into 16 proteins, namely Nsp1–Nsp16. Among these proteins, Nsp1 binds in the mRNA entry channel of the 40S ribosomal subunit and blocks the entry of mRNAs, thereby shutting down host protein synthesis. Nsp1 also induces endonucleolytic cleavage of host RNAs.
The cryo-electron microscopy (cryo-EM) structures of ribosomes from Nsp1-transfected human HEK293T cells indicate the binding of Nsp1 with 40S and 80S ribosomal subunits (Schubert et al., 2020; Thoms et al., 2020; Tidu et al., 2020; Vankadari et al., 2020; Figure 1—figure supplement 1A). Nsp1 contains 180 amino acids with N-terminal (1–127 amino acids) and C-terminal (148–180 amino acids) structured regions connected by a loop region of about 20 amino acids (Schubert et al., 2020; Thoms et al., 2020; Figure 1—figure supplement 1B). This C-terminal region of Nsp1 (Nsp1-C-ter) contains two helices that harbors a conserved positively charged motif (KH-X5-R/Y/Q-X4-R). The deposition of positive charge toward one edge of these helices enhances their ability to bind helix h18 of 18S rRNA. The other side of C-terminal helices interacts with ribosomal proteins uS3 and uS5 in mRNA entry tunnel of the 40S (Schubert et al., 2020; Thoms et al., 2020; Figure 1—figure supplement 1A, zoomed view). These interactions enable Nsp1-C-ter to bind deep into the mRNA entry tunnel and prevent the binding of mRNAs, thereby inhibiting host protein synthesis (Schubert et al., 2020; Thoms et al., 2020; Tidu et al., 2020). Thus, Nsp1 helps in hijacking the host translational machinery (Yuan et al., 2020) and renders the cells incapable of mounting an innate immune response to counter the viral infection (Narayanan et al., 2008). Mutating the positively charged residues K164 and H165 in Nsp1-C-ter to alanines leads to a decrease in binding affinity of Nsp1 with ribosome and fails to inhibit host protein synthesis (Schubert et al., 2020; Thoms et al., 2020; Tidu et al., 2020).
Nsp1 is a highly conserved protein and less than 3% of SARS-CoV-2 genomic sequences analyzed showed mutation in Nsp1 (Min et al., 2020). Further, Nsp1-C-ter showed a much reduced frequency of mutations (Min et al., 2020). The crucial role of Nsp1 in inhibiting host gene expression, suppression of host immune response (Thoms et al., 2020) and, notably, the reduced mutation frequency in Nsp1-C-ter across global SARS-CoV-2 genomes (Min et al., 2020) advocate targeting Nsp1 for therapeutics. In this study, we have employed computational, biophysical, in vitro, and mammalian cell line based studies to identify FDA-approved drugs targeting Nsp1-C-ter and check for its antiviral activity.
Results
Since repurposing a drug is a quicker way to identify an effective treatment, we screened FDA-approved drugs against Nsp1-C-ter (148–180 amino acids) which binds in the mRNA channel (Figure 1—figure supplement 1C). The drugs docked to a small region of Nsp1-C-ter consisting of residues (P153, F157, N160, K164, H165, and R171) which coincides with its ribosome-binding interface (Figure 1—figure supplement 1C). The residues in Nsp1-C-ter involved in binding drugs show minimal mutations in worldwide deposited 4,440,705 sequences of SARS-CoV-2 genome in GISAID database (Figure 1—figure supplement 1D). We identified top hits with at least three hydrogen bonds;(H-bonds) near the ribosome binding site of Nsp1-C-ter (Supplementary file 1). Further, the clash that the drugs may have against ribosome in its bound form with Nsp1-C-ter was also analyzed. Montelukast sodium hydrate (hereafter referred to as montelukast) and saquinavir mesylate (hereafter referred to as saquinavir) showed high clash scores (Supplementary file 1). Montelukast is regularly used to make breathing easier in asthma (Paggiaro and Bacci, 2011), while saquinavir is an anti-retroviral drug used in the treatment of human immunodeficiency virus (HIV) (Khan et al., 2021).
Next, all 12 drugs were tested in vitro for their ability to bind to Nsp1. The purified proteins, i.e., full-length Nsp1 and C-terminal helices truncated Nsp1 (Nsp1∆C) proteins, were loaded on the Ni-NTA sensors in bio-layer interferometry (BLI), and the compounds were screened to determine its binding to these proteins. We found that montelukast and saquinavir show binding to Nsp1 (Figure 1A) but not with Nsp1∆C (Figure 1B). This indicates that montelukast and saquinavir bind to Nsp1-C-ter. The rest of the compounds does not show binding with Nsp1 or with Nsp1∆C (Figure 1A, B). We next determined binding affinities of montelukast and saquinavir against Nsp1. Montelukast shows a binding affinity (Kd) of 10.8 ± 0.2 µM (Figure 1C) while saquinavir shows a binding affinity of 7.5 ± 0.5 µM toward Nsp1-C-ter (Figure 1D).
Figure 1 with 2 supplements see all
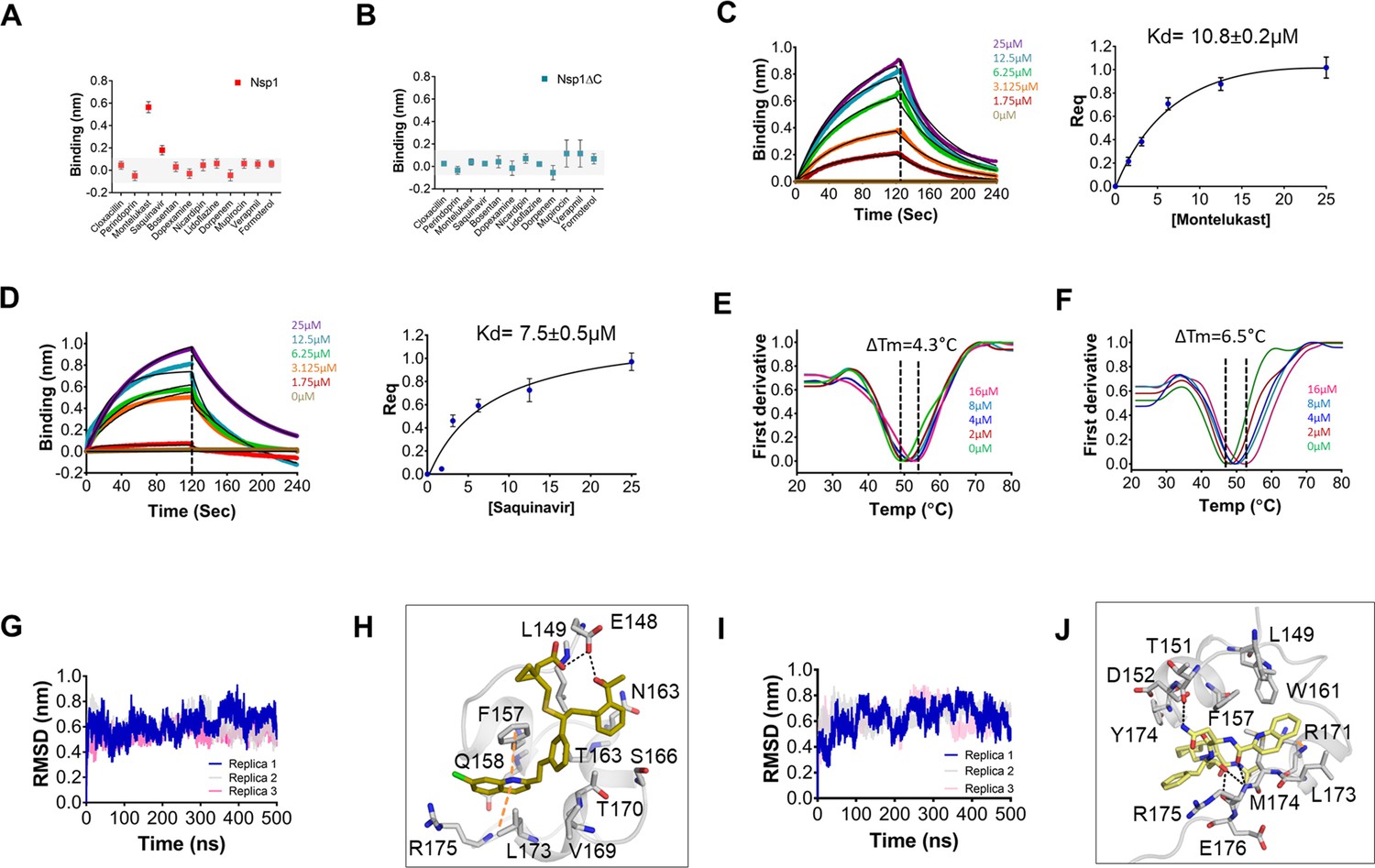
Screening and binding kinetics and molecular simulation dynamics runs of drugs against C-terminal helices of Nsp1 (Nsp1-C-ter).
(A and B) Bio-layer interferometry (BLI) analysis for the initial screening of binding of the drugs with the (A) non-structural protein 1 (Nsp1) and (B) C-terminal helices truncated Nsp1 (Nsp1∆C) proteins. (C and D) The kinetic behaviors of (C) montelukast and (D) saquinavir monitored using BLI by incubating increasing concentration of the drug molecule (0–25 µM) on the protein-bound sensors. Montelukast shows a binding constant (KD) of 10.8 ± 0.8 µM, while saquinavir binds with Nsp1-C-ter with a KD value of 7.5 ± 0.5 µM. (Error bars represent standard deviation of three replicates in (C) and (D). (E and F) Nanoscale differential scanning fluorometry (NanoDSF) experiments to evaluate the change in the melting temperature of the Nsp1 by incubating increasing concentration of (E) montelukast and (F) saquinavir. (The experiments were performed in three replicates.)) (G) Simulation runs with montelukast show stable root mean square deviation (RMSD) values for all replica throughout all molecular dynamic simulation trajectories for 500 ns. (H) The analysis of binding mode of montelukast at the end of 500 ns shows stable binding with C-terminal helices. The residues E148 and L149 form H-bonds with montelukast, while F157 and L173 forms base stacking interactions. (I) Simulation runs with saquinavir show stable pattern in RMSD values throughout in all molecular dynamic simulation trajectories for 500ns. (J) The analysis of binding mode of saquinavir at the end of 500 ns shows stable binding with the C-terminal helices. The residues T151, M174, and R175 form H-bonds with saquinavir, while R171 forms base stacking interactions.
To further validate the binding of ligands with Nsp1-C-ter, we performed NanoDSF experiments where we observed the change in the melting temperature of Nsp1 in the presence of drugs. We observed that only montelukast and saquinavir induce a change in the melting temperature of Nsp1 (Figure 1—figure supplement 1E). None of the ligands were able to change the melting temperature of the Nsp1∆C protein (Figure 1—figure supplement 1F). Next, we performed NanoDSF experiments with different concentrations of montelukast and saquinavir to determine the change in melting temperature of Nsp1. We observed that montelukast shifts the ∆Tm by 4.3°C while the saquinavir causes a ∆Tm shift by 6.5°C (Figure 1E, F). Overall, montelukast and saquinavir showed binding to Nsp1-C-ter in vitro.
To gain insights into the binding mode of montelukast and saquinavir with Nsp1-C-ter, we analyzed the docked drugs and performed molecular dynamic simulation runs. The molecular screening experiment shows the binding of montelukast with Nsp1-C-ter with a 5.61 docking score (Supplementary file 1 and Figure 1—figure supplement 2A). In the simulation runs the root mean square deviation (RMSD) of C-terminal helices bound with montelukast shows less deviation from the mean atomic positions (Figure 1G). The analysis of H-bonds and hydrophobic interactions indicate strong binding of montelukast during the simulation run. At the end of the simulation run, montelukast shows a stable complex by forming H-bonds with E148 and L149, while F157 and L173 form base stacking interactions (Figure 1H). The root mean square fluctuation (RMSF) plot shows the thermal stability of individual residues throughout the molecular dynamics run of the molecule, and it appears to be stable (Figure 1—figure supplement 2B). Saquinavir shows binding with Nsp1 with a docking score of 5.6 (Supplementary file 1 and Figure 1—figure supplement 2C). The RMSD plot of saquinavir bound C-terminal helices shows reduced deviation of the protein atoms during the simulation runs from the mean atomic position (Figure 1I). The residues T151, M174, and R175 form H-bonds with saquinavir while R171 forms base stacking interaction at the end of the run (Figure 1J). The RMSF plot show that the participating residues is also stabilized upon the binding of saquinavir (Figure 1—figure supplement 2D). Overall, the residues involved in binding montelukast and saquinavir show extremely low mutational frequency.
Furthermore, these drug-Nsp1 complexes were subjected to free binding energy calculations using end state free binding energy for 500 ns in two replicas for each complex. Montelukast and saquinavir bind with Nsp1 with binding energies of –95.8 ± 13.3 kJ/mol and –42.7 ± 5.2 kJ/mol, respectively. The average H-bonds were analyzed for the C-terminal region of Nsp1 alone and drug-bound complexes. We observed that these drugs-bound complexes show higher average H-bonds throughout different replica simulations (Figure 1—figure supplement 2E).
Since Nsp1 is known to inhibit host protein synthesis by blocking the mRNA entry tunnel on the ribosome and co-transfection of Nsp1 with capped luciferase reporter mRNA causes reduction of luciferase expression (Thoms et al., 2020). We hypothesized that binding of montelukast or saquinavir to Nsp1-C-ter may prevent inhibition of host protein synthesis. To test this hypothesis, we carried out the cell-based translational rescue of luciferase activity in the presence of montelukast and saquinavir in HEK293 cells when co-transfected with Nsp1. Co-transfection of Nsp1 decreased the luciferase activity by almost half, which is restored by the increasing amount of montelukast (Figure 2A). However, we do not observe a similar rescue of luciferase activity in the presence of saquinavir (Figure 2B). Further experiments are needed to figure out why saquinavir is unable to rescue the Nsp1-mediated translation inhibition. There was no significant change in gene expression of the firefly luciferase FLuc gene (Figure 2C, D).
Figure 2
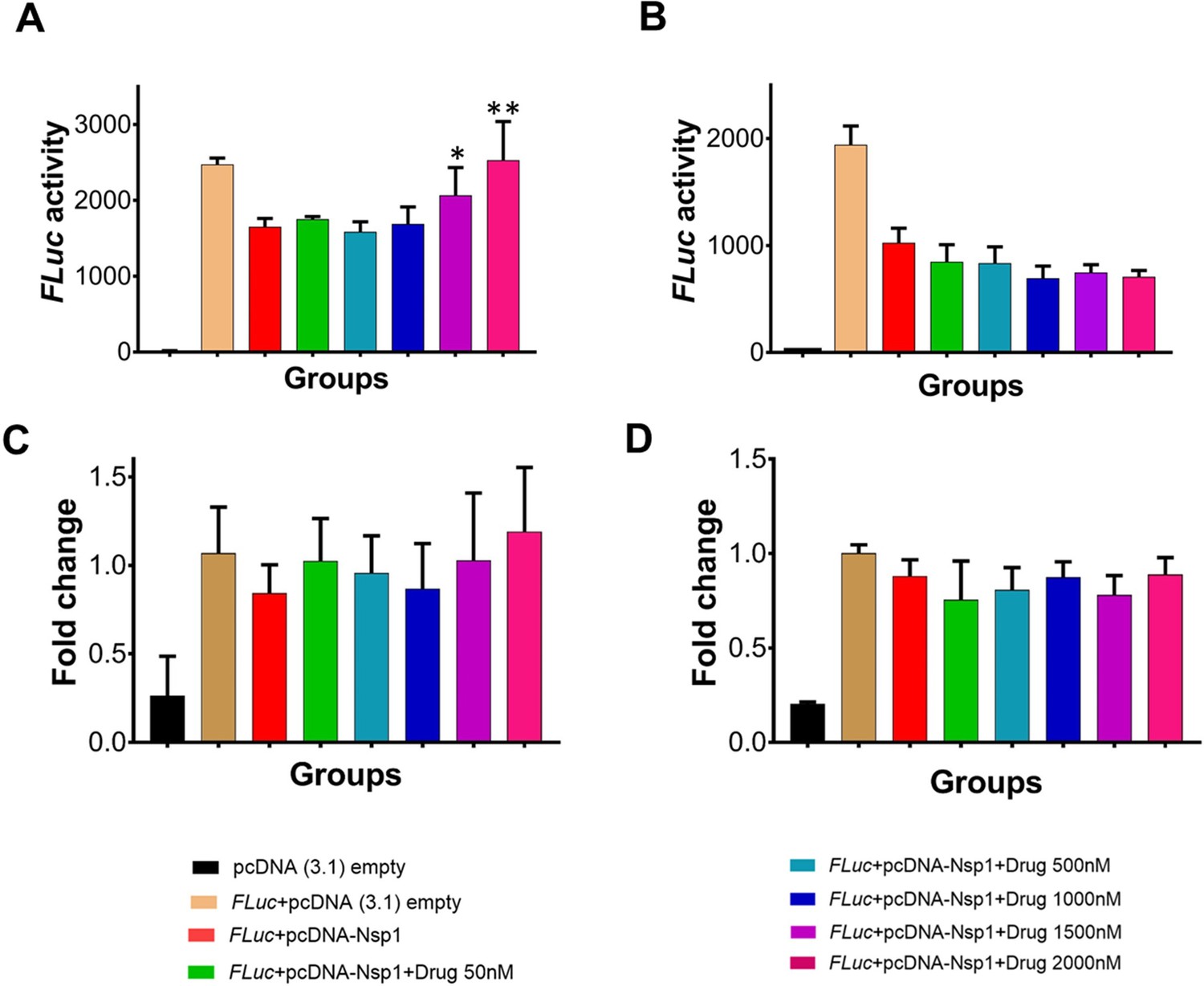
Translational rescue experiments in the presence of montelukast and saquinavir.
(A) Luciferase-based reporter assay shows translational rescue of luciferase in the presence of montelukast. (B) Luciferase-based reporter assay shows that saquinavir could not rescue the luciferase expression. Error bars represent standard deviation of three replicates in (A) and (B). (C and D) The real-time PCR to quantitate the fold change of FLuc gene in comparison to GAPDH in the presence of different concentration of the drug molecules. (A) montelukast (B) saquinavir. The panel below provides the details of experimental conditions. Error bars represent standard deviation of three replicates in (A) and (B). The significance of the data was monitored by applying the unpaired t-test through assuming Gaussian distribution parametric test by defining the statistical significance. **p < 0.01; ***p < 0.001; ****p < 0.0001. The error bars represent the standard deviation.
To test antiviral effects of montelukast and saquinavir against SARS-CoV-2, we first tested the cytotoxicity of these drugs in HEK293T-ACE2 and Vero-E6 cells. Results showed minimal toxicity up to 10 µM montelukast and saquinavir in both cell lines. However, in Vero-E6 cells, the highest concentration (20 µM) of both drugs showed an almost 80% decrease in cell viability, compared to untreated cell control (Figure 3—figure supplement 1). Based on this, a working concentration of 10 µM or lower was used for both drugs. At a concentration of 10 µM, montelukast showed significant antiviral activity, as indicated by reduced expression of viral spike protein in HEK293T-ACE2 and Vero-E6 cells (Figure 3A, D). The corresponding qRT-PCR data demonstrated up to 1-log reduction in viral copy number in both HEK293T-ACE2 and Vero-E6 cells at this concentration (Figure 3B, E), supported by a decrease in infectious virus titer measured by plaque assay (Figure 3C, F). No significant antiviral effects were observed in the presence of 10 µM saquinavir (Figure 3—figure supplement 2).
Figure 3 with 2 supplements see all
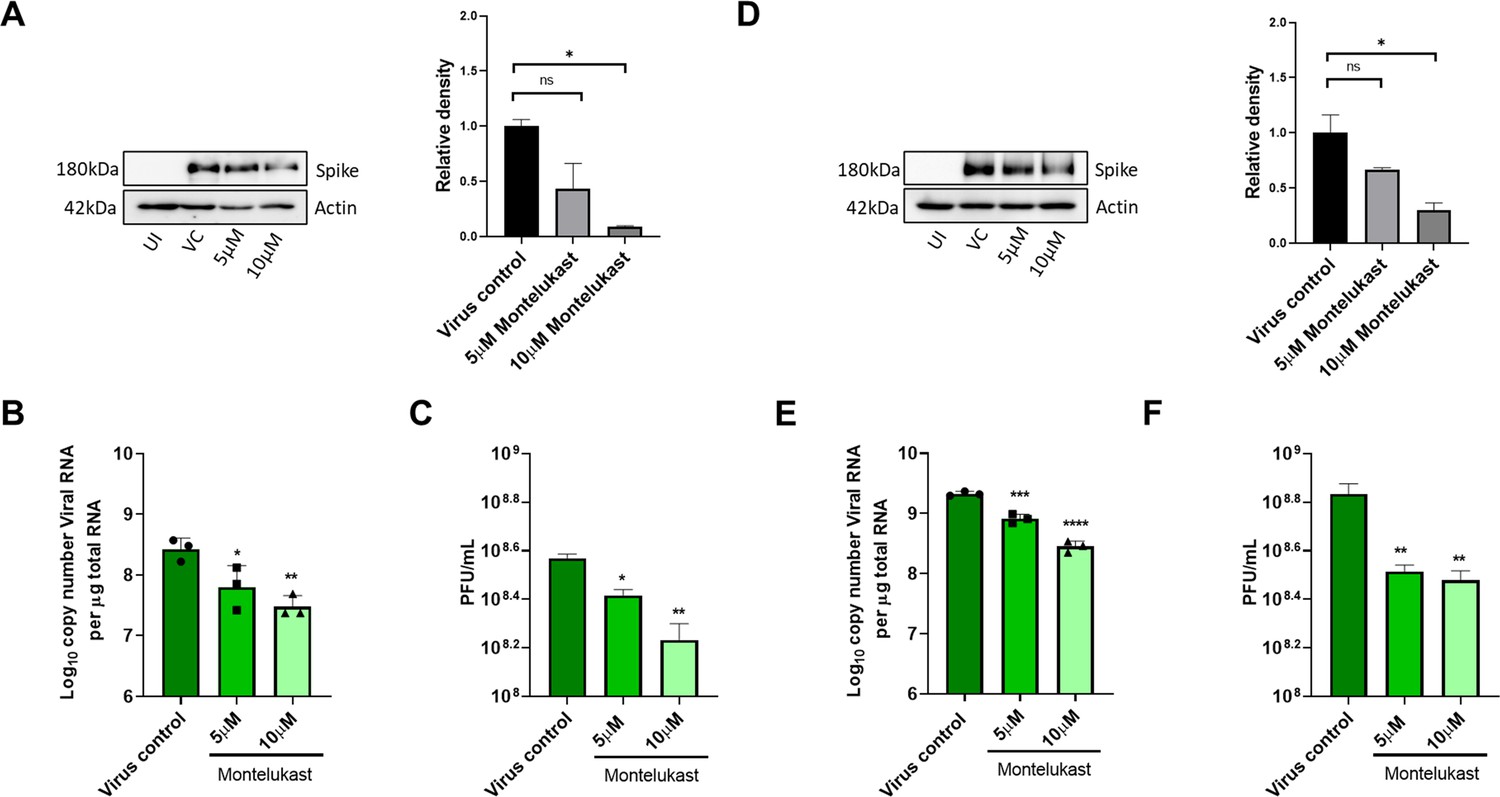
Montelukast shows antiviral activity against SARS-CoV-2.
(A) HEK ACE2 cells were pre-treated with 5 or 10 µM montelukast and infected with 0.1 MOI SARS-CoV-2 for 48 hr. Virus spike protein expression by western blot analysis, with corresponding relative density of bands are shown. (B) Viral RNA copy number from infected cells was quantified by qRT PCR and (C) infectious virus titer from cell culture supernatants by plaque assay, respectively. Vero E6 cells were pre-treated with 5 or 10 µM montelukast and infected with 0.001 MOI SARS-CoV-2 for 48 hr. (D) Virus spike protein expression by western blot analysis, with corresponding relative density of bands. (E) Viral RNA copy number from infected cells was quantified by qRT PCR and (F) infectious virus titer from cell culture supernatants by plaque assay. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns-not significant, using one-way ANOVA with Dunnett’s multiple comparison test. Error bars represent standard deviation.
Discussion
Nsp1 is a major virulence factor in SARS-CoV2 which effectively blocks the synthesis of major immune effectors (IFN-β, IFN-λ1, and interleukin-8, retinoic acid–inducible gene I), thereby aiding in establishment of the viral infection (Thoms et al., 2020). It serves as a blockage to host mRNA entry by interacting with rRNA helix 18 and ribosomal proteins-uS5 and uS3 near the mRNA entry channel of the 40S ribosomal subunit via its C-terminal helices (Thoms et al., 2020). Structural studies on 48S-like preinitiation complex on Cricket paralysis viral internal ribosomal entry site in presence of Nsp1 revealed its ability to lock the head domain of 40S ribosome in a closed conformation. In addition, it competes with eIF3j for uS3 and weakens the binding of the eIF3 to the 40S subunit (Yuan et al., 2020). While the host translation is inhibited by the C-terminal helices of Nsp1, its N-terminal domain enhances translation of viral mRNAs by binding to the 5' UTR (Gordon et al., 2020). Moreover, Nsp1 interacts with host mRNA export receptor NXF1-NXT1 heterodimer and aids in retention of cellular mRNAs in the nucleus (Zhang et al., 2021). Further, Mou et al., 2021 deciphered the frequency of mutation accumulation in the N-terminal domain was higher than that of the C-terminal domain (Mou et al., 2021). Therefore, we targeted the C-terminal helices of Nsp for this study.
Since repurposing a drug is a quicker way to identify an effective treatment, we screened FDA-approved drugs against Nsp1-C-ter and found montelukast as potential lead molecule against it. Montelukast is a leukotriene receptor antagonist and repurposing montelukast for tackling cytokine storms in COVID-19 patients has been suggested (Sanghai and Tranmer, 2020) and hospitalized COVID-19 patients that were given montelukast had significantly fewer events of clinical deterioration (Khan et al., 2021). Montelukast also appears as a hit against the SARS-CoV-2 main protease, (Mpro) protease, in computational studies (Abu-Saleh et al., 2020; Sharma et al., 2021). However, Ma and Wang demonstrated that montelukast gives false positive anti-protease activity as it cannot bind the GST-tagged-Mpro in thermal shift assay and native mass spectrometry experiments (Ma and Wang, 2021). Thus, montelukast may not be an inhibitor for Mpro protease.
Viruses employ different strategies to shutdown host translation machinery. In SARS-CoV-2, Nsp1 inhibits translation by binding to the mRNA channel. Here, we show that montelukast binds to Nsp1, rescues the Nsp1-mediated translation inhibition and has antiviral activity against SARS-CoV-2. The rescue of shutdown of host protein synthesis machinery by montelukast seems to contribute toward the antiviral activity of the drug; however, further experiments would be essential to figure out detailed mechanism of its antiviral activity. Overall, our study identifies C-terminal region of Nsp1 as a druggable target and montelukast as a starting point for designing more potent drug molecules against SARS-CoV-2.
Materials and methods
Receptor preparation for in silico studies and molecular screening of FDA-approved drugs
Request a detailed protocolThe three-dimensional coordinates of C-terminal helices of Nsp1 (Nsp1-C-ter; residue numbers 148–180) were taken from the cryo-EM structure of Nsp1-bound 40S (PDB ID: 6ZOJ). The close contacts, side chains, and bumps were fixed in Chimera (Pettersen et al., 2004). The molecule was minimized using 100 steepest descent steps and ten conjugate gradient steps using AMBERff14SB force field (Maier et al., 2015). None of the atoms were fixed during minimization, and charges were assigned using the AMBERff14SB force field on standard residues. The final structure was optimized by Powell method implemented in biopolymer program of SYBYL-X v2.1 (Tripos International, St. Louis, Missouri, 63144, USA).
The FDA-approved drug library was used to screen the drugs toward Nsp1-C-ter. The drug library containing 1,645 compounds was subjected to in silico molecular screening. Three-dimensional structure of (SDF format) compound library was optimized in SYBYL-ligand prep module at default parameters. The single lowest strain energy tautomer for each compound was searched using Surflex in ligand preparation module. Subsequently, the binding pocket for ligands on Nsp1-C-ter was determined by Computed Atlas of Surface Topography of proteins (CASTp) online server (Tian et al., 2018). The T151, P153, D156, F157, Q158, N160, K164, H165, S167, T170, R171, E172, L173, R175, and L177 were found to form the binding pocket. Finally, the compound library was screened against 18S rRNA interacting interface of Nsp1-C-ter using the Surflex-dock program, which is available in SYBYL v2.1 (Jain, 2003). Twenty conformers were generated for each molecule with 100 maximum rotatable bonds, and top potential molecules were selected based on docking score, which was calculated based on scoring function (flex C-score).
Nsp1 expression and purification
Request a detailed protocolThe gene construct encoding Nsp1 from SARS-CoV-2 in pCDNA 5–3X-Flag-Nsp1 was amplified and sub-cloned into pET28a with N-terminal His-tag (Schubert et al., 2020; Thoms et al., 2020) using appropriate primers (Supplementary file 2). The sub-cloned construct was further used to amplify and clone the C-terminal 28 amino acid deleted construct of Nsp1 (Nsp1∆C) using appropriate primers (Supplementary file 2). Then constructs were transformed into E. coli BL-21 DE3 expression system. The secondary cultures were then inoculated with 1% of the primary culture and incubated at 37°C at 180 rpm. At 0.6 O.D., the cultures were induced with 1 mM IPTG at 16°C and 120 rpm for 18 hr. Cells were harvested at 6000 rpm and resuspended in buffer A (50 mM HEPES-KOH pH 7.6, 500 mM KCl, 5 mM MgCl2, 5% Glycerol). Lysis was done by sonicating at 18% amplitude (10 sec on/off cycles for 10 min) and clarified by centrifugation at 12,000 rpm for 30 min. The clear supernatant was then loaded on the Ni-NTA beads (Qiagen) and incubated for 3 hr, and beads were washed using buffer A. The bound protein was eluted with buffer A supplemented with 300 mM imidazole, and purity was analyzed on SDS-PAGE. The fractions containing corresponding protein were concentrated and subjected to size exclusion chromatography on Superdex 200 increase 10/300 column in buffer B (50 mM HEPES-KOH pH 7.6, 150 mM KCl, 5 mM MgCl2, 2% Glycerol and 2 mM DTT). The pure protein fractions were pooled and concentrated between 2 and 8 mg/mL and stored in –80°C for further use.
Drug-binding assays
Bio-layer interferometry
Request a detailed protocolTo identify the kinetic behavior of the top selected compounds, we performed the label-free binding kinetics of protein and ligands by using bio-layer interferometry. The Ni-NTA sensors were activated by incubating in 10 mM phosphate buffer saline for 10 min. Thereafter, 2 µM of each protein was loaded on the Ni-NTA sensor and a binding response of around 1 nm was obtained. The initial screening of compounds was performed at 20 µM for all in silico selected top hits. The drug molecules that showed binding response of more than 0.2 nm were chosen for further kinetic experiments. The binding kinetics were measured by incubating protein-bound sensors with the increasing ligand concentration (0–25 µM). The data for control sensors (without protein) for each ligand concentration were also collected and subtracted from the response of proteins-bound sensors. The subtracted data was then analyzed by fitting the 1:1 stoichiometric ratio for association and dissociation by applying the global fitting. Three independent experiments were performed to evaluate the steady-state kinetics and calculate KD values.
Nanoscale differential scanning fluorometry
Request a detailed protocolIn silico identified potential hits were then subjected to evaluate the binding with His-Nsp1 and His-Nsp1∆C of SARS-CoV-2 protein. 2 µM of each protein was subjected to determine the melting temperature the in buffer B. The temperature scans ranged from 20 to 90°C with the 1°C/min ramp size using Prometheus NT.48 NanoTemper. Next, the ΔTm was determined in the presence of drug molecules (10 µM) to figure out binding of drug molecules. The top hits were selected for further evaluation in a change of the Tm by incubating with different concentrations of ligand (0–16 µM). The data was analyzed by using ThermControl software.
Molecular dynamics simulation of C-terminal helices of Nsp1 and drugs-bound complexes
Request a detailed protocolThe molecular dynamic simulations of FDA-approved drugs in complex with Nsp1-C-ter were selected based on top binding score using BLI and NanoDSF. The final docked complexes were then prepared for molecular dynamics simulation studies. The systems for molecular dynamics studies were prepared for Nsp1-C-ter alone and their complex with top hits using the Desmond v4.1implemented in Schrodinger-Maestro v11, where steric clashes and side-chain bumps were fixed. These prepared structures were then optimized by GROMOS96 54a7 force field (Schmid et al., 2011) and simple point charge water model was used to add the solvent molecules in dodecahedron box with a distance of 1 Å from the surface of protein. Additionally, four sodium ions were added to neutralize the system. The following energy minimization was performed for all the systems with 5000 steps of steepest descent and conjugate gradient algorithms with threshold energy of 100 kcal/mol. The systems were then equilibrated in two phases, first is isothermal-isochoric equilibration, where constant number, volume, and temperature (NVT) was equilibrated for 100 ps, and the temperature of the system was monitored for all constants. In second phase, isothermal-isobaric equilibration was performed where number of particles, pressure, and temperature (NPT) was equilibrated for 100 ps. After successful equilibration of the system, final molecular dynamic runs were performed for 500 ns in three replicas with 2 femtoseconds of time steps. The RMSD, RMSF, and three-dimensional coordinates for all atoms of protein and ligands were extracted to analyze the molecular dynamics runs.
Binding energy calculation
Request a detailed protocolThe binding energy for protein and ligands were calculated by applying the gmx_Molecular Mechanic and Poisson-Boltzmann Surface Area (gmx-MMPBSA) (Valdés-Tresanco et al., 2021). Two subsequent 500 ns runs from MD simulations were further subjected to perform the gmx_MMPBSA by using AmberTools21. The binding energy was decomposed into free binding energy for drug molecules for 5000 frames. This binding energy calculation quantitatively provides in silico biomolecular interaction between selected ligands and target protein. This binding energy mainly constitutes the polar solvation energy, non-polar solvation energy and potential energy. The free binding energy (∆Gbinding) of the ligand was calculated by the following equation:
Where ∆Gcomplex describes the Gibbs free energy of the complex, Greceptor and Glignad are total energy of protein and ligand, respectively.
Luciferase-based assay: translation inhibition and rescue experiments
Request a detailed protocolThe luciferase based reporter assay was used to evaluate the target-specific action of drug molecules. HEK293 cells were transfected with 100 ng/well of pGL3-Fluc plasmid using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol at around 75–90% confluency in a 96 well plate. The plasmid expressing Nsp1 protein (pcDNA 3.1-Nsp1) was co-transfected at 100 ng/well concentration. The transfection was performed in the presence of drugs montelukast and saquinavir at different concentrations. The cells were lysed 24 hr post-transfection, and luciferase activity was measured by using Luciferase Reporter assay system (Promega Corporation) in the GLoMax Explorer system (Promega Corporation).
The expression level of FLuc was measured, keeping Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the control. Total RNA from all conditions was isolated using the TRIzol as per the user manual protocol. 0.5 µg of total RNA was used as a template for cDNA synthesis (RevertAid First Strand cDNA synthesis kit using manufacturer’s protocol), which was further used as template to quantitate FLuc and GAPDH expression in the presence of appropriate primers as mentioned in Supplementary file 2. The relative Ct values were monitored in the three replicates and relative fold change in expression was calculated. The significance of the data was monitored by applying the unpaired t-test through assuming Gaussian distribution parametric test by defining the statistical significance p < 0.5.
To evaluate the total viral copy number, RNA from SARS-CoV-2 infected cells was isolated using TRIzol as per manufacturer’s instructions, and equal amount of RNA used to determine the viral load using AgPath-ID One-Step RT-PCR kit (AM1005, Applied Biosystems). The primers and probes against SARS CoV-2 N-1 gene used are mentioned in Supplementary file 1. A standard curve was made using SARS-CoV-2 genomic RNA standards, which was used to determine viral copy number from Ct values.
Cells and virus
Request a detailed protocolThe following cell lines were used in this study, namely, HEK293 (ATCC), HEK293T-ACE2 (HEK293T cells stably expressing human angiotensin-converting enzyme 2) (BEI Resources NR-52511, NIAID, NIH. RRID: CVCL_A7UK) and Vero-E6 cells (CRL-1586, ATCC, RRID: CVCL_0574). The authenticity of HEK293T-ACE2 and Vero-E6 cell lines was confirmed by Certificate of Analysis from their respective sources. HEK293T-ACE2 are human embryonic kidney 293T cells that express the human ACE2 receptor, which is required for SARS-CoV-2 entry. HEK293T-ACE2 and Vero E6 cells are of human and primate origin respectively, and express ACE2 receptor. All cell lines tested negative for mycoplasma contamination. Cells were cultured in complete media prepared using Dulbecco’s modified Eagle medium (12100–038, Gibco) supplemented with 10% HI-FBS (16140–071, Gibco), 100 U/mL Penicillin-Streptomycin (15140122, Gibco) and GlutaMAX (35050–061, Gibco).
SARS-CoV2 (Isolate Hong Kong/VM20001061/2020, NR-52282, BEI Resources, NIAID, NIH) was propagated and quantified by plaque assay in Vero-E6 cells as described before (Case et al., 2020).
Cytotoxicity assay
Request a detailed protocolHEK293T-ACE2 cells were seeded in 0.1 mg/mL poly-L-lysine (P9155-5MG, Sigma-Aldrich) coated 96-well plate to reach 70–80% confluency after 24 hr. Vero-E6 cells were seeded in a regular 96 well plate to reach similar confluency. Cells were treated with 5, 10, and 20 µM montelukast or saquinavir in triplicates and incubated at 37°C/5% CO2. After 48 hr, cytotoxicity was measured using AlamarBlue Cell Viability Reagent (DAL 1025, Thermo Fisher Scientific) as per manufacturer’s instructions.
Western blot
Request a detailed protocolCells were washed gently with 1× warm PBS (162528, MP Biomedicals), lysed using 1× Laemmli buffer (1610747, BIO-RAD), and heated at 95°C before loading on to a 10% SDS-PAGE gel. Separated proteins were transferred onto a PVDF membrane (IPVH00010, Immobilon-P; Merck) and incubated for 2 hr with blocking buffer containing 5% Skimmed milk (70166, Sigma-Aldrich) in PBST (1× PBS containing 0.05% Tween 20 (P1379, Sigma-Aldrich)) for 2 hr at RT (room temperature). The blots were then probed with SARS-CoV-2 spike antibody (NR-52947, BEI Resources, NIAID, NIH) in blocking buffer for 12 hr at 4°C, followed by secondary Goat Anti-Rabbit IgG antibody (ab6721, Abcam, RRID:AB_955447) incubation for 2 hr. Proteins were detected using Clarity Western ECL Substrate (1705061, BIO-RAD). Actin was labeled using antibody against beta-actin [AC-15] (HRP) (ab49900, Abcam, RRID: AB_867494). Relative intensity of bands was quantified using imagej/Fiji.
Virus infection
Request a detailed protocolHEK293T-ACE2 cells were seeded in poly-L-lysine coated 24-well plate to reach 80% confluency at the time of infection. Vero-E6 cells were seeded in a regular 24 well plate to reach similar confluency. Cells, in quadruplicates, were first pre-treated with 5 and 10 µM concentrations of montelukast sodium hydrate (PHR1603, Merck) or saquinavir mesylate (1609829, Merck) for 3 hr in complete media, washed and infected with 0.1 MOI (HEK ACE2) or 0.001 MOI (Vero-E6 cells) SARS-CoV-2. After 48 hr, cell culture supernatants were collected for plaque assay, and cells were harvested for western blot analysis or processed for total RNA extraction using TRIzol (15596018, Thermo Fisher Scientific). The drugs were present in the media for the entire duration of the experiment.
Plaque assay
Request a detailed protocolInfectious virus particles from cell culture supernatants were quantified by plaque assay. Briefly, Vero-E6 cells were seeded in 12-well cell culture dishes, and once confluent, cells were washed with warm PBS and incubated with dilutions of cell culture supernatants in 100 μL complete DMEM for 1 hr at 37°C/5% CO2. The virus inoculum was then removed, and cells overlaid with 0.6% Avicel (RC-591, Dupont) in DMEM containing 2% HI-FBS. After 48 hr incubation, cells were fixed with 4% paraformaldehyde, and crystal violet (C6158, Merck) staining was done to visualize the plaques.
Plasmids pLVX-EF1alpha-SARS-CoV-2-nsp1-2xStrep-IRES-Puro expressing SARS-CoV-2 NSP1 was a kind gift from Prof. Nevan Krogan (Gordon et al., 2020). Other plasmids used in this study include Plasmids pRL-TK (mammalian vector for weak constitutive expression of wild-type Renilla luciferase), pGL4 (mammalian vector expressing firefly luciferase), pIFN-β Luc (IFN beta promoter-driven firefly luciferase reporter). The plasmid pMTB242 pcDNA5 FRT-TO-3xFLAG-3C-Nsp1_SARS2 was a kind gift from Prof. Ronald Beckmann.
Supporting Information
Request a detailed protocolSupporting information contains four figures and two supplementary files.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting file.
References
-
Discovery of potent inhibitors for SARS-CoV-2’s main protease by ligand-based/structure-based virtual screening, MD simulations, and binding energy calculationsPhysical Chemistry Chemical Physics 22:23099–23106.https://doi.org/10.1039/d0cp04326e
-
GESS: a database of global evaluation of SARS-CoV-2/hCoV-19 sequencesNucleic Acids Research 49:706–714.https://doi.org/10.1093/nar/gkaa808
-
Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engineJournal of Medicinal Chemistry 46:499–511.https://doi.org/10.1021/jm020406h
-
Montelukast in hospitalized patients diagnosed with COVID-19The Journal of Asthma 1:1–7.https://doi.org/10.1080/02770903.2021.1881967
-
ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SBJournal of Chemical Theory and Computation 11:3696–3713.https://doi.org/10.1021/acs.jctc.5b00255
-
Emerging Mutations in Nsp1 of SARS-CoV-2 and Their Effect on the Structural StabilityPathogens (Basel, Switzerland) 10:1285.https://doi.org/10.3390/pathogens10101285
-
Montelukast in asthma: a review of its efficacy and place in therapyTherapeutic Advances in Chronic Disease 2:47–58.https://doi.org/10.1177/2040622310383343
-
UCSF Chimera-a visualization system for exploratory research and analysisJournal of Computational Chemistry 25:1605–1612.https://doi.org/10.1002/jcc.20084
-
Definition and testing of the GROMOS force-field versions 54A7 and 54B7European Biophysics Journal 40:843–856.https://doi.org/10.1007/s00249-011-0700-9
-
SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translationNature Structural & Molecular Biology 27:959–966.https://doi.org/10.1038/s41594-020-0511-8
-
Screening of drug databank against WT and mutant main protease of SARS-CoV-2: Towards finding potential compound for repurposing against COVID-19Saudi Journal of Biological Sciences 28:3152–3159.https://doi.org/10.1016/j.sjbs.2021.02.059
-
CASTp 3.0: computed atlas of surface topography of proteinsNucleic Acids Research 46:W363–W367.https://doi.org/10.1093/nar/gky473
-
gmx_MMPBSA: A New Tool to Perform End-State Free energy calculations with GROMACSJournal of Chemical Theory and Computation 17:6281–6291.https://doi.org/10.1021/acs.jctc.1c00645
-
Structure of the SARS-CoV-2 Nsp1/5’-Untranslated Region Complex and Implications for Potential Therapeutic Targets, a Vaccine, and VirulenceThe Journal of Physical Chemistry Letters 11:9659–9668.https://doi.org/10.1021/acs.jpclett.0c02818
-
Coronavirus biology and replication: implications for SARS-CoV-2Nature Reviews. Microbiology 19:155–170.https://doi.org/10.1038/s41579-020-00468-6
Article and author information
Author details
Sandeep M Eswarappa

Funding
Wellcome Trust/DBT India Alliance (IA/I/17/2/503313)
- Tanweer Hussain
IRPHA (IPA/2020/000094)
- Tanweer Hussain
Wellcome Trust/DBT India Alliance (IA/I/18/1/503613)
- Shashank Tripathi
Swarnajayanti Fellowship (SB/SJF/2020-21/18)
- Sandeep M Eswarappa
Biotechnology Industry Research Assistance Council (BT/CS0007/CS/02/20)
- Shashank Tripathi
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by Intermediate Fellowship from DBT-Wellcome Trust India Alliance to TH (IA/I/17/2/503313). TH also thanks SERB for funds released under IRPHA (COVID-19 Life Sciences; File Number:IPA/2020/000094). ST acknowledges funding from DBT-BIRAC grant (BT/CS0007/CS/02/20) and DBT-Wellcome Trust India Alliance Intermediate Fellowship (IA/I/18/1/503613). We acknowledge Swarnajayanti Fellowship from DST to SME (SB/SJF/2020-21/18).
Copyright
© 2022, Afsar et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Drug targeting Nsp1-ribosomal complex shows antiviral activity against SARS-CoV-2
eLife 11:e74877.
https://doi.org/10.7554/eLife.74877
Further reading
-
- Medicine
Inherited retinal degenerations (IRDs) constitute a group of clinically and genetically diverse vision-impairing disorders. Retinitis pigmentosa (RP), the most common form of IRD, is characterized by gradual dysfunction and degeneration of rod photoreceptors, followed by the loss of cone photoreceptors. Recently, we identified reserpine as a lead molecule for maintaining rod survival in mouse and human retinal organoids as well as in the rd16 mouse, which phenocopy Leber congenital amaurosis caused by mutations in the cilia-centrosomal gene CEP290 (Chen et al., 2023). Here, we show the therapeutic potential of reserpine in a rhodopsin P23H rat model of autosomal dominant RP. At postnatal day (P) 68, when males and females are analyzed together, the reserpine-treated rats exhibit higher rod-derived scotopic b-wave amplitudes compared to the controls with little or no change in scotopic a-wave or cone-derived photopic b-wave. Interestingly, the reserpine-treated female rats display enhanced scotopic a- and b-waves and photopic b-wave responses at P68, along with a better contrast threshold and increased outer nuclear layer thickness. The female rats demonstrate better preservation of both rod and cone photoreceptors following reserpine treatment. Retinal transcriptome analysis reveals sex-specific responses to reserpine, with significant upregulation of phototransduction genes and proteostasis-related pathways, and notably, genes associated with stress response. This study builds upon our previously reported results reaffirming the potential of reserpine for gene-agnostic treatment of IRDs and emphasizes the importance of biological sex in retinal disease research and therapy development.
-
- Medicine
The incidence of post-cardiac arrest myocardial dysfunction (PAMD) is high, and there is currently no effective treatment available. This study aims to investigate the protective effects of exogenous mitochondrial transplantation in Sprague-Dawley (SD) rats. Exogenous mitochondrial transplantation can enhance myocardial function and improve the survival rate. Mechanistic studies suggest that mitochondrial transplantation can limit impairment in mitochondrial morphology, augment the activity of mitochondrial complexes II and IV, and raise ATP level. As well, mitochondrial therapy ameliorated oxidative stress imbalance, reduced myocardial injury, and thus improved PAMD after cardiopulmonary resuscitation (CPR).




{kind=link}
{kind=link}
{kind=link}