Mechanism of Ca2+ transport by ferroportin
- Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, United States
Abstract
Ferroportin (Fpn) is a transporter that releases ferrous ion (Fe2+) from cells and is important for homeostasis of iron in circulation. Export of one Fe2+ by Fpn is coupled to import of two H+ to maintain charge balance. Here, we show that human Fpn (HsFpn) binds to and mediates Ca2+ transport. We determine the structure of Ca2+-bound HsFpn and identify a single Ca2+ binding site distinct from the Fe2+ binding sites. Further studies validate the Ca2+ binding site and show that Ca2+ transport is not coupled to transport of another ion. In addition, Ca2+ transport is significantly inhibited in the presence of Fe2+ but not vice versa. Function of Fpn as a Ca2+ uniporter may allow regulation of iron homeostasis by Ca2+.
Editor's evaluation
This study makes an important contribution to the function of the iron transporter Ferroportin (Fpn). By using a combination of proteoliposome assays, mutagenesis and structural studies by cryo EM, the authors are able to demonstrate that the H+-driven transporter for Fe2+-efflux is also capable of passive Ca2+ influx. The evidence supporting the conclusions is convincing, but the physiological relevance of Ca2+ entry by Fpn is yet to be established. The work will be of broad interest to cell biologists and biochemists.
https://doi.org/10.7554/eLife.82947.sa0Introduction
Ferroportin (Fpn), encoded by the SLC40A1 gene, is the only known Fe2+ exporter in human (Nemeth and Ganz, 2021; Abboud and Haile, 2000; Donovan et al., 2000; McKie et al., 2000). Fpn is highly expressed on enterocytes, hepatocytes, and macrophages, and mediates release of iron stored in cells (Drakesmith et al., 2015; Mackenzie and Garrick, 2005; Knutson et al., 2005). Activity of Fpn is regulated by hepcidin, a small peptide hormone secreted by hepatocytes, which binds to Fpn and reduces iron export by inhibiting its transport activity and triggering endocytosis of Fpn (Aschemeyer et al., 2018; Nemeth et al., 2004; Nemeth and Ganz, 2021). Mutations in Fpn cause hereditary hemochromatosis and iron-loading anemias in human (Pietrangelo, 2017; Ganz, 2013; Ginzburg, 2019; Vlasveld et al., 2019).
We have shown previously that Fpn is a Fe2+/2H+ exchanger, that is, the export of one Fe2+ is accompanied by the import of two H+ (Pan et al., 2020). The electroneutral transport mechanism is likely an adaptation to overcome the negative resting membrane potential that would have significantly hindered export of cations. Structures of mammalian Fpn show two transition-metal ion binding sites, termed Site 1 (S1) and Site 2 (S2) (Billesbølle et al., 2020; Pan et al., 2020). Curiously, each ion binding site is composed of only two residues, Asp39 and His43 for S1, and Cys326 and His507 for S2, and it remains unsettled how S1 and S2 mediate coupled transport of Fe2+ and H+. In the current study, we found that Asp39 is also part of a Ca2+ binding site, and that mutation Asp39Ala almost completely eliminates Ca2+ transport but has a modest impact on Fe2+ transport, suggesting that S1 and S2 may have different roles in Fe2+ or H+ transport.
Ca2+ is known to bind to mammalian Fpn (Deshpande et al., 2018), but Ca2+ transport by Fpn has not been demonstrated and the role of Ca2+ in Fe2+ transport remains ambiguous. Using Fpn expressed in Xenopus oocytes, Deshpande et al. found that Fe2+ export requires Ca2+, but Ca2+ is not transported by Fpn (Deshpande et al., 2018). More recently, several studies using purified Fpn reconstituted into liposomes found that Fe2+ transport occurs in the absence of Ca2+ (Li et al., 2020; Pan et al., 2020; Billesbølle et al., 2020), and that Ca2+ could potentiate Fe2+ transport under certain conditions (Li et al., 2020; Billesbølle et al., 2020). In the current study, we visualize the Ca2+ binding site in HsFpn and we demonstrate that Ca2+ is transported by Fpn. We then examine the mechanism of Ca2+ transport and its effect on Fe2+ transport.
Results
HsFpn is a Ca2+ uniporter
HsFpn was expressed, purified, and reconstituted into liposomes for transport assays (Figure 1—figure supplement 1a and Materials and methods). Significant Ca2+ uptake was observed in proteoliposomes reconstituted with HsFpn, as indicated by increased fluorescence of Fluo-4 trapped inside of the vesicles (Figure 1a and Figure 1—figure supplement 1b). In contrast, control liposomes that do not have HsFpn show minimal change in fluorescence (Figure 1a). We also found that Ca2+ transport is inhibited by hepcidin and by a monoclonal antibody (11F9) that binds to HsFpn with nanomolar affinity (Figure 1a–b), providing further support that HsFpn mediates Ca2+ transport. As the fragment of antigen-binding (Fab) of 11F9 is known to target the intracellular side of HsFpn (Figure 2a; Pan et al., 2020; Wilbon et al., 2023), we applied the Fab either to only the external side of the liposomes or to both sides of the liposomes, and found that the inhibition is significantly higher when Fab is present on both sides. This result indicates that HsFpn is incorporated into liposomes in both orientations (Figure 1—figure supplement 1c–g). We also examined HsFpn expressed in human embryonic kidney (HEK) cells and found that HsFpn mediates Ca2+ uptake (Figure 1c–d).
Figure 1 with 4 supplements see all
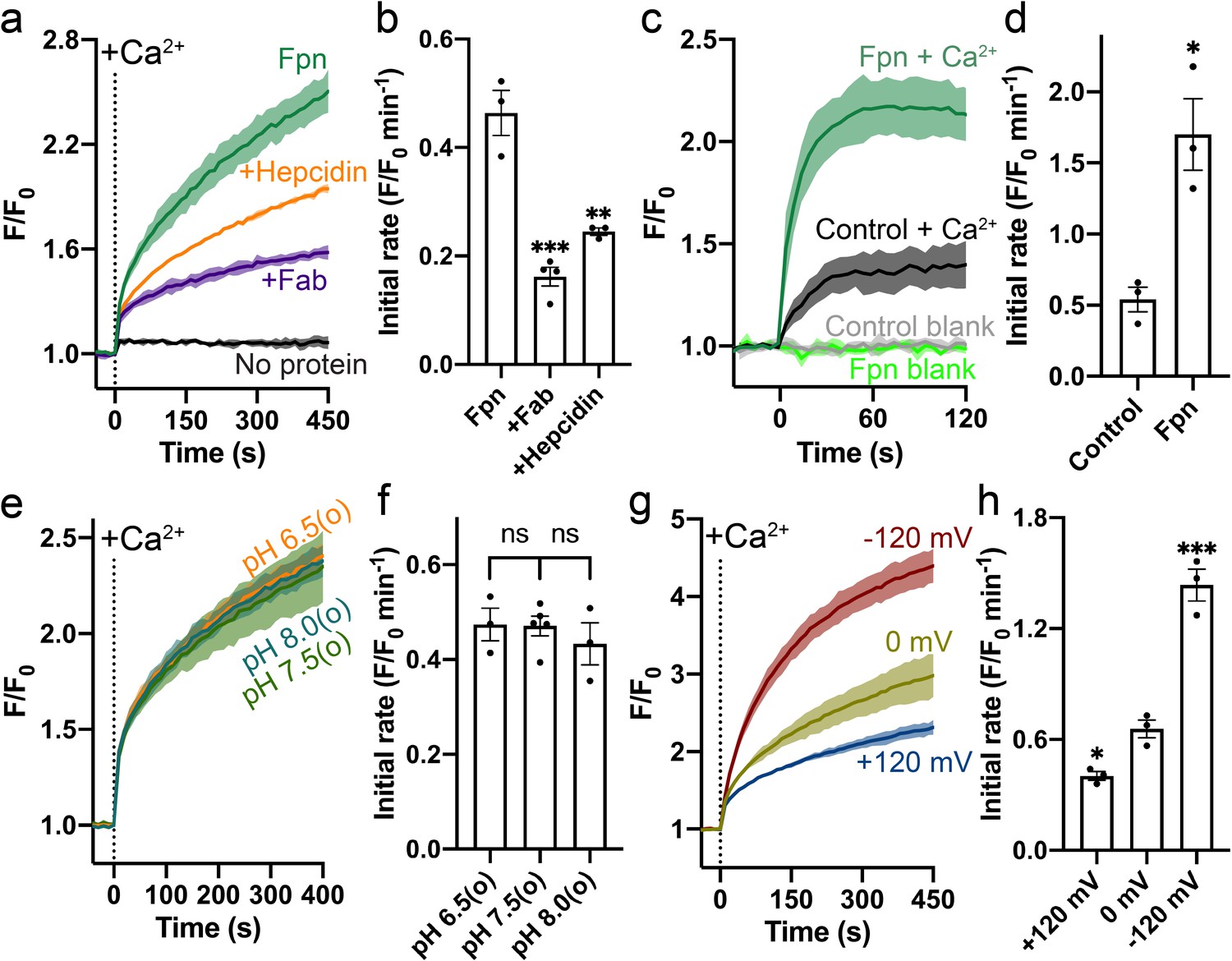
Specific Ca2+ uniport by HsFpn.
(a) Ca2+ influx by HsFpn in proteoliposomes measured by fluorescence changes (F/F0) of Fluo-4 (green). The presence of 11F9 Fab (purple) or hepcidin (orange) reduces the Ca2+ influx. (b) Comparison of initial rates of Ca2+ influx in (a). (c) Ca2+ uptake in HEK cells expressing HsFpn monitored by Fluo-4 loaded inside cells. Cells overexpressing Fpn show a significantly faster Ca2+ uptake compared to control cells transfected with an empty vector. (d) Comparison of initial rates of Ca2+ uptake in (c). (e) Ca2+ influx by HsFpn in proteoliposomes at different outside (o) pHs. The inside pH is maintained at 7.5. (f) Comparison of initial rates of Ca2+ transport at different outside pHs. (g) Ca2+ influx by HsFpn in proteoliposomes at different membrane potentials. Valinomycin was used to generate a membrane potential prior to the addition of Ca2+. (h) Comparison of initial rates of Ca2+ transport at different membrane potentials. In all the transport assays, 500 µM of Ca2+ was added at time zero. Statistical significances were analyzed with one-way analysis of variance (ANOVA) followed by Dunnett’s test for multiple comparisons. In this article, all time-dependent fluorescence traces are shown as solid lines (mean) with shaded regions (standard deviation, SD) from at least three biological repeats. For all bar graphs, a scatter plot is overlaid on each bar. The height represents the mean of at least three measurements, and the error bar standard error of the mean (SEM). Statistical significances are indicated as follows: ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p<0.0001.
Figure 2 with 3 supplements see all
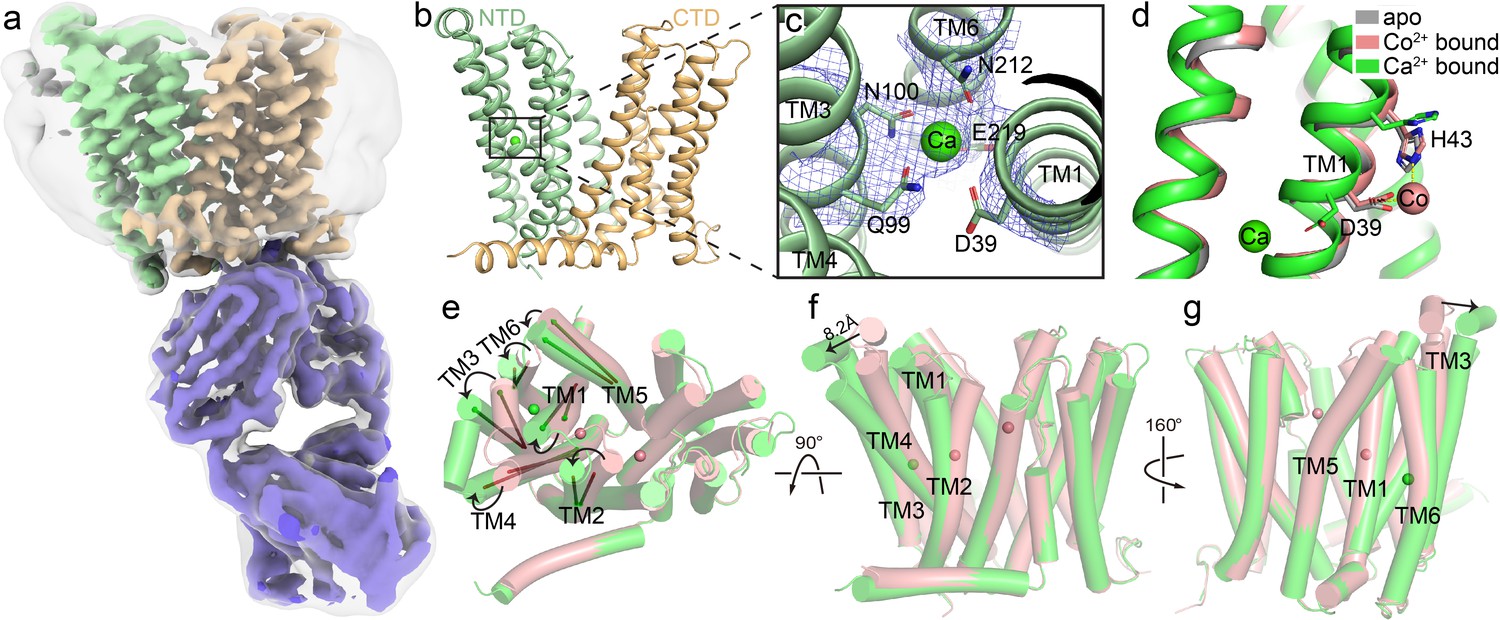
Structure of HsFpn bound to Ca2+.
(a) Cryo-EM map of HsFpn in complex with 11F9 in the presence of Ca2+. Densities for NTD, CTD, and 11F9 are colored in pale green, light orange, and slate purple, respectively. A smoothened map contoured at a low threshold (translucent grey) is overlaid to show the lipid nanodisc density around Fpn. (b) Structure of HsFpn with a bound Ca2+ in an outward-facing conformation. NTD and CTD are colored as described in (a). Ca2+ is shown as a green sphere. (c) Zoomed-in view of the Ca2+ binding site in the NTD. The five residues coordinating Ca2+ are labeled and shown as side chain sticks. The density for Ca2+ is contoured at 7.5σ as blue mesh. (d) Structural comparison of apo (grey, PDB ID 6W4S), Co2+-bound (pink, PDB ID 8DL8), and Ca2+-bound (green) HsFpn near S1. The side chains of D39 and H43 are shown as sticks. Co2+ is shown as a pink sphere. (e), (f), and (g) Three views of conformational changes in NTD induced by Ca2+ binding. The Co2+-bound (pink) and Ca2+-bound (green) structures are aligned and shown as cartoon with cylindrical helices. (e) Top view (from the extracellular side) of the alignment. The helical directions of TM1-6 are visualized by vectors inside cylinders, and the bending of these helices is indicated by black arrows. Bending of TMs viewed from the front (f) and the back (g). The displacement of the extracellular loop between TM3 and TM4 is marked with a black arrow and distance.
To understand the mechanism of Ca2+ transport by HsFpn, we first examined whether common ions, H+, Na+, K+, and Cl-, are involved or required for Ca2+ transport. We found that Ca2+ transport by HsFpn is not affected by a H+ gradient (Figure 1e–f and Figure 1—figure supplement 2), and that the absence of Na+, K+, or Cl- has no significant effect on Ca2+ transport (Figure 1—figure supplement 3). These results suggest that Fpn is a Ca2+ uniporter. If this is true, then Ca2+ transport should be electrogenic and sensitive to membrane potentials. We tested this in the following experiments. We measured Ca2+ transport at defined membrane potentials,+120 mV, 0 mV, and –120 mV, established by a K+ gradient in the presence of valinomycin, a K+ selective ionophore (Figure 1g). As shown in Figure 1g–h, Ca2+ transport is significantly enhanced at –120 mV and reduced at +120 mV, and Ca2+ transport at 0 mV is larger than that in the absence of a clamped membrane potential. These results are fully consistent with HsFpn being a Ca2+ uniporter.
We then measured Ca2+ transport at different concentrations of Ca2+ ([Ca2+]), first in the absence of a preset membrane potential and then in the presence of –120 mV membrane potential (Figure 1—figure supplement 4a). We first monitored Ca2+ influx in the presence of a 100-fold K+ concentration gradient but in the absence of valinomycin. Under this condition, import of Ca2+ would build up a positive membrane potential that slows down further Ca2+ influx. After the influx reached a steady state (~460 s), we added valinomycin to clamp the membrane potential to –120 mV and we observed a large increase of Ca2+ influx. The rate of Ca2+ uptake in the second phase is faster than that in the first phase (Figure 1—figure supplement 4a). These results are consistent with HsFpn being a Ca2+ uniporter. The Michaelis-Menten constant, KM, for Ca2+ transport, calculated based on the initial rate of fluorescence increase in the second phase, is 48.5 (27.3–88.8) µM (95% confidence interval in parentheses) (Figure 1—figure supplement 4b). We also measured Ca2+ uptake in HEK cells expressing HsFpn and obtained a KM of 85.9 (36.6–193.0) µM (Figure 1—figure supplement 4c–d).
Structure of HsFpn bound to Ca2+
We determined the structure of nanodisc-enclosed HsFpn-11F9 complex in the presence of 2 mM Ca2+ by cryo-electron microscopy (cryo-EM) to an overall resolution of 3.0 Å (Figure 2a and Figure 2—figure supplement 1, Table 1). The density map reaches a resolution of ~2.4 Å in the transmembrane (TM) regions and allows for the building and refinement of a structural model that contains residues 15–240, 289–400, and 452–555, which covers all 12 TM helices (Figure 2—figure supplement 2). Residues 1–14, 241–288, 401–451, and 556–571, which are predicted to be disordered regions located to either the N- or C-terminus or loops between TM helices, are not resolved. The 12 TMs form two well-defined domains, with the N-terminal domain (NTD) composed of TM1–6 and the C-terminal domain (CTD) TM7–12 (Figure 2b). The current structure assumes an outward-facing conformation in which the NTD and CTD make contact on the intracellular side. The overall conformation of the Ca2+-bound HsFpn is similar to that of the Co2+-bound HsFpn (Wilbon et al., 2023; Figure 2e–g and Figure 2—figure supplement 3b).
Table 1
Summary of cryo-EM data collection, processing, and refinement.
| Sample | HsFpn-Ca2+–11 F9 |
|---|---|
| Cryo-EM data collection | |
| Voltage (kV) | 300 |
| Magnification (×) | 81,000 |
| Pixel Size (Å) | 1.10 |
| Electron exposure (e-/Å2/frame) | 1.25 |
| Defocus range (µm) | [-2.5,–0.8] |
| Number of image stacks | 4,498 |
| Number of frames per stack | 40 |
| Cryo-EM data processing | |
| Initial number of particles | 2,184,301 |
| Final number of particles | 437,959 |
| Symmetry imposed | C1 |
| Map resolution (Å) | 3.0 |
| Map resolution range (Å) | 2.4–3.6 |
| FSC threshold | 0.143 |
| Model refinement | |
| Number of amino acids | 875 |
| Total non-hydrogen atoms | 6,192 |
| Average B factor (Å2) | 171.4 |
| Bond length RMSD (Å) | 0.008 |
| Bond angle RMSD (°) | 0.939 |
| Ramachandran Plot | |
| Favored (%) | 93.53 |
| Allowed (%) | 6.24 |
| Outliers (%) | 0.23 |
| Rotamer outliers (%) | 1.25 |
| MolProbity Score | 1.98 |
In the density map (Figure 2a), we noticed a non-protein density corralled by four helices (TM1, TM3, TM4, and TM6) in the NTD, and assigned it as a Ca2+ based on the following observations. This density was not present in previous structures of Co2+-bound HsFpn or a highly homologous Fpn from Tarsier monkey (Pan et al., 2020; Billesbølle et al., 2020; Wilbon et al., 2023). The side chains of five conserved residues, Asp39, Gln99, Asn100, Asn212, and Glu219, are within 4 Å of the presumed Ca2+ density (Figure 2b–c). The side chain positions of Gln99, Asn100, and Asn212 are defined by the density map, while these of Asp39 and Glu219 are deduced based on their backbone and partially resolved side chain densities. While in structures of Co2+-bound HsFpn (Wilbon et al., 2023; Billesbølle et al., 2020), the side chains of Asp39 and His43 point towards the central cavity between the NTD and CTD to form S1, the side chain of Asp39 in the current structure points towards the interior of the four-helical bundle to coordinate Ca2+ (Figure 2d). His43 also assumes a different rotamer conformation in the current structure (Figure 2d). When the NTD and CTD are aligned individually with their counterparts in the Co2+-bound HsFpn (Wilbon et al., 2023), the NTD has a root mean squared distance (RMSD) of 1.32 Å and the CTD 0.51 Å. The larger RMSD in the NTD is due to deviations of the extracellular halves of the TM helices and the extracellular loop between TM3–4 (Figure 2e–g). The bound Ca2+ is not solvent-accessible in the current structure.
Validation of the Ca2+ binding site
We validated the Ca2+ binding site by mutational studies. Both a single alanine mutation (Asp39Ala) and a triple alanine mutation (Gln99_Asn100_Glu219 to Ala) to the Ca2+ binding site show significantly reduced Ca2+ transport activity in the proteoliposome assay (Figure 3a–b). In contrast, the double alanine mutation to S2 (Cys326Ala_His507Ala) does not affect Ca2+ transport significantly (Figure 3a–b). We also measured Ca2+ uptake by HsFpn mutants expressed in HEK cells (Figure 3—figure supplement 1) and found that single alanine mutations to residues at the Ca2+ binding site significantly impair Ca2+ transport activity, while mutation to His43 (at S1), which is one helical turn away from Asp39 at Ca2+ binding site, maintains high Ca2+ transport activity (Figure 3c–d).
Figure 3 with 3 supplements see all
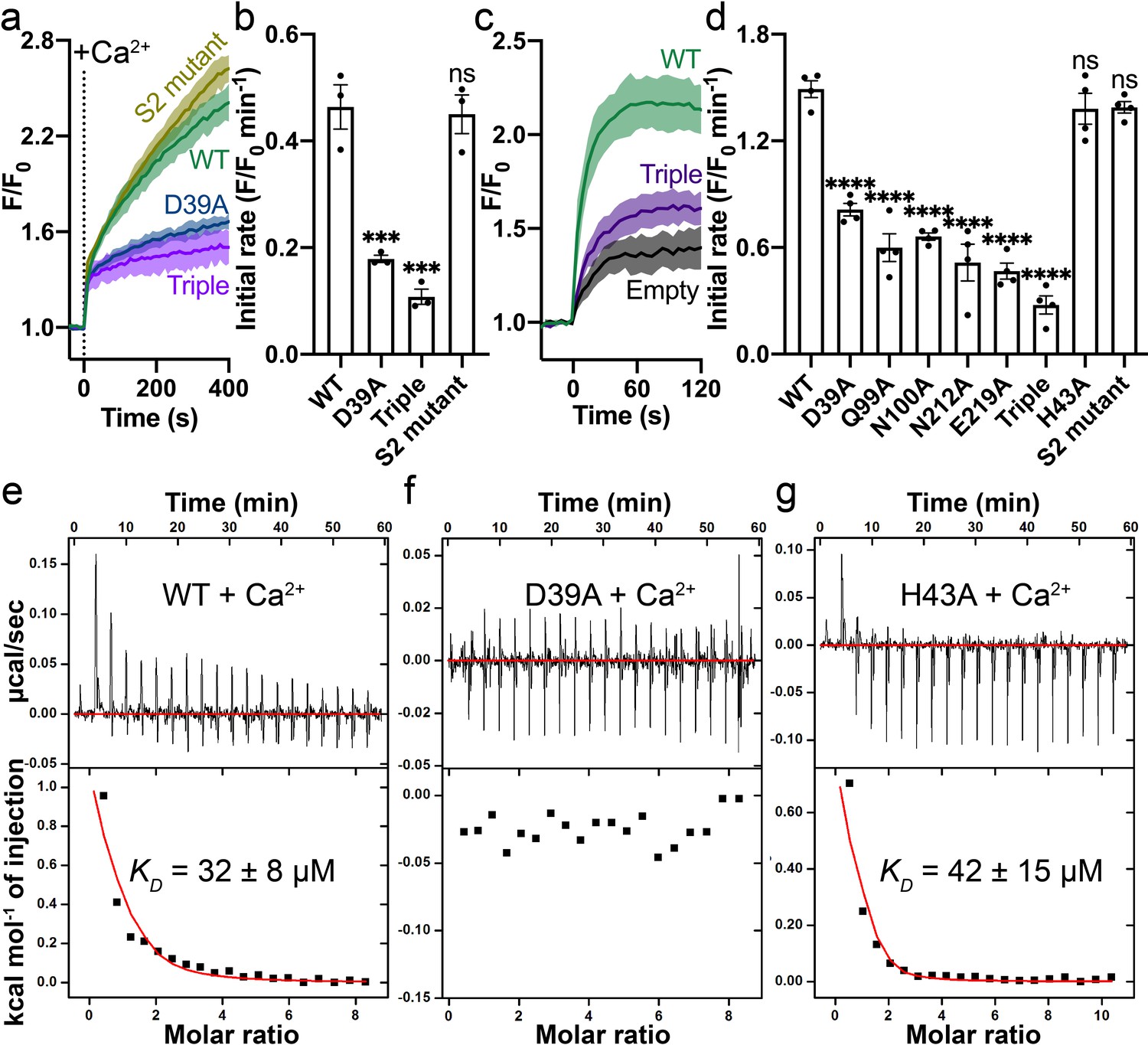
Mutations on the Ca2+ binding site.
(a) Ca2+ influx by WT (green), D39A (blue), and the triple mutant (Q99A_N100A_E219A, purple) of the Ca2+ binding site in proteoliposomes. (b) Comparison of initial rates of Ca2+ influx in (a). (c) Ca2+ uptake in HEK cells expressing WT (green) or the triple mutant (purple) Fpn. (d) Fpn-specific Ca2+ transport activities of WT and mutants. Initial rates are subtracted from the empty control. For statistical significances in (b) and (d), Dunnett’s test was used as a post hoc test following one-way ANOVA with the WT as the control. 500 µM Ca2+ was used in (a–d). Binding of Ca2+ to WT (e), D39A (f), and H43A (g) HsFpn measured by ITC. Upper plot: raw thermogram showing the heat during binding and baseline (red line). Lower plot: integrated heat of each injection (black square) and the fit of data (red line).
We estimated Ca2+ binding affinity by isothermal titration calorimetry (ITC). We found that Ca2+ binding is an endothermic process with a dissociation constant (KD) of ~32 µM (Figure 3e). Similar endothermic binding of Ca2+ was also reported in mouse Fpn and a bacterial homolog of Fpn (Deshpande et al., 2018). The 11F9 Fab does not interfere with or enhance Ca2+ binding as the HsFpn-11F9 complex has a similar Ca2+ binding affinity (KD = 20 ± 6 µM; ) (Figure 3—figure supplement 2a). We then measured Ca2+ binding to HsFpn mutants, and found that single point mutations to residues at the Ca2+ binding site abolish Ca2+ binding (Figure 3f and Figure 3—figure supplement 2e–h). As a control, we measured Ca2+ binding to the His43Ala mutant and to the S2 mutant, and found that both mutants retain Ca2+ binding with affinities similar to that of the wild-type (WT; Figure 3g and Figure 3—figure supplement 2c). In addition, a Ca2+ binding site mutant, Gln99Ala, does not significantly change Co2+ binding affinity (KD = 226 ± 23 µM; Figure 3—figure supplement 2d). Asn212Ala does not significantly change Zn2+ binding to HsFpn (KD = 224 ± 30 µM) either (Figure 3—figure supplement 2i–j). Combined, these results provide further validation to the observed Ca2+ binding site in HsFpn.
Competition between Ca2+ and Co2+ in HsFpn
Next, we measured binding of Ca2+ in the presence of Co2+, and vice versa. In the presence of 2 mM Co2+, Ca2+ binding affinity is reduced by ~fourfold as shown in Figure 4a. The reduced binding is apparent both from the reduced heat absorption during the titration, and the reduced KD from fitting of the ITC data. We interpret the reduced Ca2+ binding in the presence of Co2+ as the loss of Asp39 to the coordination of Co2+ at S1 (Figure 2d). On the other hand, Co2+ binding affinity (KD) is not affected in the presence of 2 mM Ca2+, as shown in Figure 4b, although the amount of heat release is appreciably less in the presence of Ca2+. This result is similar to the effect of the S1 mutation on Co2+ binding (Asp39_His43 to Ala, KD = 266 ± 24 µM) (Pan et al., 2020). We interpret this result as the preservation of Co2+ binding at S2, which is not affected by Ca2+ binding.
Figure 4 with 3 supplements see all
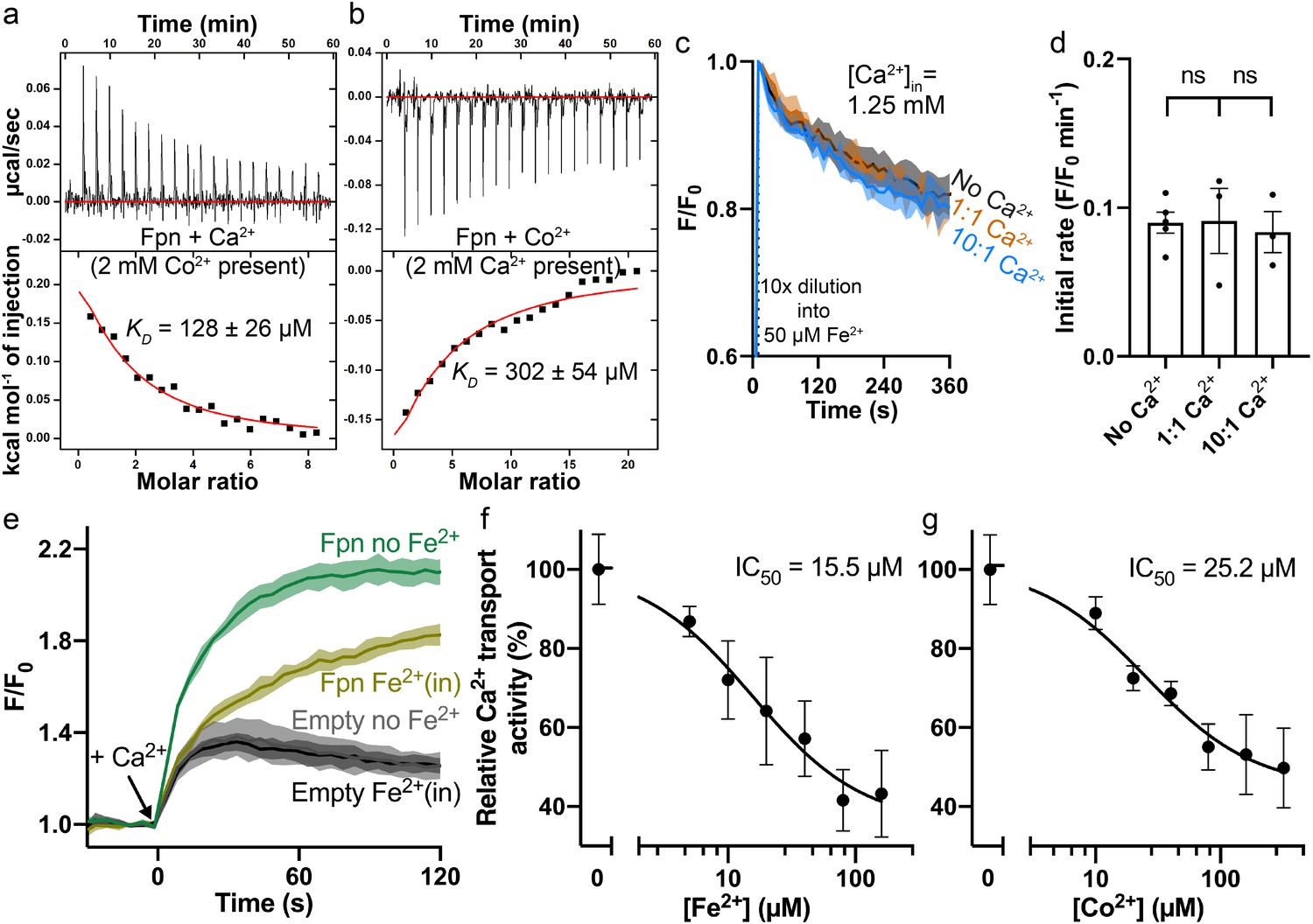
Interplay between Fe2+/Co2+ and Ca2+ in HsFpn.
(a) Ca2+ binding in the presence of 2 mM Co2+. (b) Co2+ binding in the presence of 2 mM Ca2+. (c) Fe2+ transport into proteoliposomes in the presence or absence of Ca2+. The external [Fe2+] is 50 µM. ‘1:1’ indicates symmetrical [Ca2+] at 1.25 mM and “10:1” indicates 1.25 mM Ca2+ inside and 0.125 mM Ca2+ outside. 1 mM sodium ascorbate was included in all samples. All fluorescence traces are subtracted from corresponding blank controls using vesicles with no HsFpn. (d) Comparison of initial rates of Fe2+ transport in (c). One-way ANOVA was used for statistical analysis. (e) Ca2+ (1 mM) uptake by HEK cells expressing HsFpn in the presence (dark yellow) or absence (green) of 80 µM Fe2+ that has been pre-loaded into the cells. Uptake is monitored by jGCaMP7s which is co-expressed in cytosol. Cells transfected with an empty vector (dark and light gray) serve as negative controls. Ca2+ transport at different concentrations of Fe2+ (f) or Co2+ (g). Normalized initial rates of Fpn-specific Ca2+ uptake were used to represent relative Ca2+ transport activities. Data were fitted (black solid line) to an inhibitory dose-response equation to calculate IC50 values.
We then measured Ca2+ transport in the presence of Fe2+ or Co2+, and Fe2+ or Co2+ transport in the presence of Ca2+. We found that Fe2+ or Co2+ transport is not affected significantly in the presence of either 1.25 mM or 0.5 mM Ca2+ (Figure 4c–d and Figure 4—figure supplement 1a–f), but Ca2+ transport is significantly reduced in the presence of Fe2+ or Co2+ (Figure 4e–g and Figure 4—figure supplement 2c–e). These results are consistent with the structures and our understanding of Fe2+ and Ca2+ transport in Fpn. Fe2+ or Co2+ transport is mediated by both S1 and S2, and binding of Ca2+, which renders Asp39 unavailable, does not significantly affect Fe2+ or Co2+ transport because part of S1, His43, and the entire of S2 remain available to Fe2+ or Co2+ transport. On the other hand, Ca2+ transport is mediated by a single site and in the presence of Fe2+ or Co2+, Ca2+ binding site is impaired due to the loss of Asp39 and hence significantly reduced Ca2+ binding and transport (Figure 4—figure supplement 3a–c). Indeed, Asp39Ala single mutation significantly reduces Ca2+ transport (Figure 3a) but has a modest effect on Co2+ transport (Figure 3—figure supplement 3a–b). Further studies are needed to dissect the intertwined transport pathways of Fe2+ and Ca2+ transport in Fpn.
Discussion
In summary, we show that HsFpn has a well-defined single Ca2+ binding site, and that HsFpn transports Ca2+. We also show that Ca2+ transport by HsFpn is not coupled to another ion and is diminished in the presence of Fe2+, however, Fe2+ transport is not sensitive to the presence of Ca2+.
Our report of Ca2+ transport by HsFpn contradicts the conclusion from a previous study (Deshpande et al., 2018); however, the two studies also have common grounds. Deshpande et al. identified a Ca2+ binding site in the structure of BbFpn (Bdellovibrio bacteriovorus), a bacterial homolog of human Fpn (Figure 2—figure supplement 3a), and demonstrated Ca2+ binding to mouse Fpn, which is in agreement with our demonstration of a Ca2+ binding site in HsFpn. Deshpande et al. conclude that Ca2+ may serve as a bound “cofactor” required for Fe2+ transport (Deshpande et al., 2018), and our results imply that Fpn could transport Fe2+ while bound to a Ca2+. With the structure of Ca2+-bound HsFpn, we noticed that residues forming the Ca2+ binding site are conserved between BbFpn and HsFpn, except for Asn100 in HsFpn, which is Phe85 in BbFpn. The persistence of a Ca2+ binding site from bacteria to mammals suggests a conserved functional role of Ca2+, and future studies aimed at understanding the biological relevance of Ca2+ transport through Fpn will lead to understanding of regulations in iron homeostasis.
Ca2+ binding and transport in HsFpn imposes constraint to the conceptualization of the Fe2+/2H+ antiport mechanism in the context of S1 and S2 of Fe2+ binding sites. In the absence of a bound Ca2+, export of one Fe2+ could be mediated through sequential occupation of S1 and S2 and structural changes that expose the binding sites to the extracellular side; and import of two H+ could be mediated by protonation of His43 at S1 and His507 at S2 followed by a structural change that exposes the two residues to the intracellular side. When Ca2+ is bound, Asp39 is not available to mediate Fe2+ export so that transport would likely be mediated by S2 only or by coalescing of His43 and S2 around a single Fe2+. Since we do not observe significant changes in Fe2+ transport in the presence and absence of Ca2+, it is likely that S1, or residue Asp39, has a more modest effect on Fe2+ transport, while S2 has a more prominent role. Results from our initial mutational studies on S1 and S2 are consistent with this hypothesis. Further studies are required to establish a mechanism of transport.
Our discovery of Ca2+ transport in Fpn demonstrates a novel Ca2+ entry pathway in cells expressing Fpn, and our study establishes Fpn as a transporter capable of operating with two different transport mechanisms. Although we are not able to estimate the amount of Ca2+ uptake through HsFpn under physiological conditions to provide an interpretation for its cellular function, we speculate that Ca2+ entry through HsFpn is an important component contributing to iron homeostasis.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Chemical compound, druC | n-Dodecyl-β-D-Maltopyranoside | Anatrace | Cat#D310 | |
| Chemical compound, drug | Lauryl maltose neopentyl glycol | Anatrace | Cat#NG310 | |
| Chemical compound, drug | 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | Cat#850457 C | |
| Chemical compound, drug | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) | Avanti Polar Lipids | Cat#850457 C | |
| Chemical compound, drug | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (POPG) | Avanti Polar Lipids | Cat#840457 C | |
| Chemical compound, drug | Calcein | Invitrogen | Cat#C481 | |
| Chemical compound, drug | Fluo-4, Pentapotassium Salt, cell impermeant | Invitrogen | Cat#F14200 | |
| Chemical compound, drug | Valinomycin | Sigma | Cat#V0627 | |
| Peptide, recombinant protein | Hepcidin-25 (human) | Sigma | Cat#SML1118 | |
| Commercial assay, kit | TALON Metal Affinity Resin | TaKaRa | Cat#635504 | |
| Commercial assay, kit | SRT-10C SEC-300 | Sepax Technologies | Cat#239300–10030 | |
| Commercial assay, kit | Q Sepharose Fast Flow | GE Healthcare | Cat#17051010 | |
| Commercial assay, kit | Biobeads SM2 | Bio-Rad | Cat#1528920 | |
| Commercial assay, kit | PD-10 Desalting Column | GE Healthcare | Cat#17085101 | |
| Commercial assay, kit | 400 nm NanoSizer Extruder | T&T Scientific Corporation | Cat#TT-004–0010 | |
| Commercial assay, kit | KOD Hot Start DNA polymerase | Novagen | Cat#71086–3 | |
| Commercial assay, kit | Fluo-4, AM, cell permeant | Invitrogen | Cat#F14201 | |
| Commercial assay, kit | pHrodo Red AM | Invitrogen | Cat#P35372 | |
| Recombinant DNA reagent | pGP-CMV-jGCaMP7s | Addgene | Plasmid #104463 | RRID:Addgene_104463 |
| Software, algorithm | MotionCorr2 | Zheng et al., 2017 | https://msg.ucsf.edu/em/software/motioncor2.html | |
| Software, algorithm | Gctf | Zhang, 2016 | https://www.mrc-lmb.cam.ac.uk/kzhang/Gctf/ | RRID:SCR_016500 |
| Software, algorithm | Relion 3.0 | Kimanius et al., 2016 | https://www3.mrc-lmb.cam.ac.uk/relion | RRID:SCR_016274 |
| Software, algorithm | CryoSPARC | Punjani et al., 2017 | https://cryosparc.com/ | RRID:SCR_016501 |
| Software, algorithm | ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ | RRID:SCR_015872 |
| Software, algorithm | COOT | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | RRID:SCR_014222 |
| Software, algorithm | PHENIX | Adams et al., 2010 | http://www.phenix-online.org/ | RRID:SCR_014224 |
| Software, algorithm | EMringer | Barad et al., 2015 | http://fraserlab.com/2015/02/18/EMringer/ | |
| Other | Grids: R1.2/1.3 Cu 300 mesh | Quantifoil | Cat#Q325CR1.3 | Cryo-EM grid |
Cloning, expression, and purification of human Fpn (HsFpn)
Request a detailed protocolCodon-optimized cDNA of HsFpn (UniProt ID: Q9NP59) was cloned into a pFastBac dual vector. A Tobacco Etch Virus (TEV) protease site and an octa-histidine (8×His) tag was appended to the C-terminus of the protein. HsFpn was expressed in Sf9 (Spodoptera frugiperda) cells using the Bac-to-Bac method (Invitrogen). Purification of HsFpn follows the same protocol reported for Tarsius syrichta Fpn (TsFpn) (Pan et al., 2020). Purified HsFpn was collected from size-exclusion chromatography (SEC) in the FPLC buffer containing 20 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES, pH7.5), 150 mM NaCl, and 1 mM (w/v) n-dodecyl-β-D-maltoside (DDM, Anatrace). Mutations to HsFpn were generated using the QuikChange method (Stratagene) and verified by sequencing. Mutants were expressed and purified following the same protocol for the WT.
Preparation of HsFpn-11F9 complex in nanodisc
Request a detailed protocolMembrane scaffold protein (MSP) 1D1 was expressed and purified following an established protocol (Martens et al., 2016). For lipid preparation, 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC, Avanti Polar Lipids), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE, Avanti Polar Lipids) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (POPG, Avanti Polar Lipids) were mixed at a molar ratio of 3:1:1, dried under Argon and resuspended with 14 mM DDM (Autzen et al., 2018). For nanodisc reconstitution, HsFpn, MSP1D1 and lipid mixture were mixed at a molar ratio of 1:2.5:50 and incubated on ice for 1 hr. Detergents were removed by the sequential addition of 60 mg/mL Biobeads SM2 (Bio-Rad) for three times over a 3-hr period. The sample was then incubated with Biobeads overnight at 4 °C. After removal of Biobeads, 11F9 was added to the sample at a molar ratio of 1.1:1 to HsFpn. The complex was incubated on ice for 30 min before being loaded onto a SEC column equilibrated with the FPLC buffer without detergent. The purified nanodisc sample was concentrated to 10 mg/ml and incubated with 2 mM CaCl2 for 30 min on ice before cryo-EM grid preparation.
Cryo-EM sample preparation and data collection
Request a detailed protocolThe cryo-EM grids were prepared using Thermo Fisher Vitrobot Mark IV. The Quantifoil R1.2/1.3 Cu grids were glow-discharged with air for 15 s at 10 mA using Plasma Cleaner (PELCO EasiGlow). Aliquots of 3.5 µl nanodisc sample were applied to the glow-discharged grids. After blotted with filter paper (Ted Pella, Inc) for 4.0 s, the grids were plunged into liquid ethane cooled with liquid nitrogen. A total of 4498 micrograph stacks were collected on a Titan Krios at 300 kV equipped with a K3 direct electron detector (Gatan) and a Quantum energy filter (Gatan) at a nominal magnification of 81,000×and defocus values from –2.5 µm to –0.8 µm. Each stack was exposed in the super-resolution mode with an exposing time of 0.0875 s per frame for a total of 40 frames per stack. The total dose was about 50 e-/Å2 for each stack. The stacks were motion corrected with MotionCor2 (Zheng et al., 2017) and binned 2-fold, resulting in a pixel size of 1.10 Å/pixel. In the meantime, dose weighting was performed (Grant and Grigorieff, 2015). The defocus values were estimated with Gctf (Zhang, 2016).
Cryo-EM data processing
Request a detailed protocolA total of 2,184,301 particles were automatically picked based on a reference map of TsFpn-11F9 (EMD-21460) low-pass filtered to 20 Å in RELION 3.1 (Scheres, 2015, Scheres, 2012; Kimanius et al., 2016; Zivanov et al., 2018). Particles were extracted and imported to CryoSparc (Punjani et al., 2017) for 2D classification. A total of 1,329,782 particles were selected from good classes in 2D classification, which display recognizable structural features. Four 3D references were generated by ab initio reconstruction with limited particles from the best 2D classes. Multiple rounds of heterogeneous refinement were performed with particles selected from the 2D classification and four initial reference models until no more than 5% of input particles were classified into bad classes. A final of 437,959 particles after heterogeneous refinement were subjected to non-uniform (NU) refinement with an adaptive solvent mask. After handedness correction, local refinement and CTF refinement were performed with a soft mask around the Fpn and the Fv region of the Fab. Resolutions were estimated with the gold-standard Fourier shell correlation 0.143 criterion (Rosenthal and Henderson, 2003). Local resolution of the maps was estimated in CryoSparc (Punjani et al., 2017).
Model building and refinement
Request a detailed protocolThe structure of apo HsFpn (from PDB ID 6W4S) and 11F9 Fab (from PDB ID 6VYH) were individually docked into density maps in Chimera (Pettersen et al., 2004). The docked model was manually adjusted with added ligands in COOT (Emsley et al., 2010). Structure refinements were carried out by PHENIX in real space with secondary structure and geometry restraints (Adams et al., 2010). The EMRinger Score was calculated as described (Barad et al., 2015). All structure figures were prepared in Pymol and ChimeraX (Pettersen et al., 2021).
Isothermal titration calorimetry
Request a detailed protocolThe WT and mutant HsFpn proteins were purified as described above and concentrated to 50–75 μM (3–4.5 mg/mL) in the FPLC buffer. The buffer was degassed, and all the protein samples were centrifuged at 18,000×g for 20 min to remove aggregates. The injectant of 2 mM CaCl2 or 5 mM CoCl2 was prepared in the same FPLC buffer. For competition binding, either 2 mM CoCl2 or CaCl2 was added to protein samples prior to ligand titration. The ITC measurements were performed in Auto-iTC200 (MicroCal) at 25 °C. A total of 25 injections were administered (1.01 μL for injections 1 and 2.02 μL for injections 2–25) with a 150 s interval between injections. Background-subtracted data were fitted with binding models in the Origin 8 software package (MicroCal) to extract KD, ΔH, and entropy change (ΔS).
Expression of HsFpn in HEK cells
Request a detailed protocolThe cDNAs of WT and mutant HsFpn were subcloned into a modified pEG BacMam vector with a C-terminal Strep-tag. The resulting plasmids with Fpn or the empty plasmid were transfected into HEK 293 F cells on six-well plates with 293fectin transfection reagent (Invitrogen/Thermo Fisher) per the manufacturer’s protocol. After incubation at 37 °C with 8% CO2 for 2 days, cells were harvested and solubilized in the lysis buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol) plus 1% LMNG and Protease Inhibitor Cocktail (Roche) for 1 hr at 4 °C. Insoluble fractions were pelleted by centrifugation and supernatants were run in SDS-PAGE. Proteins were visualized by western blotting with mouse anti-Strep (Invitrogen/Thermo Fisher) and IRDye-800CW anti-mouse IgG (Licor). Images were taken on an Odyssey infrared scanner (Licor).
Ca2+ uptake and H+ transport assays in HEK cells
Request a detailed protocolThe pEG BacMam plasmids with HsFpn or the empty plasmid were transfected into HEK 293 F cells on black wall 96-well microplates coated with poly-D-lysine (Invitrogen/Thermo Fisher). After 2 days, cells were washed in the live cell imaging solution (LCIS) containing 20 mM HEPES (pH 7.4), 140 mM NaCl, 2.5 mM KCl, 1.0 mM MgCl2, and 5 mM D-glucose. The loading of Fluo-4 (Invitrogen/Thermo Fisher, AM, cell-permeant) for Ca2+ uptake assays or pHrodo Red (Invitrogen/Thermo Fisher, AM) for H+ transport assays was performed following manufacturer’s protocols. After the loading finished, free dyes were washed away, and cells in each well were maintained in 90 µL LCIS. Both the Ca2+ uptake and H+ transport assays were performed in the FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices) at 37 °C. Fluorescence changes were recorded at an excitation and emission wavelength of 485 nm and 538 nm for Ca2+ uptake assays, and 544 nm and 590 nm for H+ transport assays with 5 s intervals. Transport was triggered by the addition of 10 µL ligand stock solution (CaCl2 or CoCl2) to achieve the desired concentration of extracellular Ca2+ or Co2+. The H+ transport was assayed with 500 µM Co2+ or Ca2+. To test the effect of extracellular pH on Ca2+ uptake, the extracellular buffer was changed to pre-warmed LCIS with adjusted pH soon before the addition of 500 µM Ca2+. All the mutants were assayed with 500 µM Ca2+. For Ca2+ uptake assays, the initial rate is defined by slope of a linear fit to the first 25 s of data points. For H+ transport assays, relative fluorescence changes at the equilibrium stage were averaged to represent intracellular pH changes.
Ca2+ uptake in the presence of Fe2+ or Co2+ in HEK cells
Request a detailed protocolThe WT and mutant HsFpn were expressed in HEK 293 F cells as described above except that the plasmid for mammalian expression of jGCaMP7s (pGP-CMV-jGCaMP7s) was co-transfected. The pGP-CMV-jGCaMP7s was a gift from Douglas Kim & GENIE Project (Addgene plasmid # 104463; http://n2t.net/addgene:104463; RRID:Addgene_104463) (Dana et al., 2019). GCaMP is a green fluorescence protein (GFP)-based Ca2+ indicator that contains a fused calmodulin (CaM) domain. Ca2+ binding to CaM triggers conformational changes that results in increased GFP fluorescence. Ca2+ uptake in the presence of Fe2+ or Co2+ were performed in FlexStation 3 as described above except that the excitation and emission wavelength were set at 485 nm and 513 nm. Fe2+ (NH4Fe(SO4)2) or Co2+(CoCl2) ions were loaded into cells by incubation for ~10 min at 37 °C, during which time fluorescence readings were recorded. For Fe2+ loading, 1 mM sodium ascorbate was used to protect Fe2+ ions from oxidation. To start the export of Fe2+ or Co2+, the extracellular buffer was exchanged to pre-warmed LCIS ~30 s before the addition of Ca2+. LCIS with adjusted pH was used when testing the effect of extracellular pH. The initial rate is defined as the slope of a linear fit to the first 25 s of transport data.
Reconstitution of HsFpn into liposomes
Request a detailed protocolPOPE and POPG lipid (Avanti Polar Lipids) were mixed at a 3:1 molar ratio, dried under Argon, and vacuumed overnight to remove chloroform. The dried lipid was resuspended in the reconstitution buffer (20 mM HEPES, pH 7.5, 100 mM KCl) to a final concentration of 10 mg/mL. After hydration for 2 hr, the liposome sample was sonicated to transparency and incubated with 40 mM n-decyl-β-D-maltoside (DM, Anatrace) for 2 hr at room temperature under gentle agitation. Then HsFpn protein was added at a 1:100 (w/w, protein:lipid) ratio. For the empty control, the same volume of blank buffer was added. Detergent was removed by dialysis at 4 °C against the reconstitution buffer. Dialysis buffer was changed every day for 4 days. The proteoliposome or empty liposome sample was aliquoted and frozen with liquid nitrogen, and was stored at –80 °C for future use.
Ca2+ and Fe2+/Co2+ influx in proteoliposomes
Request a detailed protocolProteoliposomes with HsFpn or empty liposomes were thawed and mixed with 100 μM Fluo-4 (Invitrogen/Thermo Fisher, cell impermeant) for Ca2+ influx assays, or with 250 μM calcein (Invitrogen/Thermo Fisher) for Fe2+/Co2+ influx assays. The dye was incorporated during three cycles of freeze-thaw. Liposomes were extruded to homogeneity with a 400 nm filter (NanoSizer Extruder, T&T Scientific Corporation). Removal of free dyes outside liposomes and exchange of outside buffer was achieved by passing samples through a desalting column (PD-10, GE Healthcare) equilibrated with buffer required for a desired transport condition. Liposome samples were transferred to a quartz cuvette for fluorescence recording in a FluoroMax-4 spectrofluorometer (HORIBA). Fluorescence changes were recorded at an excitation and emission wavelength of 494 nm and 513 nm with 10 s intervals at 37 °C.
To monitor Ca2+ influx, transport was initiated by the addition of CaCl2 to the desired concentration. When testing the inhibition by hepcidin or 11F9 Fab (i)/(o), 20 µM human hepcidin (Sigma) or purified 11F9 Fab (Pan et al., 2020) was added prior to freeze-thaw cycles, and the transport was assayed with 500 µM CaCl2. For the Fab (o) condition, 20 µM Fab was added to samples collected from desalting column. When testing the effect of different pHs, an outside buffer has the same components as the inside buffer but was adjusted to a desired pH. When testing the effect of different Na+, K+, or Cl-, the inside and outside of liposomes have symmetrical buffers with 100 mM NaCl, KCl, or K-Gluconate. For measurements with –120 mV membrane potential shown in Figure 1—figure supplement 4a, the outside buffer has 20 mM HEPES (pH 7.5), 1 mM KCl, and 99 mM NaCl while the inside buffer has 20 mM HEPES (pH 7.5) and 100 mM KCl. 40 nM valinomycin was added to clamp membrane potential at ~–120 mV. When testing the effects of different membrane potentials, valinomycin was incubated with liposome samples for 5 min prior to the addition of CaCl2. For the –120 mV group, the outside buffer contained 20 mM HEPES (pH 7.5), 1 mM KCl, and 99 mM NaCl while the inside buffer has 20 mM HEPES (pH 7.5) and 100 mM KCl. For the 0 mV group, the outside buffer was the same as the inside buffer with 20 mM HEPES (pH 7.5) and 100 mM KCl. For the +120 mV group, the inside buffer has 20 mM HEPES (pH 7.5), 1 mM KCl, and 99 mM NaCl while the outside buffer contained 20 mM HEPES (pH 7.5) and 100 mM KCl.
For Fe2+/Co2+ influx assays, symmetrical buffer (20 mM HEPES, pH 7.5, 100 mM KCl) was used and transport was initiated by dilution (10×) of 30 µL of liposome sample into 270 µL of outside buffer containing 50 µM of NH4Fe(SO4)2 or CoCl2. In the case of Fe2+ influx, 1 mM sodium ascorbate was added. To load liposomes with Ca2+, 1.25 mM or 0.5 mM CaCl2 was added prior to freeze-thaw cycles. Buffer with 1.25 mM or 0.5 mM CaCl2 was used during the desalting step. To create a 10-fold Ca2+ gradient opposite to the Fe2+ or Co2+gradient, samples were diluted into the outside buffer without Ca2+.
Data availability
The cryo-EM density map of nanodisc-encircled HsFpn-11F9 in the presence Ca2+ has been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/) under accession code EMD-27497. The corresponding atomic coordinate file has been deposited in the Protein Data Bank (http://www.rcsb.org) under ID code 8DL6. Uncropped gel and blot images are available as source files.
-
RCSB Protein Data BankID 8DL6. Cryo-EM structure of human ferroportin/slc40 bound to Ca2+ in nanodisc.
-
ArrayExpressID EMD-27497. Cryo-EM structure of human ferroportin/slc40 bound to Ca2+ in nanodisc.
References
-
A novel mammalian iron-regulated protein involved in intracellular iron metabolismThe Journal of Biological Chemistry 275:19906–19912.https://doi.org/10.1074/jbc.M000713200
-
PHENIX: a comprehensive python-based system for macromolecular structure solutionActa Crystallographica. Section D, Biological Crystallography 66:213–221.https://doi.org/10.1107/S0907444909052925
-
Features and development of cootActa Crystallographica. Section D, Biological Crystallography 66:486–501.https://doi.org/10.1107/S0907444910007493
-
Systemic iron homeostasisPhysiological Reviews 93:1721–1741.https://doi.org/10.1152/physrev.00008.2013
-
Hepcidin-ferroportin axis in health and diseaseVitamins and Hormones 110:17–45.https://doi.org/10.1016/bs.vh.2019.01.002
-
Iron imports. II. iron uptake at the apical membrane in the intestineAmerican Journal of Physiology. Gastrointestinal and Liver Physiology 289:G981–G986.https://doi.org/10.1152/ajpgi.00363.2005
-
Lipids modulate the conformational dynamics of a secondary multidrug transporterNature Structural & Molecular Biology 23:744–751.https://doi.org/10.1038/nsmb.3262
-
Hepcidin-ferroportin interaction controls systemic iron homeostasisInternational Journal of Molecular Sciences 22:6493.https://doi.org/10.3390/ijms22126493
-
Structural basis of ion transport and inhibition in ferroportinNature Communications 11:5686.https://doi.org/10.1038/s41467-020-19458-6
-
UCSF chimera--a visualization system for exploratory research and analysisJournal of Computational Chemistry 25:1605–1612.https://doi.org/10.1002/jcc.20084
-
Ferroportin disease: pathogenesis, diagnosis and treatmentHaematologica 102:1972–1984.https://doi.org/10.3324/haematol.2017.170720
-
RELION: implementation of a bayesian approach to cryo-EM structure determinationJournal of Structural Biology 180:519–530.https://doi.org/10.1016/j.jsb.2012.09.006
-
Semi-Automated selection of cryo-EM particles in RELION-1.3Journal of Structural Biology 189:114–122.https://doi.org/10.1016/j.jsb.2014.11.010
-
Gctf: real-time CTF determination and correctionJournal of Structural Biology 193:1–12.https://doi.org/10.1016/j.jsb.2015.11.003
Article and author information
Author details
Funding
National Institutes of Health (HL157473)
- Yaping Pan
National Institutes of Health (DK122784)
- Ming Zhou
National Institutes of Health (HL086392)
- Ming Zhou
National Institutes of Health (GM145416)
- Ming Zhou
Cancer Prevention and Research Institute of Texas (R1223)
- Ming Zhou
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by grants from NIH (HL157473 to YP and DK122784, HL086392, GM145416 to MZ), and Cancer Prevention and Research Institute of Texas (R1223 to MZ). We acknowledge the use of the cryo-EM core at Baylor College of Medicine (BCM) for grid preparation and screening. Cryo-EM data in this work were acquired at the Stanford-SLAC Cryo-EM Center (S2C2) supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24GM129541), and at the Pacific Northwest Center for Cryo-EM (PNCC) at Oregon Health & Science University, supported by the NIH grant U24GM129547. We acknowledge L Wang for help with grid preparation, and Z Ren for making some of mutants. We are grateful to A A R Adeosun for her instructions on the use of FlexStation 3.
Copyright
© 2023, Shen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,616
- views
-
- 281
- downloads
-
- 14
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Mechanism of Ca2+ transport by ferroportin
eLife 12:e82947.
https://doi.org/10.7554/eLife.82947
Further reading
-
- Biochemistry and Chemical Biology
- Microbiology and Infectious Disease
Teichoic acids (TA) are linear phospho-saccharidic polymers and important constituents of the cell envelope of Gram-positive bacteria, either bound to the peptidoglycan as wall teichoic acids (WTA) or to the membrane as lipoteichoic acids (LTA). The composition of TA varies greatly but the presence of both WTA and LTA is highly conserved, hinting at an underlying fundamental function that is distinct from their specific roles in diverse organisms. We report the observation of a periplasmic space in Streptococcus pneumoniae by cryo-electron microscopy of vitreous sections. The thickness and appearance of this region change upon deletion of genes involved in the attachment of TA, supporting their role in the maintenance of a periplasmic space in Gram-positive bacteria as a possible universal function. Consequences of these mutations were further examined by super-resolved microscopy, following metabolic labeling and fluorophore coupling by click chemistry. This novel labeling method also enabled in-gel analysis of cell fractions. With this approach, we were able to titrate the actual amount of TA per cell and to determine the ratio of WTA to LTA. In addition, we followed the change of TA length during growth phases, and discovered that a mutant devoid of LTA accumulates the membrane-bound polymerized TA precursor.
-
- Biochemistry and Chemical Biology
- Computational and Systems Biology
Protein–protein interactions are fundamental to understanding the molecular functions and regulation of proteins. Despite the availability of extensive databases, many interactions remain uncharacterized due to the labor-intensive nature of experimental validation. In this study, we utilized the AlphaFold2 program to predict interactions among proteins localized in the nuage, a germline-specific non-membrane organelle essential for piRNA biogenesis in Drosophila. We screened 20 nuage proteins for 1:1 interactions and predicted dimer structures. Among these, five represented novel interaction candidates. Three pairs, including Spn-E_Squ, were verified by co-immunoprecipitation. Disruption of the salt bridges at the Spn-E_Squ interface confirmed their functional importance, underscoring the predictive model’s accuracy. We extended our analysis to include interactions between three representative nuage components—Vas, Squ, and Tej—and approximately 430 oogenesis-related proteins. Co-immunoprecipitation verified interactions for three pairs: Mei-W68_Squ, CSN3_Squ, and Pka-C1_Tej. Furthermore, we screened the majority of Drosophila proteins (~12,000) for potential interaction with the Piwi protein, a central player in the piRNA pathway, identifying 164 pairs as potential binding partners. This in silico approach not only efficiently identifies potential interaction partners but also significantly bridges the gap by facilitating the integration of bioinformatics and experimental biology.




{kind=link}
{kind=link}
{kind=link}
{kind=link}