Plants: Re-evaluating the driving force behind mutations
Experiments on tropical trees suggest that new mutations in plants are driven by age rather than number of cell divisions during growth.
- GenPhySE, INRAE, INP, ENVT, Université de Toulouse, France
Despite the important role they play in our environment, plants are often perceived to be less complex than animals, particularly in regards to their functional and evolutionary processes (Jose et al., 2019). A fundamental question in evolution is how heritable mutations, which can be transmitted to future generations, accumulate in the genome. However, this question has been little explored to date in plants compared to animals.
In animals, it was initially assumed that mutations predominately came from errors during DNA replication, causing them to appear at the same rate as cell division. However, detailed investigations over the last decade have revealed that heritable mutations accumulate with age rather than with the number of cell divisions. This is supported by data showing that the maternal age at conception contributes to the number of new mutations passed to progeny, because oocytes do not divide after childhood (Figure 1; Goldmann et al., 2016; Jónsson et al., 2017). Consequently, it is now widely accepted that the rate animals acquire heritable mutations is mostly independent from replication, and instead driven by unrepaired DNA damage accumulating with age. This also explains why certain patterns of mutations are more common, such as a high proportion of cytosine-to-thymine mutations (Gao et al., 2019).
Figure 1
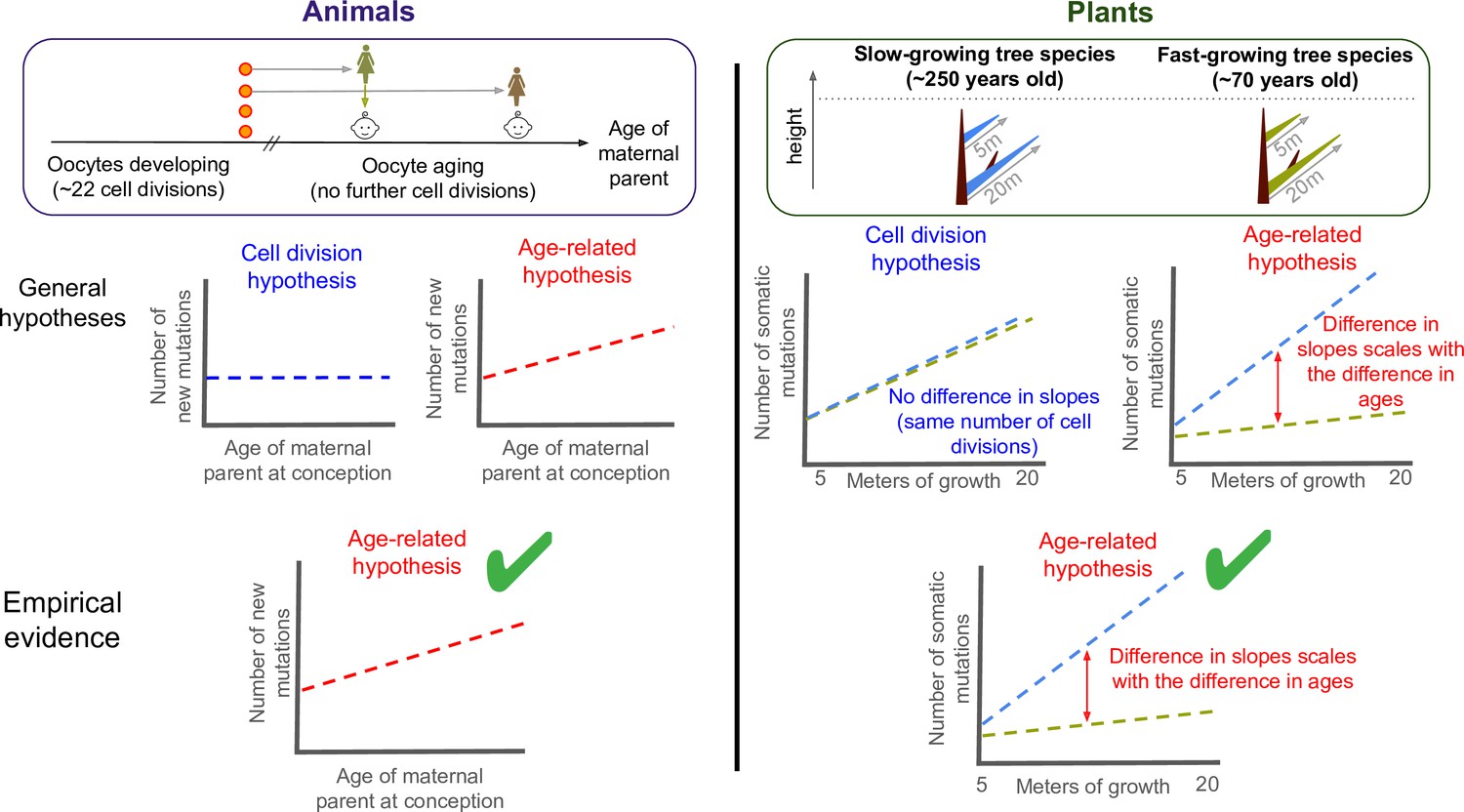
Testing what drives mutations in animals and plants.
There are two hypotheses for how mutations appear and are putatively passed down to future offspring: through errors during DNA replication (cell division hypothesis), or unrepaired damage accumulating with age (age-related hypothesis). To test what drives germline mutations in animals (left panel), previous studies compared the age of the maternal parent at conception to the number of new mutations in the offspring of mammals. This revealed a positive correlation between the two variables (bottom graph, green tick). As oocytes stop dividing in childhood once they are fully formed, this suggests that heritable mutations are caused by age-related damage, not replication errors. Despite being typically harder to observe in males, heritable mutations transmitted from the paternal parent have also recently been shown to be consistent with the age-related hypothesis (Hahn et al., 2023). To test the two hypotheses in plants (right panel), Satake et al. calculated the number of somatic mutations per metre of growth in two evolutionary related tropical trees: a slow-growing (blue) and a fast-growing (green) species that were of similar heights but different ages. The two trees acquired somatic mutations at different rates (right graph), and the gap between these slopes corresponded to the age difference between them. This suggests that the age-related hypothesis also applies to plants (bottom panel, green tick), suggesting that there are parallels in how mutations arise in plants and animals, at least between mammals and trees.
Unlike animals, it is assumed that plants generally differentiate their germline late in development, although this remains debated (Lanfear, 2018). If this assumption is true, the mutations plants accumulate in their somatic, non-reproductive cells during growth will also be present in the germline and can be inherited by future generations. This intergenerational transmission is supported by empirical experiments in trees (Plomion et al., 2018; Wang et al., 2019; Schmitt et al., 2023). Mutation rates in plants are generally assumed to scale with the number of cell divisions in tissues as they grow, as well as UV exposure and other weakly supported general hypotheses (Schmitt et al., 2023). Now, in eLife, Akiko Satake from Kyushu University and colleagues report fascinating counter-intuitive evidence showing that aging rather than number of cell divisions appears to be the major driver of new somatic mutations in trees (Satake et al., 2023).
The team (who are based at various institutes in Japan, Indonesia and Austria) sequenced and assembled the genomes of two evolutionary related tropical trees living in central Borneo, Indonesia: a fast-growing species known as Shorea leprosula, and a slow-growing species known as Shorea laevis. Two individuals from each species were selected, which were of similar heights but different ages, with the S. leprosula tree being 66 years old and the S. laevis tree being 256 years old on average. DNA was extracted from the leaves at the ends of several branches and then compared to identify somatic mutations that were specific to each tree. This revealed that the slow-growing species had far more somatic mutations (962) than the faster-growing species (174).
If cell divisions drive mutations, one would expect similar mutation rates per meter of growth, after making reasonable assumptions for two evolutionary related species (Figure 1). Instead, Satake et al. found that the slow-growing species obtained 3.7 times more mutations per meter than the fast-growing tree, after considering the physical distance between branch tips. This value, however, is remarkably similar to the ratio between the average ages of the trees studied (256/66=3.9). These findings suggest that somatic mutations in plants are mostly driven by unrepaired damage that accumulates with age rather than replication-associated mutations.
Although the experimental design used by Satake et al. only identified a small fraction of the total number of somatic mutations, their results provide sufficient evidence to draw interesting parallels between plants and animals. Satake et al. also found additional evidence in support of this similarity that confirm previous reports: for instance, that the plant genome is enriched in cytosine-to-thymine mutations at specific positions, and shares mutation signatures with human cancers (Alexandrov et al., 2020). From a more methodological perspective, methods initially developed for cancer have been demonstrated to perform better for the discovery of somatic mutations in plants (Schmitt et al., 2022). Altogether, this suggests that mutational processes in plants and animals are largely conserved, and that plant and animal research communities have much to gain from collaborating with one another in the future.
References
-
Parent-of-origin-specific signatures of de novo mutationsNature Genetics 48:935–939.https://doi.org/10.1038/ng.3597
-
Overcoming plant blindness in science, education, and societyPlants, People, Planet 1:169–172.https://doi.org/10.1002/ppp3.51
-
Do plants have a segregated germline?PLOS Biology 16:e2005439.https://doi.org/10.1371/journal.pbio.2005439
-
Oak genome reveals facets of long lifespanNature Plants 4:440–452.https://doi.org/10.1038/s41477-018-0172-3
Article and author information
Author details
Publication history
Copyright
© 2023, Leroy
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 828
- views
-
- 83
- downloads
-
- 1
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Plants: Re-evaluating the driving force behind mutations
eLife 12:e89706.
https://doi.org/10.7554/eLife.89706
Further reading
-
- Ecology
- Evolutionary Biology
Eurasia has undergone substantial tectonic, geological, and climatic changes throughout the Cenozoic, primarily associated with tectonic plate collisions and a global cooling trend. The evolution of present-day biodiversity unfolded in this dynamic environment, characterised by intricate interactions of abiotic factors. However, comprehensive, large-scale reconstructions illustrating the extent of these influences are lacking. We reconstructed the evolutionary history of the freshwater fish family Nemacheilidae across Eurasia and spanning most of the Cenozoic on the base of 471 specimens representing 279 species and 37 genera plus outgroup samples. Molecular phylogeny using six genes uncovered six major clades within the family, along with numerous unresolved taxonomic issues. Dating of cladogenetic events and ancestral range estimation traced the origin of Nemacheilidae to Indochina around 48 mya. Subsequently, one branch of Nemacheilidae colonised eastern, central, and northern Asia, as well as Europe, while another branch expanded into the Burmese region, the Indian subcontinent, the Near East, and northeast Africa. These expansions were facilitated by tectonic connections, favourable climatic conditions, and orogenic processes. Conversely, aridification emerged as the primary cause of extinction events. Our study marks the first comprehensive reconstruction of the evolution of Eurasian freshwater biodiversity on a continental scale and across deep geological time.
-
- Evolutionary Biology
Gene duplication drives evolution by providing raw material for proteins with novel functions. An influential hypothesis by Ohno (1970) posits that gene duplication helps genes tolerate new mutations and thus facilitates the evolution of new phenotypes. Competing hypotheses argue that deleterious mutations will usually inactivate gene duplicates too rapidly for Ohno’s hypothesis to work. We experimentally tested Ohno’s hypothesis by evolving one or exactly two copies of a gene encoding a fluorescent protein in Escherichia coli through several rounds of mutation and selection. We analyzed the genotypic and phenotypic evolutionary dynamics of the evolving populations through high-throughput DNA sequencing, biochemical assays, and engineering of selected variants. In support of Ohno’s hypothesis, populations carrying two gene copies displayed higher mutational robustness than those carrying a single gene copy. Consequently, the double-copy populations experienced relaxed purifying selection, evolved higher phenotypic and genetic diversity, carried more mutations and accumulated combinations of key beneficial mutations earlier. However, their phenotypic evolution was not accelerated, possibly because one gene copy rapidly became inactivated by deleterious mutations. Our work provides an experimental platform to test models of evolution by gene duplication, and it supports alternatives to Ohno’s hypothesis that point to the importance of gene dosage.




{kind=link}