Vascular Endothelium: More than just a simple barrier
Endothelial cell subpopulations are characterized by unique gene expression profiles, epigenetic landscapes and functional properties.
- Department of Pathology, Heersink School of Medicine, The University of Alabama at Birmingham, United States
- Division of Molecular and Cellular Pathology, Department of Pathology, The University of Alabama, United States
Our blood vessels are lined with a single layer of endothelial cells, known as the vascular endothelium, which provides a permeable barrier to protect the blood from toxins and other harmful substances. Endothelial cells have many important roles, including regulating the contraction and relaxation of the blood vessels, enabling exchange of molecules between blood and tissues, and regulating immune response and inflammation.
Advances in cell biology and molecular techniques have shown that the endothelium is more than just a uniform layer lining the blood vessels. Endothelial cells vary greatly among different tissues and even between individual cells within a region (Kalucka et al., 2020; Paik et al., 2020; Kalluri et al., 2019; Chen et al., 2024). These differences, which can include gene expression, epigenetic landscape, functional properties and responsiveness to stimuli, suggest distinct and specialized roles in vascular physiology and pathology (Figure 1).
Figure 1
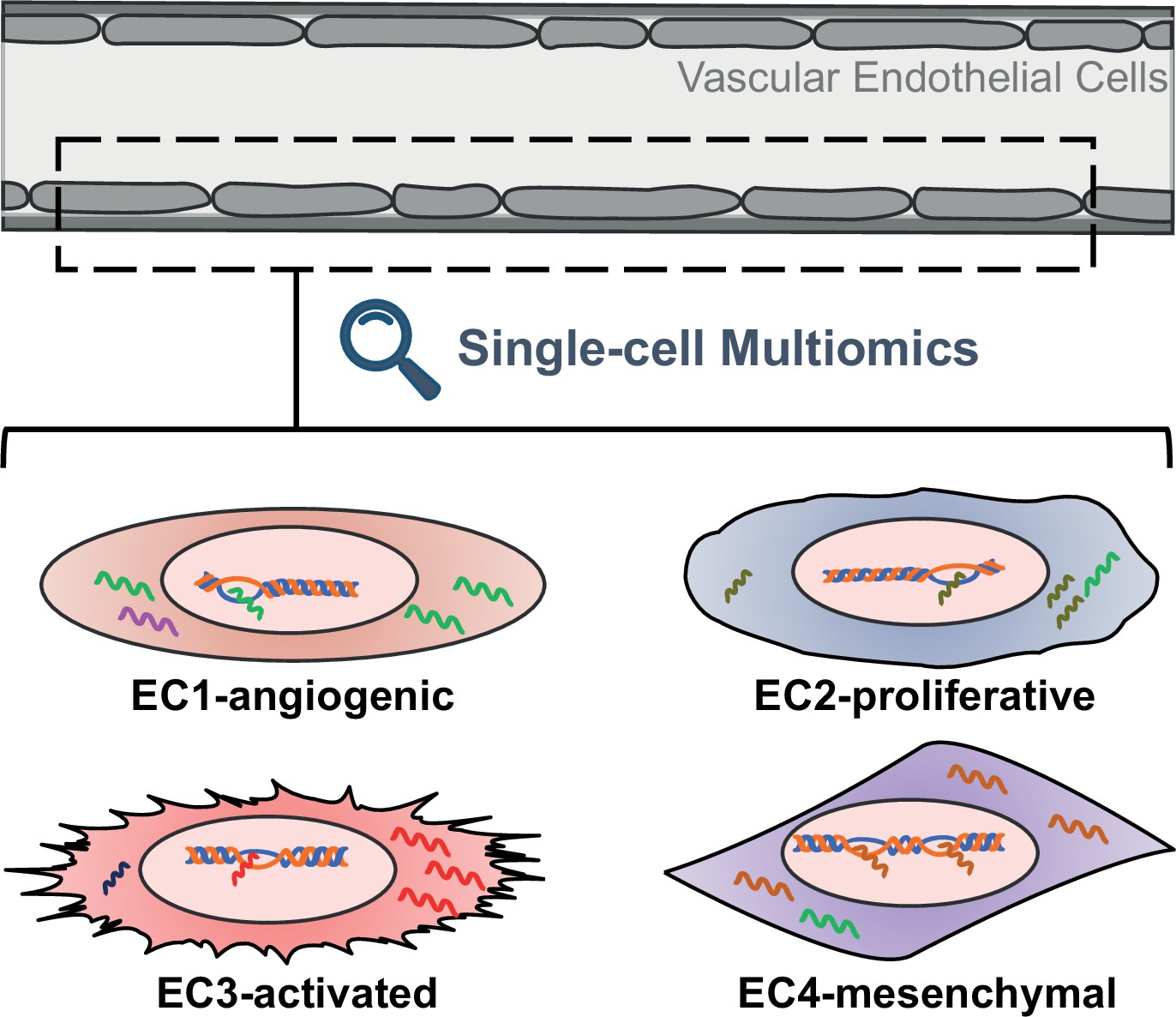
Schematic representing the different properties of endothelial cell subtypes.
The cells of the vascular endothelium are extremely heterogenous, which could affect their role in vascular function and disease. EC1 cells are involved in the formation of new blood vessels, EC2 cells have a fast cell turnover, and both EC3 cells and EC4 cells show mesenchymal properties.
Disrupted endothelial function has been linked to atherosclerosis, a thickening of the arteries caused by a build-up of plaque in the endothelium, which can lead to ischemic heart diseases. Many of the endothelial cells in atherosclerotic plaques no longer express typical endothelial cell markers and instead become more like another type of cell, called a mesenchymal cell, which acquires properties or releases molecules that overactivate the immune system, worsening the disease. Understanding the extent of the heterogeneity of endothelial cells promises to yield valuable insights into vascular biology and could provide new therapeutic targets for cardiovascular diseases. However, studies that quantify the heterogeneity of endothelial cells in live animals and cell models are lacking. Now, in eLife, Casey Romanoski and colleagues at the University of Arizona – including Maria Adelus as first author – report a comprehensive analysis of endothelial cell diversity and their unique responses under atherosclerosis-like conditions (Adelus et al., 2023).
To delve into the intricacies of endothelial cells within human aortas, the team combined experiments with human aortic cells grown in the laboratory and analysis of publicly available single-cell gene expression data from human arterial specimens. This uncovered four distinct subtypes of endothelial cells, which are characterized by unique gene expression profiles, epigenetic landscapes and functional properties. For example, the expression profile of EC1 cells suggests they are involved in the formation of new blood vessels, while EC2 cells display rapid growth characteristics. On the other hand, both EC3 and EC4 cells resemble mesenchymal cells, suggesting they have transitioned from their original cell type.
Next, Adelus et al. investigated how these different populations respond to factors known to be important for endothelial cell biology and the development of atherosclerosis. The experiments unveiled distinct reactions across endothelial populations. The EC1 and EC2 populations, which mostly represent healthy endothelial cells, were most responsive when a transcription factor required for endothelial cell specification and homeostasis was deactivated. In contrast, EC3 populations were most affected by stimulation with a cytokine to mimic inflammation, whereas EC4s reacted to activation of a specific growth factor that allows cells to transition to different cell types. Furthermore, Adelus et al. identified accessible chromatin regions unique to EC2 and EC4 populations that are enriched with genetic variations associated with coronary artery disease. This suggests that EC2 and EC4 populations could be involved in mechanisms underlying the development and progression of coronary artery disease.
Taken together, these findings offer valuable insights into the underlying pathophysiology of atherosclerosis and suggest that the characteristics of individual endothelial cells enable them to respond uniquely to various pathological processes, potentially playing distinct roles in the progression of vascular diseases. The identification of heterogeneous endothelial subtypes underscores the complexity of endothelial cell populations and emphasizes the necessity for further investigation into their roles in both health and disease. By integrating laboratory-based experiments with publicly available datasets, Adelus et al. enhanced the reliability of the findings and established a solid foundation for future research endeavors.
Thus far, single-cell studies have paved the way for unraveling the diversity of vascular endothelial biology. For future studies, it will be crucial to standardize how different populations of endothelial cells are characterized and identified to enable more consistent evaluations based on uniform criteria. A next step would be to profile gene expression and epigenetic landscapes of the endothelial cell clusters to define their unique roles and to distinguish athero-protective from athero-prone cells. Once the latter are identified, more accurate cell-targeting strategies, such as nanoparticles, can be developed to ultimately improve the treatment of cardiovascular diseases with limited side effects.
References
Article and author information
Author details
Erandi Velazquez-Miranda

Publication history
Copyright
© 2024, Velazquez-Miranda and He
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 750
- views
-
- 71
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Vascular Endothelium: More than just a simple barrier
eLife 13:e97247.
https://doi.org/10.7554/eLife.97247
Further reading
-
- Chromosomes and Gene Expression
- Evolutionary Biology
Repression of retrotransposition is crucial for the successful fitness of a mammalian organism. The domesticated transposon protein L1TD1, derived from LINE-1 (L1) ORF1p, is an RNA-binding protein that is expressed only in some cancers and early embryogenesis. In human embryonic stem cells, it is found to be essential for maintaining pluripotency. In cancer, L1TD1 expression is highly correlative with malignancy progression and as such considered a potential prognostic factor for tumors. However, its molecular role in cancer remains largely unknown. Our findings reveal that DNA hypomethylation induces the expression of L1TD1 in HAP1 human tumor cells. L1TD1 depletion significantly modulates both the proteome and transcriptome and thereby reduces cell viability. Notably, L1TD1 associates with L1 transcripts and interacts with L1 ORF1p protein, thereby facilitating L1 retrotransposition. Our data suggest that L1TD1 collaborates with its ancestral L1 ORF1p as an RNA chaperone, ensuring the efficient retrotransposition of L1 retrotransposons, rather than directly impacting the abundance of L1TD1 targets. In this way, L1TD1 might have an important role not only during early development but also in tumorigenesis.
-
- Chromosomes and Gene Expression
RNA interference (RNAi) is a conserved pathway that utilizes Argonaute proteins and their associated small RNAs to exert gene regulatory function on complementary transcripts. While the majority of germline-expressed RNAi proteins reside in perinuclear germ granules, it is unknown whether and how RNAi pathways are spatially organized in other cell types. Here, we find that the small RNA biogenesis machinery is spatially and temporally organized during Caenorhabditis elegans embryogenesis. Specifically, the RNAi factor, SIMR-1, forms visible concentrates during mid-embryogenesis that contain an RNA-dependent RNA polymerase, a poly-UG polymerase, and the unloaded nuclear Argonaute protein, NRDE-3. Curiously, coincident with the appearance of the SIMR granules, the small RNAs bound to NRDE-3 switch from predominantly CSR-class 22G-RNAs to ERGO-dependent 22G-RNAs. NRDE-3 binds ERGO-dependent 22G-RNAs in the somatic cells of larvae and adults to silence ERGO-target genes; here we further demonstrate that NRDE-3-bound, CSR-class 22G-RNAs repress transcription in oocytes. Thus, our study defines two separable roles for NRDE-3, targeting germline-expressed genes during oogenesis to promote global transcriptional repression, and switching during embryogenesis to repress recently duplicated genes and retrotransposons in somatic cells, highlighting the plasticity of Argonaute proteins and the need for more precise temporal characterization of Argonaute-small RNA interactions.




{kind=link}