Cellular Energy Production: Mycofactocin and the mycobacterial electron transport chain
In the bacterium M. smegmatis, an enzyme called MftG allows the cofactor mycofactocin to transfer electrons released during ethanol metabolism to the electron transport chain.
- Laboratory of Microbial Biochemistry and Biotechnology, School of Biological Sciences, The University of Auckland, New Zealand
To survive, bacteria must adapt to ever-changing environments and, as a result, have developed diverse strategies to respond to this challenge. In particular, species from the Mycobacterium genus can flexibly adjust their metabolism (Cook et al., 2009), allowing them to break down a wide range of nutrients to produce energy (de Carvalho et al., 2010).
This metabolic flexibility, displayed by both environmental and pathogenic species, is partly due to a variety of unusual cofactors – molecules that help enzymes to catalyse reactions (Peña-Ortiz et al., 2020). Among these is mycofactocin, a peptide-derived cofactor that has been linked to the metabolism of cholesterol and fatty acids in M. tuberculosis (Krishnamoorthy et al., 2019; Mendauletova and Latham, 2022) and ethanol in M. smegmatis. Previous work showed that an enzyme known as Mdo/Mno, which breaks down alcohol and uses mycofactocin as a cofactor, is essential for M. smegmatis to consume ethanol (Krishnamoorthy et al., 2019). Now, in eLife, Gerald Lackner and colleagues – including Ana Patrícia Graça as first author – report that by carrying electrons released when ethanol is broken down, mycofactocin links ethanol metabolism to the generation of cellular energy in the form of ATP (Graça et al., 2025).
Previous studies have shown that multiple enzymes encoded in a gene cluster known as mft contribute to the production of bioactive mycofactocin molecules (Ayikpoe et al., 2018; Haft, 2011). However, one gene in the cluster, which encodes an enzyme known as MftG, remained uncharacterised. Graça et al. (who are based at the Leibniz Institute for Natural Products and Infection Biology, the Hans Knöll Institute and various other research institutes in Germany) began by deleting the gene encoding MftG in M. smegmatis. When provided with ethanol as a nutrient source, strains lacking this gene showed impaired growth, displaying features of mild starvation, such as disrupted cell division and energy production.
Analysis of gene expression in cells lacking the MftG gene revealed that processes related to cell division, including DNA replication and peptidoglycan biosynthesis, were downregulated. Additionally, cellular respiratory machinery had been remodelled to compensate for a shortage of electron donors. These observations highlight the modified bacterial strain’s ability to compensate for impaired mycofactocin metabolism.
Graça et al. next investigated how deleting the gene for MftG leads to starvation-like features. Measuring bacterial respiration showed that cells lacking MftG consumed only around 45% of the oxygen consumed by the parent cells. This indicates that fewer electrons were being delivered to the electron transport chain, the final stage of energy production, where they are ultimately accepted by oxygen.
Analysing the contents of the cells using mass spectrometry revealed that strains lacking MftG accumulated higher levels of reduced mycofactocin (those that have gained electrons) while having almost no detectable oxidised mycofactocin (those that have lost electrons). In contrast, a strain containing an additional copy of the MftG gene showed a substantial increase in oxidised mycofactocin. These findings indicate that MftG catalyses the oxidation of mycofactocin. Additionally, Graça et al. suggest that the oxidation of mycofactocin, which allows it to be recycled, is the rate-limiting step in ethanol metabolism.
This prompted the team to investigate whether MftG and mycofactocin could transfer electrons to the electron transport chain, which is embedded in the mycobacterial membrane. While known electron donors to the electron transport chain, such as NADH and succinate, were able to contribute to the production of energy in M. smegmatis membrane preparations when MftG was absent, reduced mycofactocin could only do so when MftG was present. This indicates that MftG facilitates electron transfer from reduced mycofactocin to the electron transport chain, thereby directly linking the electrons generated during ethanol metabolism to energy production (Figure 1).
Figure 1
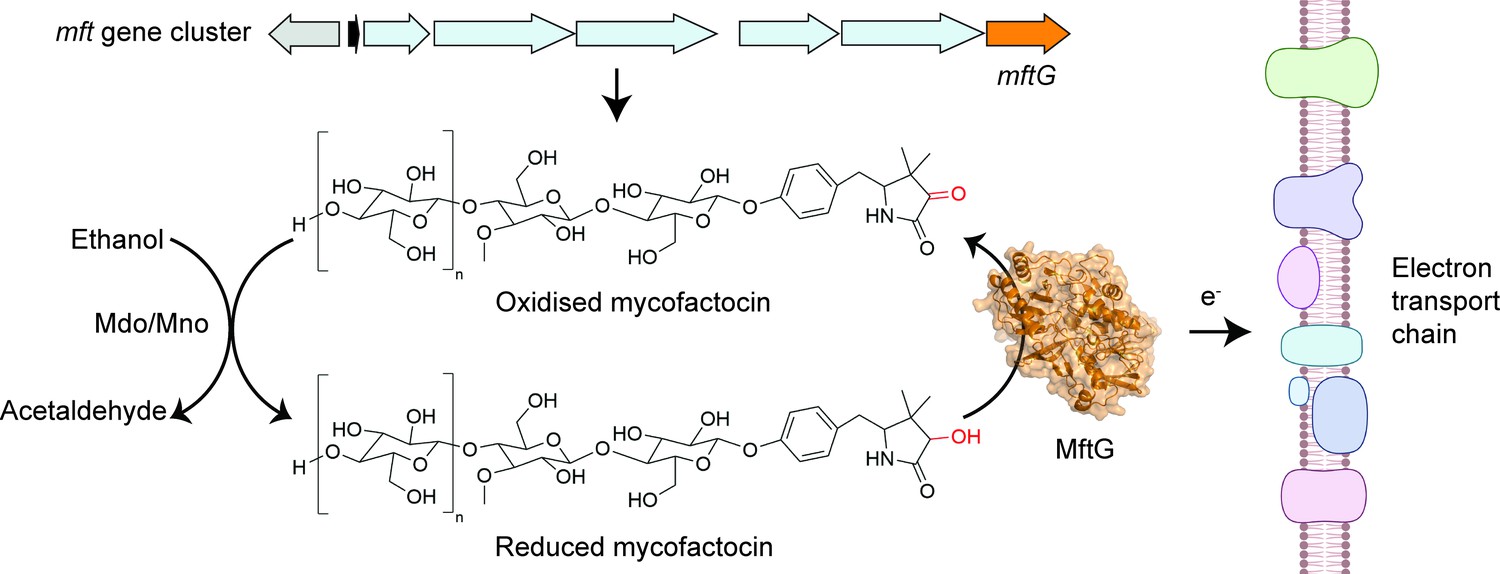
The enzyme MftG links ethanol metabolism to the electron transport chain.
The cofactor mycofactocin, which is produced by the mft gene cluster (top arrows), links ethanol metabolism to the electron transport chain. This process begins when an enzyme known as Mdo/Mno catalyses the conversion of ethanol into acetaldehyde (left), transferring electrons to mycofactocin. Experiments by Graça et al. in M. smegmatis revealed that a previously uncharacterised gene in this cluster, known as mftG (orange arrow), encodes an enzyme (MftG; orange structure) that catalyses the transfer of electrons from the reduced mycofactocin to the electron transport chain. Electrons are passed along this chain of protein complexes in the mycobacterial membrane (right), which ultimately generates energy in the form of ATP. This process allows M. smegmatis to use ethanol as an energy source when other nutrients are not available. This figure was created with BioRender.com.
Image credit: Adapted from Figure 1 of Graça et al., 2025 (CC BY 4.0). The AlphaFold (Jumper et al., 2021) structure prediction of MftG was retrieved from the AlphaFold Protein Structure Database (Varadi et al., 2022) and visualised using PyMOL (Schrödinger, LLC, 2024).
Based on these findings, Graça et al. propose a model in which MftG regenerates mycofactocin by catalysing the transfer of electrons to the electron transport chain. In this model, MftG functions alongside the enzyme Mdo/Mno (which catalyses the initial transfer of electrons from ethanol to mycofactocin) to shuttle electrons from alcohol substrates to the electron transport chain. As Graça et al. note, a key question for future research is which specific component(s) of the electron transport chain MftG interacts with and delivers electrons to. In summary, Graça et al. have demonstrated an important new role for the mycofactocin cofactor in supporting the metabolic adaptability of mycobacterial cells under complex environmental conditions.
References
-
Physiology of mycobacteriaAdvances in Microbial Physiology 55:81–182.https://doi.org/10.1016/S0065-2911(09)05502-7
-
Biosynthesis of the redox cofactor mycofactocin is controlled by the transcriptional regulator MftR and induced by long-chain acyl-CoA speciesThe Journal of Biological Chemistry 298:101474.https://doi.org/10.1016/j.jbc.2021.101474
Article and author information
Author details
Stephanie M Stuteley

Publication history
Copyright
© 2025, Stuteley and Bashiri
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 213
- views
-
- 28
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cellular Energy Production: Mycofactocin and the mycobacterial electron transport chain
eLife 14:e106286.
https://doi.org/10.7554/eLife.106286
Further reading
-
- Biochemistry and Chemical Biology
- Computational and Systems Biology
The spike protein is essential to the SARS-CoV-2 virus life cycle, facilitating virus entry and mediating viral-host membrane fusion. The spike contains a fatty acid (FA) binding site between every two neighbouring receptor-binding domains. This site is coupled to key regions in the protein, but the impact of glycans on these allosteric effects has not been investigated. Using dynamical nonequilibrium molecular dynamics (D-NEMD) simulations, we explore the allosteric effects of the FA site in the fully glycosylated spike of the SARS-CoV-2 ancestral variant. Our results identify the allosteric networks connecting the FA site to functionally important regions in the protein, including the receptor-binding motif, an antigenic supersite in the N-terminal domain, the fusion peptide region, and another allosteric site known to bind heme and biliverdin. The networks identified here highlight the complexity of the allosteric modulation in this protein and reveal a striking and unexpected link between different allosteric sites. Comparison of the FA site connections from D-NEMD in the glycosylated and non-glycosylated spike revealed that glycans do not qualitatively change the internal allosteric pathways but can facilitate the transmission of the structural changes within and between subunits.
-
- Biochemistry and Chemical Biology
- Genetics and Genomics
Deep Mutational Scanning (DMS) is an emerging method to systematically test the functional consequences of thousands of sequence changes to a protein target in a single experiment. Because of its utility in interpreting both human variant effects and protein structure-function relationships, it holds substantial promise to improve drug discovery and clinical development. However, applications in this domain require improved experimental and analytical methods. To address this need, we report novel DMS methods to precisely and quantitatively interrogate disease-relevant mechanisms, protein-ligand interactions, and assess predicted response to drug treatment. Using these methods, we performed a DMS of the melanocortin-4 receptor (MC4R), a G-protein-coupled receptor (GPCR) implicated in obesity and an active target of drug development efforts. We assessed the effects of >6600 single amino acid substitutions on MC4R’s function across 18 distinct experimental conditions, resulting in >20 million unique measurements. From this, we identified variants that have unique effects on MC4R-mediated Gαs- and Gαq-signaling pathways, which could be used to design drugs that selectively bias MC4R’s activity. We also identified pathogenic variants that are likely amenable to a corrector therapy. Finally, we functionally characterized structural relationships that distinguish the binding of peptide versus small molecule ligands, which could guide compound optimization. Collectively, these results demonstrate that DMS is a powerful method to empower drug discovery and development.




{kind=link}