VE-cadherin enables trophoblast endovascular invasion and spiral artery remodeling during placental development
- Cardiovascular Institute, Department of Medicine, University of Pennsylvania, United States
- Department of Radiology, Hospital of the University of Pennsylvania, United States
- University Laboratory Animal Resources, University of Pennsylvania, United States
- Department of Molecular Pharmacology and Physiology, University of South Florida, United States
Abstract
During formation of the mammalian placenta, trophoblasts invade the maternal decidua and remodel spiral arteries to bring maternal blood into the placenta. This process, known as endovascular invasion, is thought to involve the adoption of functional characteristics of vascular endothelial cells (ECs) by trophoblasts. The genetic and molecular basis of endovascular invasion remains poorly defined, however, and whether trophoblasts utilize specialized endothelial proteins in an analogous manner to create vascular channels remains untested. Vascular endothelial (VE-)cadherin is a homotypic adhesion protein that is expressed selectively by ECs in which it enables formation of tight vessels and regulation of EC junctions. VE-cadherin is also expressed in invasive trophoblasts and is a prime candidate for a molecular mechanism of endovascular invasion by those cells. Here, we show that VE-cadherin is required for trophoblast migration and endovascular invasion into the maternal decidua in the mouse. VE-cadherin deficiency results in loss of spiral artery remodeling that leads to decreased flow of maternal blood into the placenta, fetal growth restriction, and death. These studies identify a non-endothelial role for VE-cadherin in trophoblasts during placental development and suggest that endothelial proteins may play functionally unique roles in trophoblasts that do not simply mimic those in ECs.
Editor's evaluation
Understanding molecular and cellular pathways for endovascular invasion and pathogenesis of preeclampsia are important topics for current vascular biology and Ob/Gyn biology, making this study timely and important. It shows for the first time a causal link for the need of VE-cadherin on trophoblasts in vivo for the invasion of these cells into the decidua and for their role in vascular remodeling. The conclusions of this paper are well supported by data. It may provide a novel avenue for the prevention or treatment of preeclampsia.
https://doi.org/10.7554/eLife.77241.sa0Introduction
During placental development in mice and humans, fetal trophoblasts invade the maternal decidua by a process known as endovascular invasion to remodel and connect to maternal spiral arteries (SAs) (Velicky et al., 2016; Soares et al., 2014; Hu and Cross, 2011). This connection allows the flow of maternal blood through trophoblast-lined sinuses characteristic of hemochorial placentation (Soares et al., 2018; Rai and Cross, 2014). Shallow trophoblast invasion, deficient SA remodeling, and poor remodeling of the maternal decidua are features of placental dysfunction such as preeclampsia, a hypertensive condition of pregnancy that can lead to maternal and fetal complications (Lyall et al., 2013; Roberts and Escudero, 2012). Despite the broad and clinically significant impacts of placental dysfunction, the mechanisms controlling trophoblast endovascular invasion and SA remodeling remain poorly defined in vivo.
Invasive trophoblasts are believed to adopt an endothelial-like state by expressing endothelial specific genes (Nelson et al., 2016; Zhou et al., 1997b; Govindasamy et al., 2021), a process that has also been termed ‘vascular mimicry’ (reviewed in Rai and Cross, 2014). Invasive trophoblasts in human and mouse placentas express vascular endothelial (VE)-cadherin (gene name Cdh5) during remodeling of SAs (Zhou et al., 1997b; Govindasamy et al., 2021; Zhou et al., 1997a). Invasive trophoblasts in preeclamptic placentas lack VE-cadherin (Zhou et al., 1997a), and loss of VE-cadherin reduces trophoblast invasion in vitro (Cheng et al., 2013). These studies suggest a functional role for VE-cadherin in endovascular invasion and vessel formation. VE-cadherin is a well-studied cell-cell adhesion protein in the vascular endothelium where it regulates vascular integrity and growth and endothelial barrier function (Corada et al., 1999; Crosby et al., 2005; Carmeliet et al., 1999). In vitro studies have suggested that VE-cadherin may regulate trophoblast-endothelial interactions (Bulla et al., 2005), but the requirement for VE-cadherin in trophoblasts during placental development in vivo remains unknown.
In the present study, we functionally tested the role of VE-cadherin in trophoblasts during placental development in mice. We find that conditional deletion of VE-cadherin from trophoblasts disrupts trophoblast invasion into the decidua and SA remodeling, resulting in placental insufficiency and fetal growth restriction. We show that VE-cadherin is important for trophoblasts to interact with and displace SA endothelium. Additionally, trophoblast invasion is important for triggering multiple changes in the decidual extracellular matrix (ECM) and immune cell microenvironment. These studies identify a molecular mechanism by which fetal trophoblasts use VE-cadherin to invade and remodel the maternal environment for successful pregnancy that is relevant to preeclampsia pathogenesis. They also provide a first functional test of the concept of trophoblasts utilizing endothelial programs during endovascular invasion in vivo, and suggest that canonical endothelial proteins may be used by vascular trophoblasts in the placenta in ways that are specific for their function and do not merely mimic endothelial cell (EC) use.
Results
Trophoblast-specific deletion of VE-cadherin restricts placental and fetal growth and causes embryonic lethality
To understand the role of VE-cadherin in trophoblasts, we generated CYP19A1(Tg)Cre; Cdh5fl/fl (‘Cdh5 knockout’) placentas and mice in which VE-cadherin (encoded by Cdh5) is deleted specifically in fetal trophoblasts. Immunostaining for VE-cadherin and the trophoblast marker Cytokeratin 8 (CK8) demonstrated efficient deletion of VE-cadherin in trophoblasts but not ECs in the Cdh5 knockout placenta (Figure 1—figure supplement 1A, B). Cdh5 knockout embryos were present at the expected Mendelian ratio at E10.5–12.5 (Table 1, p = 1.0000). However, there was an almost complete loss of trophoblast Cdh5 knockout embryos at E14.5–16.5 and postnatal day 21 (Table 1, p < 0.05 and p < 0.005, respectively). Examination of placentas at E12.5 revealed that trophoblast Cdh5 knockout placentas were smaller and paler than those of control littermates in the same uterus (Figure 1A–D). Cdh5 knockout embryos exhibited marked fetal growth restriction and variable degrees of hemorrhage at E12.5 (green arrowheads, Figure 1E–H). No differences in weights were observed between Cre-negative (Cdh5fl/+ or Cdh5fl/fl) and Cre-positive heterozygous (CYP19A1(Tg)Cre; Cdh5fl/+) placentas and embryos (Figure 1—figure supplement 1C, D). Histological and immunofluorescence analysis of E12.5 knockout embryos showed growth defects in numerous organs, including the heart, brain, and liver (Figure 1—figure supplement 2A-D). Immunofluorescence staining for VE-cadherin in CYP19A1(Tg)Cre; Cdh5fl/fl embryos show that VE-cadherin is retained in the embryonic vasculature of affected organs, including the brain, heart, liver, lungs, and thorax (Figure 1—figure supplement 3A-D). These data demonstrate that trophoblast-specific loss of VE-cadherin confers fetal growth restriction and lethality. Importantly, since CYP19A1(Tg)Cre activity is present only in placental trophoblasts (Wenzel and Leone, 2007; López-Tello et al., 2019), these embryonic defects are secondary to placental defects.
Table 1
Decreased survival of CYP19A1(Tg)Cre; Cdh5fl/fl mutants in late gestation.
Cdh5fl/fl male mice were crossed with CYP19A1(Tg)Cre; Cdh5fl/+ females to generate litters with mixed genotypes. The expected percentage is listed under the genotype label. The observed number of each genotype is shown with the corresponding percentage given in parentheses. P-values were calculated using Fisher’s exact test at stages E10.5-12.5 (pooled), E14.5-16.5 (pooled), and P21. E designates embryonic day and P designates postnatal day.
| CYP19A1(Tg)Cre; Cdh5fl/fl (25%) | CYP19A1(Tg)Cre; Cdh5fl/+ (25%) | Cdh5fl/fl (25%) | Cdh5fl/+ (25%) | Total (100%) | Fisher Exact Test | |
|---|---|---|---|---|---|---|
| E10.5-E12.5 | 13* (26%) | 13 (26%) | 12 (24%) | 12 (24%) | 50 (100%) | P=1.0000 |
| E14.5-E16.5 | 1 (2.9%) | 13 (38.2%) | 12 (35.3%) | 8 (23.5%) | 34 (100%) | P=0.0033 |
| P21 | 1 (2.2%) | 11 (24.4%) | 15 (33.3%) | 18 (40.0%) | 45 (100%) | P=0.0333 |
-
*
One embryo at E12.5 was dead.
Figure 1 with 3 supplements see all
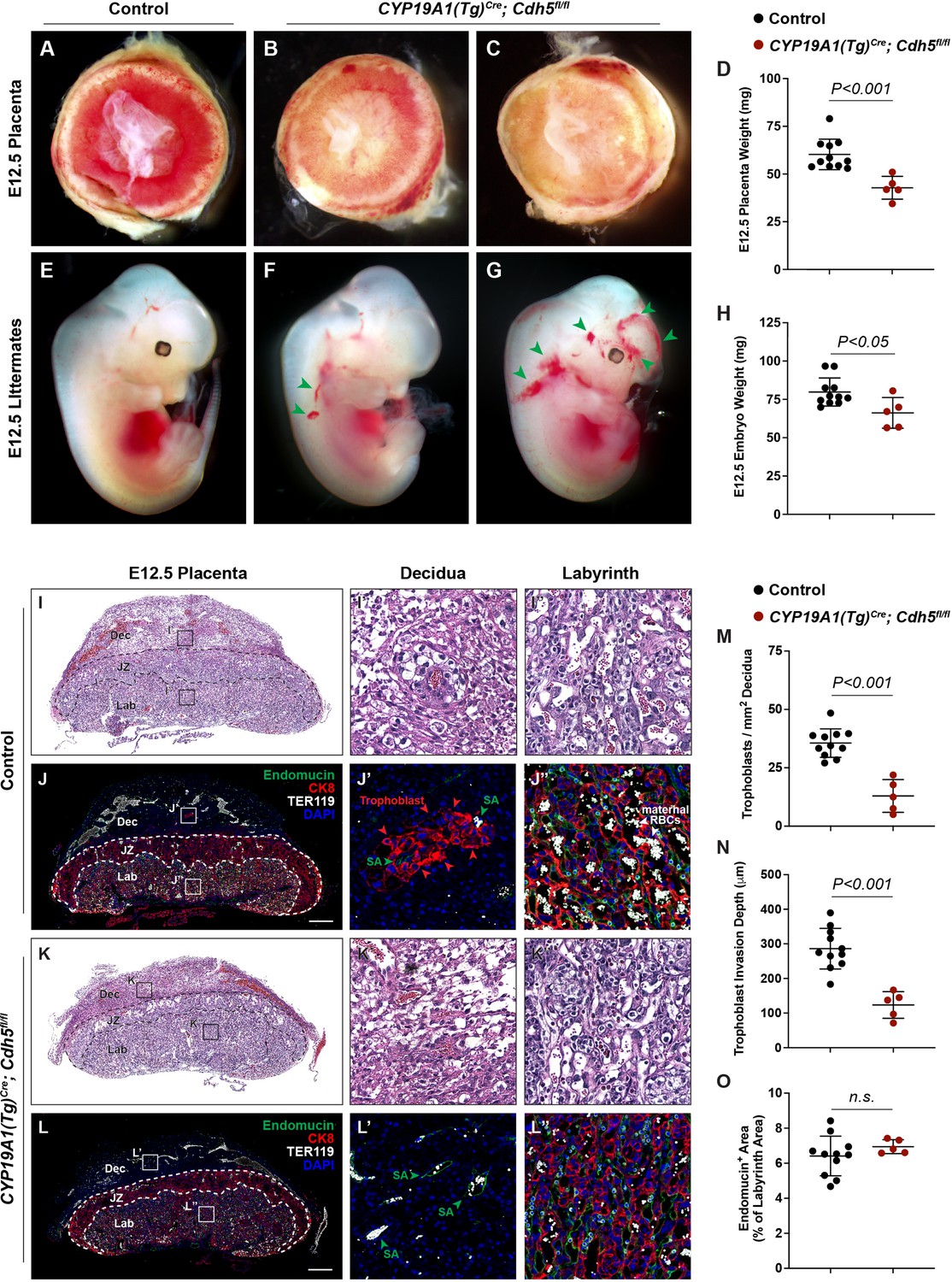
Deletion of VE-cadherin in trophoblasts disrupts cell migration resulting in placental and fetal growth restriction.
(A–D) Gross examination and quantification of E12.5 Control and CYP19A1(Tg)Cre; Cdh5fl/fl placentas and weights. Control n = 11, CYP19A1(Tg)Cre; Cdh5fl/fl n = 5. (E–H) Gross examination and quantification of E12.5 Control and CYP19A1(Tg)Cre; Cdh5fl/fl embryos and weights. Control n = 11, CYP19A1(Tg)Cre; Cdh5fl/fl n = 5. Green arrowheads point to areas of hemorrhage. (I–L) Hematoxylin and eosin (H&E) staining and immunofluorescence staining for Endomucin (green), CK8 (red), and TER119 (gray) of E12.5 Control (I, J) and CYP19A1(Tg)Cre; Cdh5fl/fl (K, L) serial placenta sections. Dotted lines demarcate the different placental regions. Red arrowheads indicate trophoblasts. Green arrowheads indicate spiral arteries (SA). White arrowheads indicate maternal red blood cells (RBCs). Note fewer non-nucleated, maternal TER119+ cells in the labyrinth region of CYP19A1(Tg)Cre; Cdh5fl/fl placentas. Boxes on the left correlate with magnified images on the right, and boxes in H&E and immunofluorescence images are of the same region. Scale bars = 500 μm. Dec (decidua), JZ (junctional zone), Lab (labyrinth). (M–O) Quantification of number of trophoblasts in the decidua (M), trophoblast invasion depth (N), and percent labyrinth Endomucin+ area (O). Control n = 11, CYP19A1(Tg)Cre; Cdh5fl/fl n = 5. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test. Data are shown as means ± SD.
-
Figure 1—source data 1
Excel file containing quantification for embryo weights, placenta weights, trophoblast density, trophoblast migration distance, and fetal labyrinth vasculature in Figure 1.
- https://cdn.elifesciences.org/articles/77241/elife-77241-fig1-data1-v2.xlsx
Loss of VE-cadherin disrupts trophoblast endovascular invasion but not formation of the fetal vasculature
To characterize the placental defects conferred by trophoblast loss of VE-cadherin, we performed histological staining with hematoxylin and eosin (H&E) and immunofluorescence staining for CK8 (trophoblasts), Endomucin (endothelial cells), and TER119 (erythrocytes) on serial control and Cdh5 knockout placenta sections (Figure 1I–L). H&E and immunofluorescence staining both showed abundant trophoblasts, marked by CK8 positivity, surrounding Endomucin+ SAs within the decidua in control placentas (red arrowheads, Figure 1J and J’). Notably, trophoblasts surrounding maternal SAs were absent in knockout placentas (Figure 1L and L’), and quantification of number of trophoblasts and invasion depth into the decidua showed fewer and shallower invasion of trophoblasts overall (decidua, Figure 1M and N).
The findings described above suggested that Cdh5 knockout placentas were less able to carry maternal blood to nourish the growing embryo. Fetal and maternal red blood cells (RBCs) can be differentiated by the presence of nuclei in fetal RBCs. The labyrinth in knockout placentas had fewer enucleated TER119+ RBCs, indicating less maternal blood and consistent with paler placentas observed by gross examination (white arrowheads, Figure 2J” vs. L”). To determine whether loss of VE-cadherin affects the fetal placental vasculature, we quantified Endomucin+ vascular area in the labyrinth. We detected no differences in Endomucin+ staining, demonstrating that loss of VE-cadherin from trophoblasts does not disrupt formation of the fetal placental capillary plexus (Figure 1J, L and O). These findings suggest that VE-cadherin is important for trophoblast migration and for the association with SAs required to channel maternal blood into the placenta.
Figure 2

VE-cadherin is required in trophoblasts to remodel spiral arteries (SAs) and to displace SA endothelium.
(A) Immunofluorescence staining of E12.5 Control and CYP19A1(Tg)Cre; Cdh5fl/fl placentas for Endomucin (green) and alpha-smooth muscle actin (αSMA) (red). White arrowheads indicate αSMA+ cells. Scale bars = 25 μm. (B, C) Quantification of αSMA+ cells per SA and SA diameter. Control n = 11, CYP19A1(Tg)Cre; Cdh5fl/fl n = 5. (D) Immunofluorescence staining of E12.5 Control and CYP19A1(Tg)Cre; Cdh5fl/fl placentas for Endomucin (green) and CK8 (red). White arrowheads indicate Endomucin+ SA endothelial cells (ECs). Positive signal in small, rounded cells in the lumen is the result of erythrocyte autofluorescence. (E) Schematic demonstrating differences in trophoblast and SA EC contact with the vessel lumen. Scale bars = 25 μm. (F) Quantification of the percent trophoblast-lumen contact, which was calculated by measuring the circumference of the vessel lumen and then measuring the length of CK8+ trophoblasts in contact with the lumen. Each point represents the average of at least three SAs from an individual placenta. Control n = 10, CYP19A1(Tg)Cre; Cdh5fl/fl n = 5. (G, H) Maximum intensity projections of whole-mount immunofluorescence of the decidua (G) and labyrinth (H) from 200 μm thick placenta sections stained for Endomucin (green), TER119 (red), and CK8 (magenta). Double-headed arrows indicate differences in lumen size. White arrowheads indicate Endomucin+ SA ECs. Red arrowheads indicate maternal red blood cells within the trophoblast-lined vessels. Yellow arrowheads indicate fetal red blood cells within fetal capillaries. Dotted white line demarcates the decidua from the junctional zone. Scale bars = 50 μm. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test. Data are shown as means ± SD.
-
Figure 2—source data 1
Excel file containing quantification for smooth muscle cells per spiral artery, spiral artery diameter, and trophoblast-endothelial cell displacement in Figure 2.
- https://cdn.elifesciences.org/articles/77241/elife-77241-fig2-data1-v2.xlsx
Loss of trophoblast VE-cadherin blocks displacement of SA ECs and SA remodeling
A critical early step in establishing maternal circulation to the placenta is trophoblast invasion of the decidua and its SAs. The observation that there were fewer VE-cadherin-deficient trophoblasts adjacent to SAs and decreased maternal blood within the labyrinth suggested that trophoblast VE-cadherin may play a requisite role in SA remodeling. Since loss of vascular smooth muscle is a key step in SA remodeling, we stained control and knockout placentas for alpha-smooth muscle actin (αSMA). SAs in Cdh5 knockout placentas exhibited persistent vascular smooth muscle coverage and reduced SA diameter compared to control placentas (white arrowheads, Figure 2A–C). These studies reveal that loss of trophoblast VE-cadherin disrupts SA remodeling, which likely contributes to reduced maternal blood within the placenta.
During the process of SA remodeling, invasive trophoblasts displace the endothelial layer of SAs to direct maternal blood flow through trophoblast-lined sinuses into the labyrinth. In vitro studies have suggested that trophoblast expression of VE-cadherin may enable these cells to adhere to SA ECs (Bulla et al., 2005). The finding that trophoblast Cdh5 knockout placentas fail to remodel SAs suggested that there may be defects in trophoblast-SA interactions. To address the role of VE-cadherin at the site of trophoblast-SA connection, we first sought to image the site at which trophoblasts connect to SAs. Close inspection of this region in control placentas revealed a clear demarcation from luminal Endomucin+ SA endothelium to luminal CK8+ trophoblasts (white arrowheads, Figure 2D and E). In contrast, we found that SAs in VE-cadherin knockout placentas maintained a layer of intact ECs despite being surrounded by trophoblasts (white arrowheads, Figure 2D and E). Quantification of the percent of trophoblasts immediately in contact with the lumen demonstrated that knockout placentas had a lower percentage of trophoblasts and higher percentage of ECs covering the lumen compared to controls (Figure 2F). To better appreciate differences in placental architecture, we additionally performed whole-mount immunofluorescence of thick placental sections. Consistent with our data above, Cdh5 knockout placentas maintained a layer of ECs in SAs surrounded by trophoblasts and had smaller lumens (Figure 2G). Fewer maternal RBCs were seen in trophoblast-lined sinuses in the labyrinth of Cdh5 knockout placentas, but we found no differences in the fetal capillary plexus (Figure 2H). Together, these data suggest that VE-cadherin is required for trophoblast displacement of SA ECs during endovascular invasion for efficient maternal-fetal circulatory connection.
Defective trophoblast invasion and SA remodeling cause placental insufficiency and fetal distress
The finding that Cdh5 knockout placentas have less maternal blood and are associated with mid-gestation embryonic lethality suggested that failed SA remodeling restricts maternal blood flow into the placenta, thus causing fetal demise. Human placental insufficiency is typically assessed with ultrasound measurements of placental hemodynamics and fetal heart rate, a readout for overall fetal health. We therefore utilized Doppler ultrasound to measure peak systolic (PSV) and end diastolic velocities (EDV) in the umbilical arteries (Figure 3A) and calculated resistance and pulsatility indices (RI and PI) and fetal heart rates to assess placental vascular resistance and fetal wellbeing in individual concepti. Elevated RI and PI values are clinical indicators of placental insufficiency in humans and are associated with conditions such as preeclampsia and fetal growth restriction. Fetal heart rate is used as a clinical parameter for fetal wellbeing, with fetal bradycardia indicative of fetal distress. While control embryos had RIs, PIs, and fetal heart rates within range of previously published values (Galaz et al., 2020), we found that trophoblast Cdh5 knockout embryos exhibited significantly increased RIs and PIs (Figure 3B–D) and significantly reduced fetal heart rates (Figure 3B and E), consistent with placental insufficiency and fetal distress. Knockout embryos also exhibited reversal of end-diastolic flow as shown by the directional change of velocity from negative (peak systole) to positive (end of diastole) (Figure 3B), indicative of high vascular resistance. These hemodynamic data demonstrate placental insufficiency that contributes to fetal growth restriction and fetal demise following loss of trophoblast invasion and SA remodeling.
Figure 3
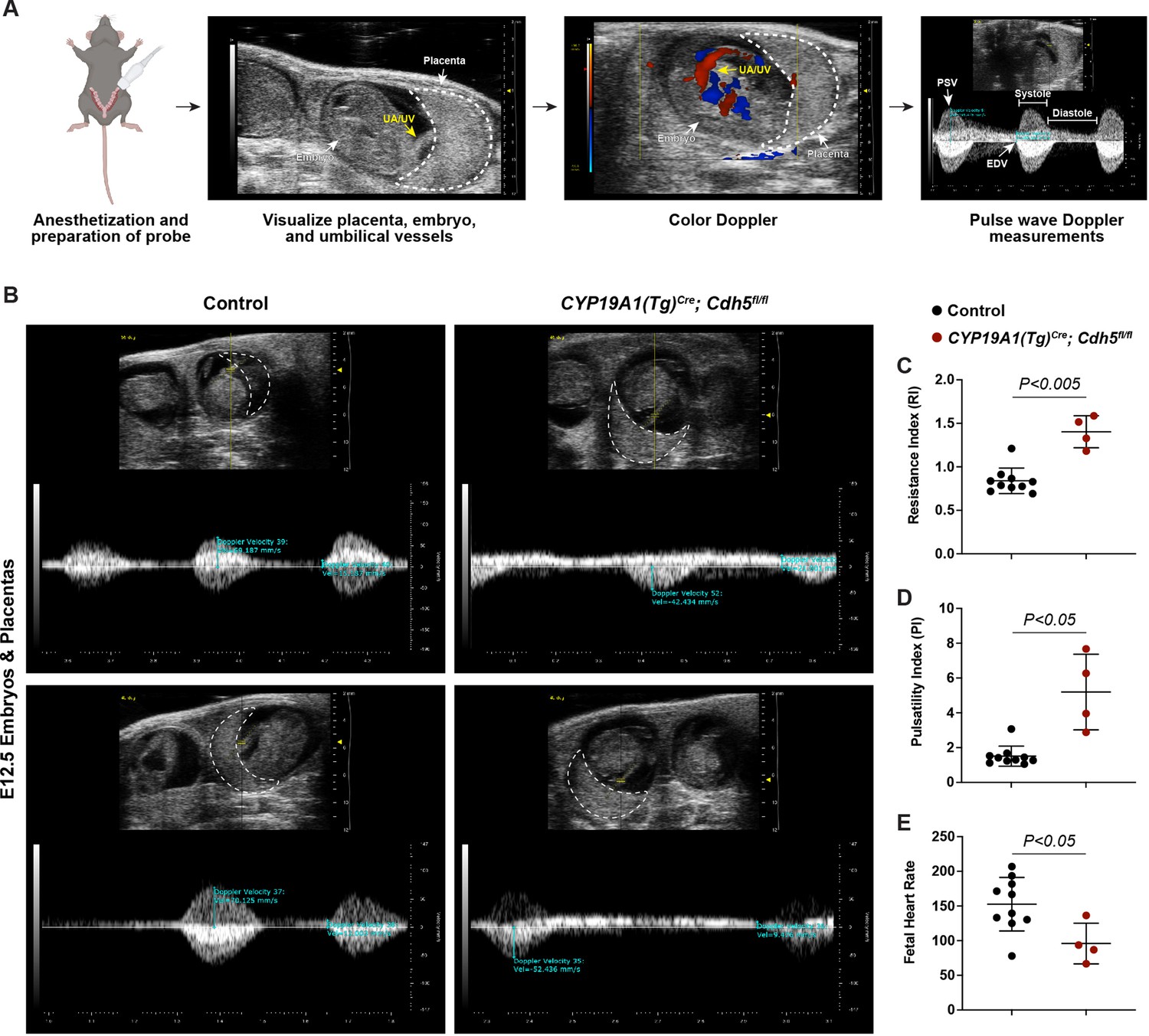
Spiral artery remodeling defects result in placental insufficiency and fetal distress.
(A) Schematic of workflow for Doppler ultrasound of pregnant dams. The embryo, UA/UV (yellow arrow), and placenta (dotted white outline) are labeled. (B) Representative umbilical artery Doppler waveforms from two Control and two CYP19A1(Tg)Cre; Cdh5fl/fl placentas. The placenta is outlined in a dotted white line. Reversal of end-diastolic flow is evident by the change of directional velocity at the end of diastole compared to peak systole (i.e., negative to positive velocity). (C–E) Quantification of resistance index (RI), pulsatility index (PI), and fetal heart rate. PSV (peak systolic velocity), EDV (end diastolic velocity), UA/UV (umbilical artery/umbilical vein). Note that red/blue colors in color Doppler images do not indicate UA/UV, which can only be differentiated based on the Doppler waveform. Control n = 10, CYP19A1(Tg)Cre; Cdh5fl/fl n = 4. Statistical analysis was performed using two-tailed, unpaired Welch’s t-test. Data are shown as means ± SD.
-
Figure 3—source data 1
Excel file containing quantification for ultrasound studies (resistance index, pulsatility index, fetal heart rate) in Figure 3.
- https://cdn.elifesciences.org/articles/77241/elife-77241-fig3-data1-v2.xlsx
Loss of trophoblast VE-cadherin alters decidual ECM remodeling and uterine natural killer cell clearance
Since trophoblast invasion occurs in conjunction with decidual changes, we hypothesized that failed trophoblast migration might affect other placental processes involved in that process. To identify such effects, we performed bulk RNA-sequencing (RNA-seq) on deciduas of E12.5 knockout and control placentas (Figure 4A). Analysis of the top 100 differentially expressed genes showed that genes highly expressed in invasive trophoblasts (Prl4a1, Pla2g4f, Pla2g4d, Nos1, Ncam1, Aldh1a3, Ascl2, Car2, Tfap2c) (Nelson et al., 2016; Müller et al., 1999; Simmons et al., 2008; Marsh and Blelloch, 2020; Outhwaite et al., 2015; Varberg et al., 2021; Bogutz et al., 2018; Sharma et al., 2016) were downregulated in knockout placentas (Figure 4B), consistent with reduced trophoblasts present in the decidua. Genes previously associated with trophoblast invasion (Gabrp, Mmp15) (Lu et al., 2016; Majali-Martinez et al., 2020) and differentiation (Cdx2) (Ralston and Rossant, 2008) were also differentially expressed, as were multiple genes related to defective decidualization (Ccl28, Slc27a2, Klk1, Csf1, Tmem132e, Ermap, Pappa2, Tmc5) (Woods et al., 2017; Goolam et al., 2020; Sun et al., 2013; Figure 4B). Thus RNA-seq data are consistent with defects in trophoblast invasion and suggest the presence of non-cell autonomous effects in the decidua that may contribute to a preeclamptic-like phenotype.
Figure 4 with 2 supplements see all
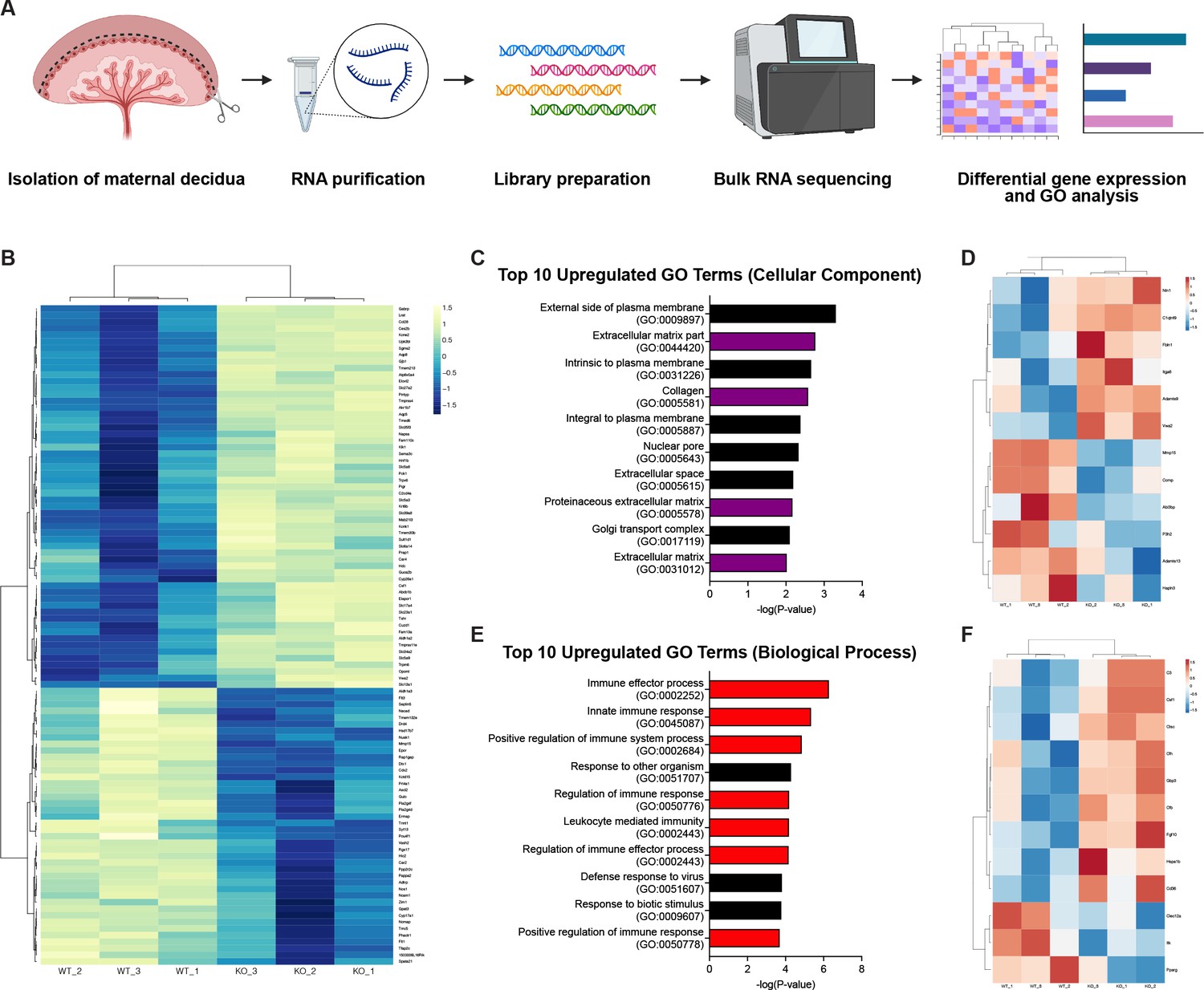
RNA-sequencing reveals defects in the decidual extracellular matrix and immune microenvironment.
(A) Schematic of bulk RNA-sequencing (RNA-seq) workflow on deciduas from Control (WT) and CYP19A1(Tg)Cre; Cdh5fl/fl (KO) E12.5 placentas. (B) Heatmap of the top 100 differentially regulated genes shown by z-score (n = 3 biological replicates). (C) Gene ontology (GO) term analysis of top 10 upregulated cellular components in CYP19A1(Tg)Cre; Cdh5fl/fl placentas. Purple bars indicate GO terms related to the extracellular matrix. (D) Heatmap of significantly differentially expressed genes from GO terms related to the extracellular matrix. (E) GO term analysis of top 10 upregulate biological processes in CYP19A1(Tg)Cre; Cdh5fl/fl placentas. Red bars indicate GO terms related to immune processes. (F) Heatmap of significantly differentially expressed genes from GO terms related to immune processes.
We next analyzed gene ontology (GO) related to cellular components and biological processes. We found that four of the top ten upregulated cellular component GO terms in knockout placentas were related to the ECM (Figure 4C). Many of the significantly upregulated genes were secreted ECM proteins (Vwa2, Ntn1, Fbln1) (Figure 4D). Several extracellular proteases previously linked to abnormal human pregnancies were also differentially expressed. These included increased expression of Adamts9 (associated with preterm birth; Li et al., 2020), reduced Adamts13 (associated with preeclampsia; Aref and Goda, 2013; Stepanian et al., 2011), and reduced Mmp15 (associated with reduced trophoblast invasion; Majali-Martinez et al., 2020). GO analysis of biological processes revealed that seven of the top ten upregulated GO terms were related to the immune system and immune activation, specifically the innate immune response (Figure 4E). Many of the significantly upregulated genes were involved in the complement factor pathway (C3, Cfb, Cfh) (Figure 4F). Thus GO analysis suggests that defects in ECM remodeling and changes in immune cells may contribute to the placental defects associated with reduced trophoblast invasion of the decidua.
We next aimed to directly assess the impact of trophoblast-specific deletion of VE-cadherin on (1) ECM remodeling and (2) immune cells. Our RNA-seq data showed a >2.5-fold decrease in Mmp15 (Figure 4D and Figure 4—figure supplement 1A). MMP15 (also known as MT2-MMP) is a membrane metalloprotease that is expressed in invasive trophoblasts in first trimester human placentas that promotes trophoblast invasion and degrades laminin as gestation progresses (Majali-Martinez et al., 2020; Pollheimer et al., 2014; Majali-Martinez et al., 2016; Turpeenniemi-Hujanen et al., 1995). Examination of MMP15 expression and its target laminin using immunofluorescence revealed increased laminin in the decidual stroma of Cdh5 knockout placentas but no differences in MMP15 expression in trophoblasts (Figure 4—figure supplement 1B, C). Additionally, we evaluated vinculin, a focal adhesion protein that regulates cell-matrix adhesion and associates with VE-cadherin (Huveneers et al., 2012). Vinculin is required for cell polarization and invasion (Thievessen et al., 2015; Mierke et al., 2010), and vinculin levels were indeed decreased in invasive trophoblasts of Cdh5 knockout placentas (Figure 4—figure supplement 1D). Together, these results suggest that VE-cadherin cell autonomously controls focal adhesions but not MMP activity and that persistent laminin in the decidua of Cdh5 knockout placentas is likely a consequence of fewer MMP15-expressing trophoblasts.
Since uterine natural killer (uNK) cells are the most abundant innate immune cell in the decidua, we stained placenta sections with the uNK cell-specific marker DBA. We found markedly increased uNK cells in knockout placentas compared to controls (Figure 4—figure supplement 2A, B). In order to determine the cause of increased uNK cells, we stained for the apoptosis marker cleaved caspase-3 and found numerous apoptotic uNK cells in control placentas; however almost no uNK cells in knockout placentas were positive for cleaved caspase-3 (Figure 4—figure supplement 2A, C). Lastly, we also found multiple significantly upregulated NK cell-related GO terms reflecting increased NK cell activity (Figure 4—figure supplement 2D). Together, these results indicate that loss of trophoblast invasion has non-cell autonomous effects that impact that maternal microenvironment of the placenta.
Discussion
The invasion of the maternal decidua by trophoblasts and their fusion to maternal SAs is a critical step in establishing placental circulation. However, the mechanisms by which trophoblast migration and endovascular invasion are accomplished remain largely unknown. Trophoblasts express endothelial molecular and genetic programs during invasion of SAs (Soares et al., 2018). However, the function of specific endothelial genes in trophoblasts has not been functionally assessed in vivo, and this model remains untested. In blood and lymphatic ECs, VE-cadherin is used to maintain vascular integrity (Corada et al., 1999; Crosby et al., 2005; Carmeliet et al., 1999), restrict endothelial migration (Crosby et al., 2005; Hägerling et al., 2018), and regulate angiogenic growth (Gaengel et al., 2012). While it is an attractive concept that trophoblasts may form vascular sinuses using similar genetic programs, the findings that loss of VE-cadherin decreases trophoblast cell migration and prevents SA remodeling suggest that trophoblasts utilize VE-cadherin in a manner distinct from ECs. Our work characterizing mechanisms of endovascular invasion in the placenta suggests that the use of endothelial proteins by trophoblasts may be relatively specific to their role in the placenta and not a simple reflection of vascular EC function.
VE-cadherin is primarily an adhesive receptor that acts in a homotypic manner to establish strong EC-cell junctions. Evidently, VE-cadherin is required in trophoblasts to invade the maternal decidua and remodel the maternal microenvironment. Our findings that loss of VE-cadherin decreases vinculin but not MMP15 in trophoblasts suggest that VE-cadherin mainly regulates cell invasion and that ECM remodeling defects are likely secondary consequences of decreased trophoblast invasion. A second interesting aspect of Cdh5 knockout placentas is persistent innate immune cells within the decidua. uNK cells (called decidual NK cells or dNK cells in humans) are present in the mouse decidua at E6.5, prior to formation and invasion of trophoblasts, and decline in number beginning at E12.5 (Rajagopalan, 2014; Sojka et al., 2018). uNK cells also secrete factors such as VEGF-C that promote SA remodeling (Pawlak et al., 2019). Significantly, increased dNK cells is also characteristic of preeclamptic placentas (Zhang et al., 2019), similar to our mouse model. Our findings raise the possibility that trophoblast migration into the decidua may coordinate decidual matrix and immune changes that promote SA remodeling.
In humans, defective SA remodeling and shallow trophoblast invasion are hallmarks of preeclampsia. Preeclampsia is a complex and heterogeneous disease with maternal and fetal contributions to its pathogenesis, and many in vitro models fail to fully recapitulate many aspects of its pathophysiology. Most rodent models of preeclampsia utilize maternal genetic or pharmacological perturbations (Gatford et al., 2020; Marshall et al., 2018), and there have been few in vivo models in which preeclamptic features are recapitulated with fetal modulation of trophoblasts. Previous studies of human placentas showed that invasive trophoblasts in severely preeclamptic placentas exhibit reduced expression of VE-cadherin (Zhou et al., 1997a) however whether this is a cause or consequence of placental dysfunction has been unclear. Our mouse model utilizing trophoblast-specific knockout of VE-cadherin exhibits many histopathological and clinical features of preeclampsia and suggests that loss of VE-cadherin in trophoblasts may be a primary contributor to preeclampsia pathogenesis. Additionally, we observe secondary defects in organogenesis and vascular development in the embryo (Perez-Garcia et al., 2018), which has also been linked to placentation defects. Interestingly, VEGF-A is known to induce VE-cadherin expression in cultured trophoblasts (Chang et al., 2005) and may therefore be a useful strategy in treating preeclampsia. Trophoblast-specific loss of VE-cadherin may serve as a useful model for studying fetal contributions to preeclampsia.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (Mus musculcus) | CYP19A1(Tg)-Cre | Wenzel and Leone, 2007 | ||
| Genetic reagent (Mus musculcus) | Cdh5 flox | Yang et al., 2019 | ||
| Antibody | Anti-Endomucin (goat polyclonal) | R&D | AF4666 | IF(1:400) |
| Antibody | Anti-Endomucin (rat monoclonal) | Abcam | ab106100 | IF(1:300) |
| Antibody | Anti-TER119 (rat monoclonal) | Abcam | ab91113 | IF(1:300) |
| Antibody | Anti-VE-cadherin (goat polycloncal) | R&D | AF1002 | IF(1:200) |
| Antibody | Anti-CK8 (rabbit monoclonal) | Abcam | ab53280 | IF(1:300) |
| Antibody | Anti-CK8 (rat monoclonal) | DSHB | TROMA-1 | IF(1:400) |
| Antibody | Anti-αSMA-Cy3 (mouse monoclonal) | Sigma | C6198 | IF(1:300) |
| Antibody | Anti-Cleaved Caspase-3 (rabbit polyclonal) | Millipore Sigma | AB3623 | IF(1:100) |
| Antibody | Anti-MMP15 (rabbit polyclonal) | Thermo Fisher Scientific | PA5-13184 | IF(1:200) |
| Antibody | Anti-Laminin (rabbit polyclonal) | Sigma | L9393 | IF(1:200) |
| Antibody | Anti-Vimentin (goat polyclonal) | R&D | AF2105 | IF(1:300) |
| Antibody | Anti-Vinculin (mouse monoclonal) | Sigma | V9131 | IF(1:200) |
| Commercial assay or kit | Direct-zol RNA Miniprep Kits | Zymo Research | R2053 | |
| Software, algorithm | ImageJ | NIH, Bethesda, MD, USA | RRID:SCR_003070 | |
| Software, algorithm | GraphPad Prism | GraphPad | RRID:SCR_002798 | |
| Software, algorithm | Picard v2.17.11 | Picard | RRID:SCR_006525 | |
| Other | DBA-Biotin | Vector Labs | B-1035 | (1:500) |
Generation of mutant mice
Request a detailed protocolCYP19A1(Tg)-Cre mice have been previously described (Wenzel and Leone, 2007) in which the transgene relies on Cre expression under a 501 bp region with the first exon of human CYP19A1 containing regulatory elements for trophoblast-specific expression, as CYP19A1 is not endogenously expressed in trophoblasts. VE-cadherin (Cdh5) floxed mice have been previously described (Yang et al., 2019) and were generated with LoxP sites flanking exons 3 and 4. Mice were bred according to standard protocols and maintained on a mixed background. Male Cdh5fl/fl mice were mated to female CYP19A1(Tg)Cre; Cdh5fl/+ mice due to the influence of parental inheritance on Cre expression, with maternal inheritance providing the most robust and consistent expression (Wenzel and Leone, 2007). Mating pairs were set up in the afternoon and vaginal plugs checked in the morning. Presence of a vaginal plug indicated embryonic day (E)0.5. Cre-negative (Cdh5fl/+ and Cdh5fl/fl) and Cre-positive heterozygous (CYP19A1(Tg)Cre; Cdh5fl/+) littermates were used as controls. All procedures were conducted using an approved animal protocol (806811) in accordance with the University of Pennsylvania Institutional Animal Care and Use Committee.
Intrauterine Doppler ultrasound
Request a detailed protocolIn utero Doppler ultrasound was performed by a trained technician using the VEVO2100 Ultrasound System equipped with the MS-400 transducer (30 MHz). E12.5 pregnant mice were lightly anesthetized using 2% isoflurane. Hair was removed from the abdomen using chemical hair remover (Nair), and the animals were placed on a warming pad. Maternal heart rate and temperature were continuously monitored and consistently within 400–500 bpm and 37°C. Ultrasound gel was applied to the abdomen and the transducer applied to visualize embryos and placentas using the maternal bladder as an anatomical landmark. Color Doppler was used to visualize the umbilical vessels, and pulse wave (angle of insonation <60°) measurements were made at the point where the umbilical artery inserts into the placenta. From the Doppler waveforms, PSV and EDV were measured and used to calculate resistance index [RI = 1 – PSV/EDV] and pulsatility index [PI = (PSV – EDV)/mean velocity, where mean velocity = (PSV +EDV)/2]. Heart rate was calculated as beats per minute by dividing 60 s by the systolic + diastolic time.
Histology and immunofluorescence staining and analysis
View detailed protocolWhole mouse embryos or placentas were collected and fixed in 4% paraformaldehyde (PFA) overnight at 4°C prior to dehydration in alcohol and paraffin embedding. Tissue sections underwent to dewaxing and rehydration through xylene and ethanol treatment and were then subject to H&E staining or processed for immunofluorescence. For immunodetection, 10 mM citrate buffer (pH 6) was used for antigen retrieval, and sections were blocked with 10% donkey serum in 1% BSA prior to primary antibody treatment overnight at 4°C. A list of antibodies can be found below. Fluorescence-conjugated Alexa Fluor secondary antibodies were used (1:500, Invitrogen) according to the primary antibody species and counterstained with DAPI (1:1000). Sections or tissues were mounted on slides with ProLong Gold Antifade reagent. Signals were detected and images collected using a Zeiss LSM 880 confocal microscope and Zeiss Axio Observer 7 widefield microscope. Images were visualized using ImageJ/FIJI software (NIH).
Whole-mount immunofluorescence
Request a detailed protocolWhole mouse placentas were collected and fixed in 4% PFA overnight at 4°C and then placed in 1× PBS. Placentas were embedded in 3% low-melt agarose and cut into 200 μm thick sections using a vibratome. Sections were permeabilized with 0.2% Triton X-100 and blocked with 10% donkey serum in 1% BSA prior to primary antibody treatment overnight at 4°C. Fluorescence-conjugated Alexa Fluor secondary antibodies were used (1:500, Invitrogen) according to the primary antibody species and counterstained with DAPI (1:1000). Sections were mounted on a glass slide in a silicone isolator and filled with ProLong Gold Antifade reagent. Signals were detected and images collected using a Zeiss Axio Observer 7 widefield microscope with the Apotome 3 attachment for optical sectioning. Raw images were deconvoluted using ZEN Blue. Maximum intensity projections were generated using ImageJ/FIJI software (NIH).
Quantification of immunofluorescence images
Request a detailed protocolNumber of CK8+ trophoblasts were manually counted and divided by total decidual area. Trophoblast invasion depth was measured as the distance the farthest CK8+ trophoblast was found in the decidua relative to the junctional zone. Percent labyrinth EC coverage was measured by quantifying Endomucin-positive area as a percentage of total labyrinth area. Number of αSMA+ smooth muscle cells were manually counted and divided by number of SAs. SA diameter was manually measured using only circular SAs, taking the average of the long- and short-axis diameters, and averaging at least five SAs per section per placenta. Trophoblast-endothelial displacement was quantified by measuring the circumference of the vessel lumen and then measuring the length of CK8+ trophoblasts in contact with the lumen. Percent displacement was calculated by dividing the trophoblast length by the lumen circumference. VE-cadherin deletion was quantified by measuring VE-cadherin+CK8+ area (determined by Colocalization Threshold plugin in FIJI) and calculating it as a percent of CK8+ area. The Colocalization Threshold plugin was also used to generate colocalization images in Figure 1—figure supplement 3C. Laminin and vinculin mean fluorescence intensity was calculated by dividing total fluorescence intensity by area. All images were analyzed using ImageJ/FIJI software.
Bulk RNA-seq
Request a detailed protocolTotal RNA was extracted from E12.5 deciduas (n = 3 per genotype) using Direct-zol RNA miniprep kit (Zymo Research). Quality assessment, cDNA library synthesis, and sequencing using Illumina HiSeq with a 2 × 150 configuration were conducted through GeneWiz. Fastq files were assessed for quality control using the FastQC program (v0.11.7). They were then aligned against the mouse reference genome (mm39) using the STAR aligner (v2.7.8a) (Dobin et al., 2013). Duplicate reads were flagged using the MarkDuplicates program from Picard tools (v2.17.11) (http://broadinstitute.github.io/picard/). Per gene read counts for Ensembl (GRCm39) gene annotations were computed using the R package Rsubread (Liao et al., 2019) and duplicate reads were removed. Gene counts were normalized as counts per million (CPM) using the R package edgeR (Robinson et al., 2010) and genes with CPM < 1 in 25% of samples were filtered out. The data was transformed using the VOOM function from the limma R package (Law et al., 2014). Differential gene expression was performed as a paired analysis using limma. p-Values were adjusted for multiple comparisons using Benjamini-Hochberg procedure. Genes with adjusted p-values less than 0.05 and an absolute log2-fold change >1 were considered significantly differentially expressed genes. The RNA-seq data set has been deposited in the NCBI GEO under accession ID number GSE189408.
Statistical analysis
Request a detailed protocolAll data are reported as means with n ≥ 3 independent experiments or mice, and error bars represent standard deviation. Each data point in the figures represents one individual placental or embryo. The explicit number of samples is indicated in the figure legends. No explicit power analyses were used to predetermine sample size, and no randomization was used. No samples were excluded for analysis. Statistical significance was determined using Welch’s t-test. Differences between means were considered significant at p < 0.05. Significant differences in expected genotypes was calculated using two-tailed Fisher’s exact test and considered statistically significant at p < 0.05.
Data availability
Source Data files have been included for Figure 1, Figure 1—figure supplement 1, Figure 1—figure supplement 2, Figure 1—figure supplement 3, Figure 2, Figure 3, Figure 4—figure supplement 1, and Figure 4—figure supplement 2. All reagents have been listed in the Methods section in this paper. The RNA-seq data set has been deposited in the NCBI GEO under accession ID number GSE189408. Investigators interested in the animals used in this study should contact Dr. Jeremy Veenstra-Vanderweele (Columbia University), Dr. Gustsavo Leone (Medical University of South Carolina), and Dr. Joshua Scallan (University of South Florida).
-
NCBI Gene Expression OmnibusID GSE189408. VE-cadherin is essential for trophoblast migration and endovascular invasion.
References
-
Increased VWF antigen levels and decreased ADAMTS13 activity in preeclampsiaHematology (Amsterdam, Netherlands) 18:237–241.https://doi.org/10.1179/1607845412Y.0000000070
-
VE-cadherin is a critical molecule for trophoblast-endothelial cell interaction in decidual spiral arteriesExperimental Cell Research 303:101–113.https://doi.org/10.1016/j.yexcr.2004.09.015
-
Transforming growth factor-β1 inhibits trophoblast cell invasion by inducing Snail-mediated down-regulation of vascular endothelial-cadherin proteinThe Journal of Biological Chemistry 288:33181–33192.https://doi.org/10.1074/jbc.M113.488866
-
STAR: ultrafast universal RNA-seq alignerBioinformatics (Oxford, England) 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Animal Models of Preeclampsia: Causes, Consequences, and InterventionsHypertension (Dallas, Tex 75:1363–1381.https://doi.org/10.1161/HYPERTENSIONAHA.119.14598
-
Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodelingThe Journal of Cell Biology 196:641–652.https://doi.org/10.1083/jcb.201108120
-
GABA A receptor π subunit promotes apoptosis of HTR-8/SVneo trophoblastic cells: Implications in preeclampsiaInternational Journal of Molecular Medicine 38:105–112.https://doi.org/10.3892/ijmm.2016.2608
-
Placental membrane-type metalloproteinases (MT-MMPs): Key players in pregnancyCell Adhesion & Migration 10:136–146.https://doi.org/10.1080/19336918.2015.1110671
-
Animal models of preeclampsia: translational failings and whyAmerican Journal of Physiology. Regulatory, Integrative and Comparative Physiology 314:R499–R508.https://doi.org/10.1152/ajpregu.00355.2017
-
Vinculin facilitates cell invasion into three-dimensional collagen matricesThe Journal of Biological Chemistry 285:13121–13130.https://doi.org/10.1074/jbc.M109.087171
-
Lymphatic mimicry in maternal endothelial cells promotes placental spiral artery remodelingThe Journal of Clinical Investigation 129:4912–4921.https://doi.org/10.1172/JCI120446
-
HLA-G-mediated NK cell senescence promotes vascular remodeling: implications for reproductionCellular & Molecular Immunology 11:460–466.https://doi.org/10.1038/cmi.2014.53
-
The placenta in preeclampsiaPregnancy Hypertension 2:72–83.https://doi.org/10.1016/j.preghy.2012.01.001
-
edgeR: A Bioconductor package for differential expression analysis of digital gene expression dataBioinformatics (Oxford, England) 26:139–140.https://doi.org/10.1093/bioinformatics/btp616
-
Tpbpa-Cre-mediated deletion of TFAP2C leads to deregulation of Cdkn1a, Akt1 and the ERK pathway, causing placental growth arrestDevelopment (Cambridge, England) 143:787–798.https://doi.org/10.1242/dev.128553
-
Adaptive mechanisms controlling uterine spiral artery remodeling during the establishment of pregnancyThe International Journal of Developmental Biology 58:247–259.https://doi.org/10.1387/ijdb.140083ms
-
Hemochorial placentation: development, function, and adaptationsBiology of Reproduction 99:196–211.https://doi.org/10.1093/biolre/ioy049
-
Cutting Edge: Local Proliferation of Uterine Tissue-Resident NK Cells during Decidualization in MiceJournal of Immunology (Baltimore, Md 201:2551–2556.https://doi.org/10.4049/jimmunol.1800651
-
Von Willebrand factor and ADAMTS13: A candidate couple for preeclampsia pathophysiologyArteriosclerosis, Thrombosis, and Vascular Biology 31:1703–1709.https://doi.org/10.1161/ATVBAHA.111.223610
-
Chemokine CCL28 induces apoptosis of decidual stromal cells via binding CCR3/CCR10 in human spontaneous abortionMolecular Human Reproduction 19:676–686.https://doi.org/10.1093/molehr/gat038
-
Function and control of human invasive trophoblast subtypes: Intrinsic vs. maternal controlCell Adhesion & Migration 10:154–162.https://doi.org/10.1080/19336918.2015.1089376
-
Expression of Cre recombinase in early diploid trophoblast cells of the mouse placentaGenesis (New York, N.Y 45:129–134.https://doi.org/10.1002/dvg.20276
-
Preeclampsia Is Associated with Failure of Human Cytotrophoblasts to Mimic a Vascular Adhesion PhenotypeThe Journal of Clinical Investigation 99:2152–2164.https://doi.org/10.1172/JCI119388
-
Human cytotrophoblasts adopt A vascular phenotype as they differentiate: A strategy for successful endovascular invasion?The Journal of Clinical Investigation 99:2139–2151.https://doi.org/10.1172/JCI119387
Article and author information
Author details
Funding
National Institutes of Health (T32 HL007439)
- Derek C Sung
National Institutes of Health (F30 HL158014)
- Derek C Sung
American Heart Association (Postdoctoral Fellowship 35200213)
- Xiaowen Chen
National Institutes of Health (T32 HL007971)
- Thomas C Stevenson Keller
American Heart Association (Postdoctoral fellowship 836238)
- Thomas C Stevenson Keller
National Institutes of Health (HL142905)
- Joshua P Scallan
National Institutes of Health (HL145397)
- Ying Yang
National Institutes of Health (HL142976)
- Mark L Kahn
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank members of the Kahn laboratory and Mainigi laboratory for thoughtful discussions during the course of these studies, the CDB Microscopy Core for support with microscopy, and the Small Animal Imaging Facility Core for their support with ultrasound imaging. We thank Dr Jeremy Veenstra-Vanderweele (Columbia University) and Dr Gustsavo Leone (Medical University of South Carolina) for kindly providing the CYP19A1(Tg)Cre mice. This work was supported by NIH grants T32 HL007439 and F30 HL158014 (to DCS), American Heart Association Postdoctoral Fellowship No. 35200213 (to XC), NIH grant T32 HL007971 and American Heart Association Postdoctoral Fellowship No. 836,238 (to TCSK), NIH R01 grant HL142905 (to JPS), NIH R01 grant HL145397 (to YY), and NIH R01 grant HL142976 (to MLK).
Ethics
All procedures were conducted using an approved animal protocol (806811) in accordance with the University of Pennsylvania Institutional Animal Care and Use Committee.
Copyright
© 2022, Sung et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,727
- views
-
- 557
- downloads
-
- 24
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
VE-cadherin enables trophoblast endovascular invasion and spiral artery remodeling during placental development
eLife 11:e77241.
https://doi.org/10.7554/eLife.77241
Further reading
-
- Cancer Biology
- Developmental Biology
Missense ‘hotspot’ mutations localized in six p53 codons account for 20% of TP53 mutations in human cancers. Hotspot p53 mutants have lost the tumor suppressive functions of the wildtype protein, but whether and how they may gain additional functions promoting tumorigenesis remain controversial. Here, we generated Trp53Y217C, a mouse model of the human hotspot mutant TP53Y220C. DNA damage responses were lost in Trp53Y217C/Y217C (Trp53YC/YC) cells, and Trp53YC/YC fibroblasts exhibited increased chromosome instability compared to Trp53-/- cells. Furthermore, Trp53YC/YC male mice died earlier than Trp53-/- males, with more aggressive thymic lymphomas. This correlated with an increased expression of inflammation-related genes in Trp53YC/YC thymic cells compared to Trp53-/- cells. Surprisingly, we recovered only one Trp53YC/YC female for 22 Trp53YC/YC males at weaning, a skewed distribution explained by a high frequency of Trp53YC/YC female embryos with exencephaly and the death of most Trp53YC/YC female neonates. Strikingly, however, when we treated pregnant females with the anti-inflammatory drug supformin (LCC-12), we observed a fivefold increase in the proportion of viable Trp53YC/YC weaned females in their progeny. Together, these data suggest that the p53Y217C mutation not only abrogates wildtype p53 functions but also promotes inflammation, with oncogenic effects in males and teratogenic effects in females.
-
- Developmental Biology
Stem cell self-renewal often relies on asymmetric fate determination governed by niche signals and/or cell-intrinsic factors but how these regulatory mechanisms cooperate to promote asymmetric fate decision remains poorly understood. In adult Drosophila midgut, asymmetric Notch (N) signaling inhibits intestinal stem cell (ISC) self-renewal by promoting ISC differentiation into enteroblast (EB). We have previously shown that epithelium-derived Bone Morphogenetic Protein (BMP) promotes ISC self-renewal by antagonizing N pathway activity (Tian and Jiang, 2014). Here, we show that loss of BMP signaling results in ectopic N pathway activity even when the N ligand Delta (Dl) is depleted, and that the N inhibitor Numb acts in parallel with BMP signaling to ensure a robust ISC self-renewal program. Although Numb is asymmetrically segregated in about 80% of dividing ISCs, its activity is largely dispensable for ISC fate determination under normal homeostasis. However, Numb becomes crucial for ISC self-renewal when BMP signaling is compromised. Whereas neither Mad RNA interference nor its hypomorphic mutation led to ISC loss, inactivation of Numb in these backgrounds resulted in stem cell loss due to precocious ISC-to-EB differentiation. Furthermore, we find that numb mutations resulted in stem cell loss during midgut regeneration in response to epithelial damage that causes fluctuation in BMP pathway activity, suggesting that the asymmetrical segregation of Numb into the future ISC may provide a fail-save mechanism for ISC self-renewal by offsetting BMP pathway fluctuation, which is important for ISC maintenance in regenerative guts.




{kind=link}
{kind=link}
{kind=link}
{kind=link}