Protein Kinase A: Probing ligand selectivity in pathogens
Why does protein kinase A respond to purine nucleosides in certain pathogens, but not to the cyclic nucleotides that activate this kinase in most other organisms?
- Department of Chemistry and Chemical Biology, McMaster University, Canada
- Department of Chemistry and Chemical Biology, and the Department of Biochemistry and Biomedical Sciences, McMaster University, Canada
Protein kinase A (PKA) is involved in a wide range of intracellular processes within eukaryotic organisms, and it also has an important role in the life cycles of a number of pathogens that cause various deadly diseases. These include Trypanosoma brucei, which causes sleeping sickness (Bachmaier et al., 2019); Trypanosoma cruzi, which causes Chagas disease (Baker et al., 2021); and the parasites that cause leishmaniosis (Cayla et al., 2022). An ability to target PKA in these pathogens – which are collectively known as trypanosomatid pathogens – could lead to new treatments for these diseases, but only if this can be done without affecting PKA in the body of the person with the disease.
Such selective targeting and inhibition may be possible because, in most organisms, PKA is activated by a cyclic nucleotide called cAMP (Rinaldi et al., 2010; Haste et al., 2012; Kurokawa et al., 2011; Taylor et al., 2012; Khamina et al., 2022). However, PKA in T. brucei does not respond to cAMP, and is activated instead by purine nucleosides. These nucleosides are similar in structure to cyclic nucleotides, but lack a phosphate group (Figure 1A and B).
Figure 1
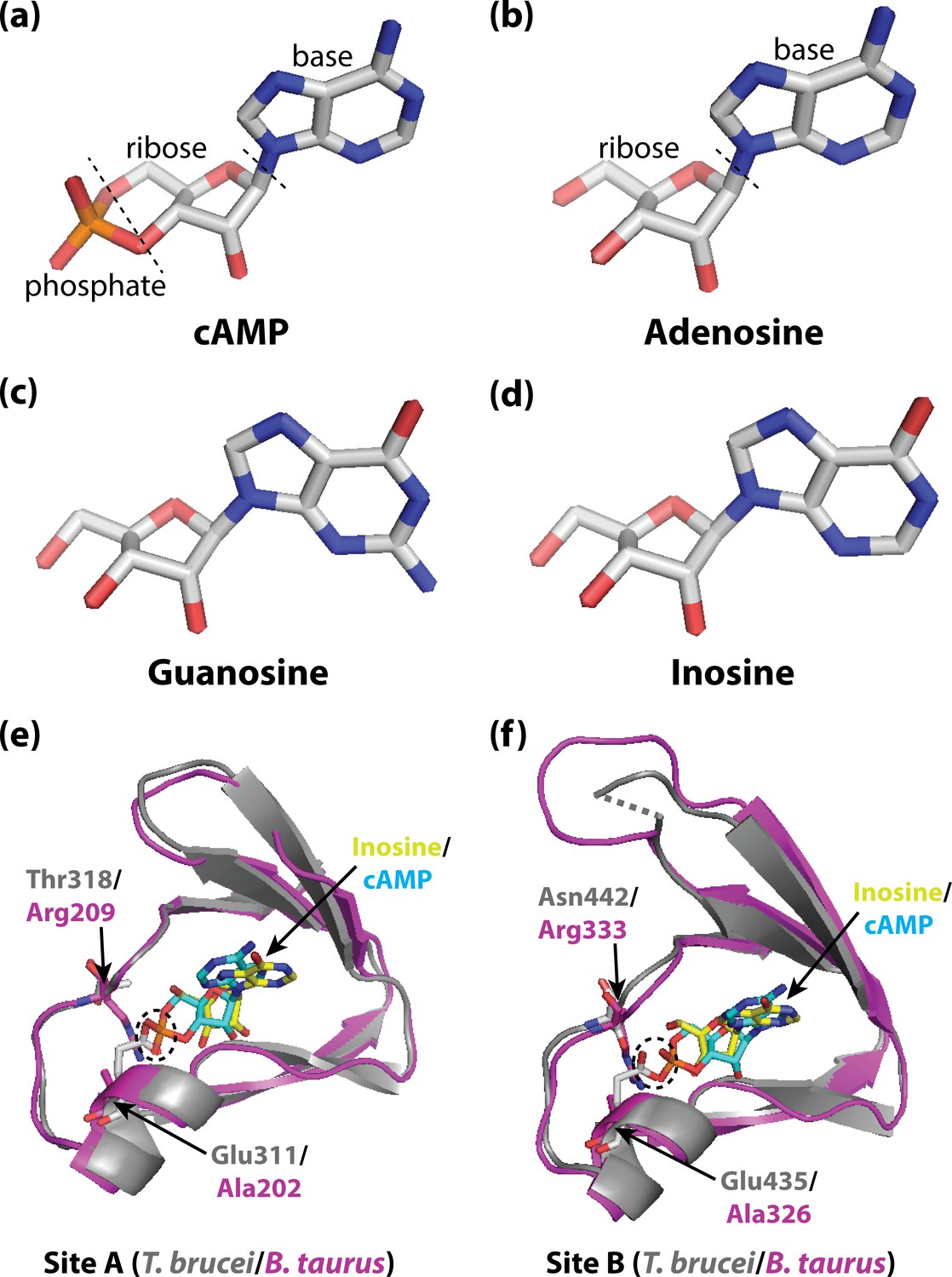
Activating PKA in mammals and trypanosomatid pathogens.
(A–D) Stick representations of the molecular structures of the cyclic nucleotide cAMP (A), and three purine nucleosides: adenosine (B), guanosine (C) and inosine (D). The main difference between these molecules is that cAMP contains a phosphate group which the purine nucleosides lack. In most organisms, an important kinase called PKA is activated when two cAMP molecules bind to two tandem binding sites in the regulatory subunit of the kinase (Su et al., 1995; Kim et al., 2007; Akimoto et al., 2015). In trypanosomatid pathogens, on the other hand, PKA is activated when purine nucleosides bind to these sites. (E) Ribbon/stick structures showing a purine nucleoside (inosine; yellow sticks) bound to T. brucei PKA at site A (grey ribbons), overlaid with cAMP (cyan sticks) bound to the same site for mammalian (Bos taurus) PKA (purple ribbons). (F) Ribbon/stick structures showing inosine and cAMP bound to PKA at site B. The amino acids that differ between mammalian and trypanosomatid PKA are labelled: Arg209 and Arg333 in mammalian PKA are replaced by Thr318 and Asn442 in trypanosomatid PKA, while Ala202 and Ala326 are replaced by Glu311 and Glu435. For clarity, other parts of the PKA structures have been omitted. The proposed clash between Glu311/Glu435 of trypanosomatid PKA and the phosphate group of cAMP that prevents cAMP from binding is also indicated (dashed ovals). PKA: protein kinase A; the protein structures used for PKA can be found at PDB ID 6FLO (T. brucei) and PDB ID 1RGS (B. taurus).
Now, in eLife, Michael Boshart (Ludwig-Maximilians-University Munich) and colleagues – including Veronica Ober, George Githure and Yuri Volpato Santos as joint first authors – report the results of experiments which shed light on the activation of PKA in T. brucei and other trypanosomatid pathogens, opening new opportunities to develop novel treatments for the diseases caused by such pathogens (Ober et al., 2023).
To start, Ober et al. assessed structure-activity relationships for kinase activation by various ligands, and found that three purine nucleosides – inosine, guanosine and adenosine (Figure 1B–D) – were direct activators of PKA in three trypanosomatid pathogens (T. brucei, T. cruzi and Leishmania donovani), with inosine being the most potent of the ligands. Moreover, no activation by cAMP was observed, in agreement with previous findings for T. brucei (Bachmaier et al., 2019). The results suggest that activation by purine nucleosides, and insensitivity to cAMP, are universal features of these pathogens.
Consistent with these results, when Ober et al. measured ligand binding affinities for T. brucei and L. donovani PKA, inosine exhibited the highest affinity, and there was no significant binding of cAMP. For human PKA, on the other hand, cAMP exhibited high-affinity binding, and there was no significant binding of the three nucleosides. Furthermore, analysis of PKA-bound ligands purified from T. brucei cells at various stages of their life cycle confirmed the presence of all three nucleosides – inosine, guanosine and adenosine – in vivo.
To explore these interactions in detail, Ober et al. – who are based at institutes across Germany, and also in Denmark – solved three-dimensional crystal structures of inosine bound to the regulatory subunits of PKA from both T. cruzi and T. brucei. From these structures, they identified key amino acid residues in the binding sites of PKA that interact with inosine, including interactions with the ribose moiety of inosine that were conserved in both structures.
Comparison of these structures with the structure of cAMP bound to mammalian PKA (Su et al., 1995) highlighted two key differences in each binding site: a conserved arginine residue in mammalian PKA (which interacts with the phosphate group of cAMP) is replaced with a polar amino acid in trypanosomatid PKA; and a conserved alanine residue is replaced with a conserved glutamate residue (which interacts with the ribose of the nucleosides; Figure 1E and F). Notably, the structure comparison suggested that the conserved glutamate residue in trypanosomatid PKA would clash with the negatively charged phosphate of bound cAMP, which would explain why cAMP does not bind and activate trypanosomatid PKA. Furthermore, when these residues were mutated to the corresponding residues found in mammalian PKA, cAMP was able to activate the trypanosomatid PKA.
The work of Ober et al. provides valuable insights into the activation of trypanosomatid PKA at the molecular level. In the future, it would be interesting to explore the complex formed by the regulatory and catalytic subunits of trypanosomatid PKA, and its nucleoside binding affinities, as this complex constitutes the resting state of PKA in other organisms (Kim et al., 2007). Furthermore, there remains the question of how the activation of trypanosomatid PKA by nucleosides is terminated in vivo, as the mechanism responsible is likely different from that found in human PKA (which relies on enzymes called phosphodiesterases).
The more we learn about the differences between trypanosomatid PKA and mammalian PKA, the better chances we have of being able to selectively target trypanosomatid PKA in medical treatments for sleeping sickness and other diseases caused by trypanosomatid pathogens.
References
-
Distinguishing functions of trypanosomatid protein kinasesTrends in Parasitology 38:950–961.https://doi.org/10.1016/j.pt.2022.08.009
-
Non-canonical allostery in cyclic nucleotide dependent kinasesJournal of Molecular Biology 434:167584.https://doi.org/10.1016/j.jmb.2022.167584
-
Assembly of allosteric macromolecular switches: lessons from PKANature Reviews Molecular Cell Biology 13:646–658.https://doi.org/10.1038/nrm3432
Article and author information
Author details
Publication history
Copyright
© 2023, VanSchouwen and Melacini
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 448
- views
-
- 48
- downloads
-
- 1
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Protein Kinase A: Probing ligand selectivity in pathogens
eLife 12:e94720.
https://doi.org/10.7554/eLife.94720
Further reading
-
- Biochemistry and Chemical Biology
- Structural Biology and Molecular Biophysics
The two identical motor domains (heads) of dimeric kinesin-1 move in a hand-over-hand process along a microtubule, coordinating their ATPase cycles such that each ATP hydrolysis is tightly coupled to a step and enabling the motor to take many steps without dissociating. The neck linker, a structural element that connects the two heads, has been shown to be essential for head–head coordination; however, which kinetic step(s) in the chemomechanical cycle is ‘gated’ by the neck linker remains unresolved. Here, we employed pre-steady-state kinetics and single-molecule assays to investigate how the neck-linker conformation affects kinesin’s motility cycle. We show that the backward-pointing configuration of the neck linker in the front kinesin head confers higher affinity for microtubule, but does not change ATP binding and dissociation rates. In contrast, the forward-pointing configuration of the neck linker in the rear kinesin head decreases the ATP dissociation rate but has little effect on microtubule dissociation. In combination, these conformation-specific effects of the neck linker favor ATP hydrolysis and dissociation of the rear head prior to microtubule detachment of the front head, thereby providing a kinetic explanation for the coordinated walking mechanism of dimeric kinesin.
-
- Biochemistry and Chemical Biology
- Computational and Systems Biology
The spike protein is essential to the SARS-CoV-2 virus life cycle, facilitating virus entry and mediating viral-host membrane fusion. The spike contains a fatty acid (FA) binding site between every two neighbouring receptor-binding domains. This site is coupled to key regions in the protein, but the impact of glycans on these allosteric effects has not been investigated. Using dynamical nonequilibrium molecular dynamics (D-NEMD) simulations, we explore the allosteric effects of the FA site in the fully glycosylated spike of the SARS-CoV-2 ancestral variant. Our results identify the allosteric networks connecting the FA site to functionally important regions in the protein, including the receptor-binding motif, an antigenic supersite in the N-terminal domain, the fusion peptide region, and another allosteric site known to bind heme and biliverdin. The networks identified here highlight the complexity of the allosteric modulation in this protein and reveal a striking and unexpected link between different allosteric sites. Comparison of the FA site connections from D-NEMD in the glycosylated and non-glycosylated spike revealed that glycans do not qualitatively change the internal allosteric pathways but can facilitate the transmission of the structural changes within and between subunits.




{kind=link}